
Abstract
Background
As clinical exome sequencing (CES) becomes more common, understanding which patients are most likely to benefit and in what manner is critical for the general pediatrics community to appreciate.
Methods
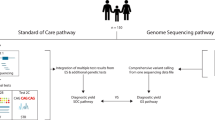
Five hundred and twenty-three patients referred to the Pediatric Genetics clinic at Michigan Medicine were systematically phenotyped by the presence or absence of abnormalities for 13 body/organ systems by a Clinical Genetics team. All patients then underwent CES.
Results
Overall, 30% of patients who underwent CES had an identified pathogenic mutation. The most common phenotypes were developmental delay (83%), neuromuscular system abnormalities (81%), and multiple congenital anomalies (42%). In all, 67% of patients had a variant of uncertain significance (VUS) or gene of uncertain significance (GUS); 23% had no variants reported. There was a significant difference in the average number of body systems affected among these groups (pathogenic 5.89, VUS 6.0, GUS 6.12, and no variant 4.6; P < 0.00001). Representative cases highlight four ways in which CES is changing clinical pediatric practice.
Conclusions
Patients with identified variants are enriched for multiple organ system involvement. Furthermore, our phenotyping provides broad insights into which patients are most likely to benefit from genetics referral and CES and how those results can help guide clinical practice more generally.
Similar content being viewed by others

Introduction
Despite the growing availability of genomic diagnostic tests, many children with suspected genetic diseases remain undiagnosed until adulthood.1 However, with the decreasing cost and increased availability of sequencing technologies, clinical exome sequencing (CES) is quickly becoming a leading modality in pediatric patients referred for clinical genetics evaluation.2,3,4 Early results from centers that have piloted the use of CES in pediatric genetic clinics have been promising, with several groups showing up to a 50% diagnosis rate,2,3,4,5 compared to non-CES-based genetic tests such as chromosomal microarray (CMA), which typically yields a diagnosis in only about 15–20% of patients.6 In addition, detailed cost analyses are beginning to show that, when used in appropriate patients, CES is cost-effective and is of greater diagnostic utility as compared to traditional chromosomal and microarray-based testing.7,8 However, while CES is quickly becoming a first test of choice for many genetic evaluations, it is most useful as a diagnostic tool when applied to the appropriate patient population.9 A comprehensive clinical assessment of patients to place genetic test results into clinical context, such as performed in a general genetics clinic, remains essential, as diagnosis of trinucleotide repeat disorders, mitochondrial diseases, copy number variants, and imprinting disorders require molecular testing methods other than CES.9
In an attempt to increase the diagnostic yield of CES, a number of approaches have been taken. Genetic information sharing platforms such as Matchmaker Exchange have been developed,10 which emphasizes comprehensive patient phenotyping with the use of standardized vocabulary such as from the Human Phenotype Ontology database.11 Many groups have also advocated for re-sequencing/re-analysis of patients with initially negative CES and a high suspicion of having a de novo variant based on expert clinical opinion.12,13 Furthermore, prior studies have shown a higher diagnostic yield of CES in patients with neurologic deficits,2,14,15 multiple congenital anomalies,16 and more severe symptomatology,16,17 further highlighting the importance of patient selection.
Here we describe the CES clinical pipeline instituted at the University of Michigan Pediatric Genetics Clinic. Specifically, we report on the first 523 cases evaluated in our clinic. We show that our local population is similar to those previously reported by other genetics clinics and laboratories.2,4 Moreover, through representative examples, we highlight how CES is changing pediatric clinical practice and furthering our understanding of the pathogenesis of many genetic diseases.
Patients and methods
The institutional review board of the University of Michigan approved this retrospective study. Patients who were referred to the Michigan Medicine Pediatric Genetics clinic for any reason and who underwent CES as part of their diagnostic workup were consecutively included in this study. Clinical evaluations of patients were performed in the comprehensive Pediatrics Genetics Clinic and included detailed evaluation of each individual and construction of a pedigree. Patients were clinically assessed by a physician (medical geneticist) who reviewed prior history and medical workup, performed a patient interview and physical exam, and ordered diagnostic testing. Patients selected for this cohort included all consecutive patients in whom CES was deemed clinically indicated by the assessing physician who presented for evaluation from October 1, 2012 through April 30, 2018.
Based on physician clinical evaluation and reviews of prior workup and diagnostic testing, a binary yes/no phenotype dataset was curated for all patients across all body systems and other developmental/neurodevelopmental categories. This phenotyping was performed by the Medical Geneticist and then entered into the phenotype database. This was done upon the patient’s initial presentation to clinic before results from genetic testing had returned. The database was created in Excel format, and data were entered by genetic counselors after the patient had been evaluated in clinic. The database was subsequently updated with results of genetic testing when available. The clinical system phenotypes assessed for abnormalities were: ear/nose/throat, ophthalmologic, neuromuscular, cardiac, pulmonary, gastrointestinal (GI), renal, skeletal/connective tissue/vascular, skin/hair/teeth, endocrine, hematologic/immunologic, disorders of sexual development/ambiguous genitalia, metabolic/inborn errors of metabolism. A number of other non-body system categories were also assessed in a similar binary way: multiple congenital anomalies, dysmorphic features, cancer, developmental delay, autism spectrum disorder (ASD) or autism-like features, and any other behavioral phenotype not otherwise specified. In addition to these binary-curated features, all patients had a descriptive phenotypic summary, as well as demographic information (age, sex) and collation of any prior genetic/molecular testing including karyotype, CMA, or other single gene or panel-based molecular testing. Each clinical phenotype was assigned its corresponding Human Phenotype Ontology term (Supplementary Table 2).
All patients included in this study underwent CES as part of their clinical workup, based on physician clinical decision making. All patients were also required to have a separate genetic counseling appointment for pre-test counseling performed by a certified genetic counselor. CES was performed in a “send-out” manner, with 424 to GeneDx, 96 to Ambry, 2 to UCLA, and 1 to Fulgent. Clinical CES results were reported using the ACMG classification scheme; however, for research purposes, we modified reporting for analysis here by collapsing pathogenic and likely pathogenic variants into the same category (pathogenic variants). We have also included genes of uncertain significance (GUS)—i.e., genes whose biologic function is not known or disease association has not been established—as these may be informative for future gene discovery research.
Clinical phenotype data were analyzed by comparing summative analysis of the binary phenotypes. Analysis of variance was used to compare differences between groups, with P value < 0.05 considered significant.
Results
Data collection for this study began October 1, 2012 and ended April 30, 2018, at which time 562 consecutive patients had been clinically evaluated and had CES testing. Patients with incomplete clinical phenotyping or CES data not yet returned were excluded, resulting in a total of 523 patients included in this analysis. One-hundred and fifty-seven out of the 523 patients (30%) had a pathogenic or likely pathogenic variant identified that established a genetic diagnosis (Table 1, Supplementary Table 1), which is in line with prior CES cohorts.2,4,5 Interestingly, the remaining 365 patients did not have a pathogenic or likely pathogenic variant identified that could clearly establish a clinical genetic diagnosis; however, 243/523 (67%) of patients had at least 1 variant of uncertain significance (VUS) or 1 GUS. Only 120/523 (23%) of patients who were referred for evaluation had no variants reported. There were no major discrepancies observed between the CES laboratory report and our clinical interpretation of the data in the context of the patient’s clinical features.
In our clinical phenotype analysis of these patients, we discovered a significant difference between the number of body systems affected and the likelihood of having a pathogenic variant by CES (Table 2). Among patients with a pathogenic variant identified, 128/157 (81%) had neuromuscular dysfunction and 131/157 (83%) had developmental delay, consistent with the nervous system being the most common body system affected. Thirty-one out of 157 (20%) of patients with pathogenic variants had a clinical diagnosis of ASD. The next most common systems affected were ophthalmologic (76/157, 48%), GI (63/157, 40%), and skeletal/connective tissue/vascular (63/157, 40%). Forty-two percent of patients (67/157) had multiple congenital anomalies. The frequencies of individual systems affected were not significantly different between patients with pathogenic variants and those with one or more VUS or GUS. Surprisingly, only 26/157 (16%) of patients with an identifiable pathogenic variant had dysmorphic features on clinical exam.
Medical genetics clinical phenotyping in correlation with CES results shows that patients with more body system abnormalities on clinical exam more frequently have an identifiable pathogenic mutation and that neurologic dysfunction and/or neurodevelopmental abnormalities are the clinical phenotypes most commonly found to have a pathogenic variant. Placement of these results in the context of lessons learned is informative for the ways in which clinical CES can be used in pediatric patients with unclear symptoms or diagnoses, and how use of CES is changing clinical care more generally, as discussed next. Patient IDs are given for reference in Supplementary Tables 1 and 2.
CES can diagnose very rare or newly described disorders
A 3-year-11-month-old girl (ID #43) with multiple congenital anomalies returned to our pediatric genetics clinic for re-evaluation. She had dysmorphic facial features, including severe midface hypoplasia, hypertelorism, hypoplastic low-set ears, and microretrognathia. She also had relative macrocephaly, agenesis of the corpus callosum, colpocephaly, pachygyria, bilateral choanal atresia, subglottic stenosis requiring tracheostomy, metatarsus adductus, and bilateral camptodactyly. Global developmental delay was present. Her parents are first cousins. Prior testing included karyotype, CMA, and SOX9 (campomelic dysplasia) and POR (Antley–Bixler syndrome) sequencing, which were all negative. CES identified a homozygous missense variant, c.12085G>A(p.Glu4029Lys), in FAT4; the parents were both heterozygous for the variant. Biallelic mutations in FAT4 have been identified in association with Van Maldergem syndrome (OMIM 615546), an autosomal-recessive disorder with physical features very similar to our patient. Although the original description of Van Maldergem syndrome was published in 1992,18 the molecular basis of the disorder was not discovered until recently,19 and clinical testing for FAT4 was not available in the United States at the time CES was performed. In this case, CES allowed for identification of a rare diagnosis not otherwise considered and for which clinical testing was not available. Notably, Yang et al. reports that, in their series of 2000 clinical exomes, approximately 30% of diagnoses were made in disease genes first identified after 2011.6
CES allows for simultaneous consideration of multiple possible diagnoses
An 8-month-old boy (ID #79) was referred for microcephaly and hypotonia. The child also had gastroesophageal reflux as well as myringotomy tube placement for chronic otitis media. An echocardiogram showed thickened aortic valve leaflets. Brain magnetic resonance imaging showed no structural abnormalities. Physical exam was notable for weight at the 9th percentile, length at the 5th percentile, and head circumference less than the 3rd percentile (and 50th percentile for a 4-month-old). A bifid lower incisor was noted as well as highly arched palate, anteriorly placed anus, and truncal hypotonia. Prior to referral to our clinic, karyotype, CMA, and Fragile X testing were all negative. Differential diagnoses included a primary microcephaly disorder (ASPM and 18 additional genes), Kabuki syndrome (KMT2D, KDM6A), Smith–Lemli–Opitz syndrome (DHCR7), and Dubowitz syndrome (molecular basis not yet known). In lieu of single-gene testing, CES was sent and identified a heterozygous de novo pathogenic variant in KMT2D, c.8856delC (p.Lys2953Argfs*51). This confirmed a diagnosis of Kabuki syndrome (OMIM 147920). In this case, CES allowed all differential diagnoses to be considered simultaneously, allowing for more rapid establishment of the correct diagnosis and appropriate clinical guidelines in this patient.
CES promotes recognition of rare presentations of known diseases
A 19-month-old boy (ID #86) was evaluated for central hypoventilation requiring tracheostomy and home ventilation as well as hypotonia and global developmental delay. Previous testing included karyotype, CMA, methylation PCR for Prader–Willi/Angelman syndromes, metabolic screening laboratories (ammonia, plasma amino acids, urine organic acids), PHOX2B sequencing and deletion/duplication analysis, next-generation sequencing panel of 12 congenital myasthenia genes, and next-generation sequencing panel of 50 neuromuscular disorder-related genes. All testing returned negative with the exception of the sequencing panels, which identified a few VUSs. CES revealed a de novo hemizygous nonsense pathogenic variant in MECP2, c.808C>T (p.Arg270*), consistent with a diagnosis of atypical Rett syndrome. This diagnosis was not previously considered for this patient; however, the clinical phenotype is consistent with those rare males with pathogenic MECP2 variants.20 The patient has since developed medically refractory seizures. In this case, CES allowed us to make a diagnosis that otherwise was not being considered yet in retrospect is consistent with rarely described cases.
Discussion
In this manuscript, we present data from 523 patients evaluated at the University of Michigan Pediatric Genetics clinic who underwent CES and systematic clinical phenotyping. We identified pathogenic or likely pathogenic variants in 30% of patients, similar rates to those previously reported.2,4,5 Interestingly, 67% of patients without an identified pathogenic or likely pathogenic variant had at least one VUS or GUS, suggesting that our diagnostic rate will increase over time with variant reclassification and reanalysis. Also similar to prior reports, we found that a higher percentage of patients with a pathogenic or likely pathogenic variant had developmental delay and/or multiple congenital anomalies as compared to patients without any identified pathogenic or likely pathogenic variant,2,4,16 suggesting that these patients may benefit the most from evaluation by a clinical geneticist and CES. Interestingly, among individuals with VUS or GUS, there were similar percentages of patients with developmental delay and multiple congenital anomalies, highlighting the importance of re-evaluation over time.
In our patient population, 120/523 (23%) patients referred had no variants reported. Surprisingly, 33% of those had ASD and 34% had behavioral abnormalities, whereas ASD was present in only 20% of patients with a pathogenic/likely pathogenic variant and behavioral abnormalities in 27% of cases with a pathogenic/likely pathogenic variant. This discrepancy suggests that other mechanisms such as epigenetic factors may be driving these neurobehavioral phenotypes in our patient population. Re-analysis of exome data in this population will likely increase diagnostic yield. While whole-genome sequencing (WGS) may help resolve some of these negative results, the diagnostic utility of WGS does not currently exceed CES, suggesting that genomic analysis at levels other than DNA variants may be clinically helpful, especially in targeted phenotypic groups such as these.7
The increased availability and decreasing cost of advanced sequencing technologies have led to CES becoming an invaluable diagnostic tool for patients referred for general genetics evaluations. In particular, our cohort highlights three ways in which CES has influenced the clinical trajectory of our patients, by allowing for (a) simultaneous consideration of multiple diagnoses, (b) recognition of rare presentations of known disorders, and (c) diagnosis of very rare or newly defined disorders. Identification of new disease genes is also possible through further investigation of reported VUS and GUS. Widespread use of CES promises to increase the number of pediatric patients with a genetic diagnosis. Our results add to the growing body of literature showing that appropriate patient selection is essential to ensure maximal diagnostic utility of advanced sequencing techniques and help guide the general pediatrics community as to the types of patients who may benefit from CES.
Comprehensive clinical phenotyping remains of critical importance when working up patients for possible genetic disorders. As demonstrated here, CES and phenotyping is beneficial not just for the individual patient’s diagnosis. In aggregate, this information can be used to help guide higher-level decisions about CES. As our results show, for instance, patients presenting with a neurodevelopmental phenotype or multiple system anomalies have a higher pre-test probability of having a pathogenic variant than those with fewer organ systems involved.
This study has a number of limitations. As with many clinical exome studies, our sample size is relatively small and limited to those patients being part of routine clinical evaluations. In addition, our clinical phenotyping pipeline employed several different clinical providers to perform assessments, which has the potential to increase variability in phenotyping. Also, while >80% of CES testing was performed at one laboratory (GeneDx), there were a number of other laboratories/companies used (Ambry, UCLA, Fulgent).
Establishment of a comprehensive clinical genotype–phenotype dataset as described here will help provide a basis for many future studies. For instance, further evaluation of patients with pathogenic or likely pathogenic variants is useful for determining how frequently genetic diagnoses change medical or surgical management, assisting with cost–benefit analyses for CES. Incorporation of other types of genomic data that are not currently used or very new in clinical testing, such as metabolomics and epigenomics, could also provide for new clinical correlations. Further, basic science investigations into the biologic roles of the identified VUS and GUS may also lead to discovery of new causative variants.
In conclusion, we present clinical phenotyping data coupled with CES information on 523 pediatric patients referred to a pediatric genetics clinic at the University of Michigan. Our results support prior studies showing that about one-third of such cases currently receive a molecular diagnosis via an identified pathogenic or likely pathogenic variant. Furthermore, our phenotype analysis suggests that patients with multiple congenital abnormalities are most likely to have an identified pathogenic or likely pathogenic variant and that neurologic and neurodevelopmental system abnormalities are the most common in patients with a pathogenic or likely pathogenic variant on CES. Three representative case studies place into perspective for the general pediatrics community how CES is rapidly changing clinical care.
References
Gahl, W. A. et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet. Med. 14, 51–59 (2012).
Lee, H. et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887 (2014).
Need, A. C. et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 49, 353–361 (2012).
Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879 (2014).
Valencia, C. A. et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front. Pediatr. 3, 67 (2015).
Yang, Y. et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511 (2013).
Clark, M. M. et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 3, 16 (2018).
Tan, T. Y. et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 171, 855–862 (2017).
Pena, L. D. M. et al. Looking beyond the exome: a phenotype-first approach to molecular diagnostic resolution in rare and undiagnosed diseases. Genet. Med. 20, 464–469 (2018).
Sobreira, N. L. M. et al. Matchmaker exchange. Curr. Protoc. Hum. Genet. 95, 9.31. 31–39.31.15 (2017).
Boycott, K. M. et al. International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum. Genet. 100, 695–705 (2017).
Bick, D. et al. Successful application of whole genome sequencing in a medical genetics clinic. J. Pediatr. Genet. 6, 61–76 (2017).
Eldomery, M. K. et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 9, 26 (2017).
Cordoba, M. et al. Whole exome sequencing in neurogenetic odysseys: an effective, cost- and time-saving diagnostic approach. PLoS ONE 13, e0191228 (2018).
Posey, J. E. et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet. Med. 18, 678–685 (2016).
Trujillano, D. et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182 (2017).
Soden, S. E. et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 6, 265ra168 (2014).
van Maldergem, L., Wetzburger, C., Verloes, A., Fourneau, C. & Gillerot, Y. Mental retardation with blepharo-naso-facial abnormalities and hand malformations: a new syndrome? Clin. Genet. 41, 22–24 (1992).
Cappello, S. et al. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat. Genet. 45, 1300–1308 (2013).
Belligni, E. F., Palmer, R. W. & Hennekam, R. C. MECP2 duplication in a patient with congenital central hypoventilation. Am. J. Med. Genet. A. 152a, 1591–1593 (2010).
Acknowledgements
We thank the patients and their families, as well as the clerical staff in the Genetics Clinic, and the research coordinators of the University of Michigan Children’s Clinical Trial Support Unit: Chaandini Jayachandran, MS, CCRP; Alyssa Paul, MS, CCRC; Jonathon Wilbur, BS; Ciara Ivantics, MS; Amy Hurst, BS; Andy Brosius, BS. All phases of this study were supported by The University of Michigan, Department of Pediatrics and the Alfred A. Taubman Medical Research Institute. D.M.M. is also funded by the National Institutes of Health (NIH) DC R01-014456 and by the Donita B. Sullivan, MD, Research Professorship in Pediatrics. J.W.I. is supported by the Morton S. and Henrietta K. Sellner Professorship in Human Genetics.
Author information
Authors and Affiliations
Contributions
D.M.M., M.N.Z., J.W.I., and J.A.B. conceptualized and designed the study, coordinated and supervised data collection and data analysis, drafted the initial manuscript, and reviewed and revised the manuscript. A.A., M.C.H., C.E.K., and S.C.Q. directly evaluated patients, coordinated and supervised data collection, and critically reviewed the manuscript for intellectual content. N.W., R.F., M.G., J.E.J., K.N.L., T.B.M., J.D.O., B.C.O., L.S., L.T., S.B., and N.L.H. collected data and reviewed and revised the manuscript for intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ziats, M.N., Ahmad, A., Bernat, J.A. et al. Genotype–phenotype analysis of 523 patients by genetics evaluation and clinical exome sequencing. Pediatr Res 87, 735–739 (2020). https://doi.org/10.1038/s41390-019-0611-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0611-5
This article is cited by
-
Atypical Sotos syndrome caused by a novel splice site variant
Human Genome Variation (2022)
-
A Novel De Novo TUBB3 Variant Causing Developmental Delay, Epilepsy and Mild Ophthalmological Symptoms in a Chinese Child
Journal of Molecular Neuroscience (2022)
