
Abstract
Background
Although prenatal and postnatal programming of metabolic diseases in adulthood is well established, the mechanisms underpinning metabolic programming are not. Dlk1, a key regulator of fetal development, inhibits adipocyte differentiation and restricts fetal growth.
Methods
Assess DLk1 expression in a Wistar rat model of catch-up growth following intrauterine restriction. Dams fed ad libitum delivered control pups (C) and dams on a 50% calorie-restricted diet delivered pups with low birth weight (R). Restricted offspring fed a standard rat chow showed catch-up growth (R/C) but those kept on a calorie-restricted diet did not (R/R).
Results
Decreased Dlk1 expression was observed in adipose tissue and skeletal muscle of R/C pups along with excessive visceral fat accumulation, decreased circulating adiponectin, increased triglycerides and HOMA-IR (from p < 0.05 to p < 0.0001). Moreover, in R/C pups the reduced Dlk1 expression in adipose tissue and skeletal muscle correlated with visceral fat (r = −0.820, p < 00001) and HOMA-IR (r = −0.745, p = 0.002).
Conclusions
Decreased Dlk1 expression relates to visceral fat expansion and insulin resistance in a rat model of catch-up growth following prenatal growth restriction. Modulation of Dlk1 expression could be among the targets for the early prevention of fetal programming of adult metabolic disorders.
Similar content being viewed by others


Introduction
Fetal programming is a process by which the fetus’ adaptations to the adverse conditions during gestation and early postnatal life have long-term effects on the newborn.1 Clinical studies, and subsequent work in animal models, have demonstrated that adverse conditions in these critical developmental windows can program physiological processes and cause metabolic alterations leading to future adult diseases, such as obesity, type 2 diabetes, metabolic syndrome and cardiovascular disease.1,2,3
Fetal development is sensitive to the nutritional environment. Adaptations to adverse conditions serve to optimize and improve the chances of fetal survival. However, when there is a mismatch between fetal and postnatal nutrition, this adapted thrifty phenotype is inappropriate, becomes detrimental and contributes to metabolic disorders in adulthood.3,4 Consequently, the major risk of developing these disorders later in life has been associated with postnatal catch-up growth following low birth weight, a condition occurring in ~8% of pregnancies worldwide.1,2,5
The impact of early life insults on childhood and adult health is certainly apparent; however, the mechanisms underlying the alterations in metabolic programming remain largely unknown. The study of genes that regulate fat storage, whose alterations lead to insulin resistance, are of central importance for understanding the pathophysiology of catch-up growth.
Delta-like protein 1 (Dlk1) gene is a key regulator of fetal development and it is involved in energy and nutrient metabolism. In adults, its expression is limited to endocrine tissues: Dlk1 inhibits adipocyte differentiation in adipose tissue and is involved in regeneration processes in skeletal muscle.6,7,8 Our group has previously linked prenatal and postnatal growth with the placental imprinted DLK1-DIO3 domain in human studies;9 others have described that impairments in fetally derived DLK1 cause fetal growth restriction.10 Studies, however, linking Dlk1 gene expression and catch-up growth following prenatal restriction are lacking.
The aim of the study was to assess whether changes in Dlk1 gene expression relate to metabolic risk markers in an animal model of postnatal catch-up growth following fetal weight restriction.
Methods
Rat model of catch-up growth following prenatal growth restriction
A Wistar rat model of intrauterine growth restriction induced by calorie restriction throughout gestation was used. Studies were approved by the Animal Research Ethic Committee (C.E.E.A., #DAAM 6911) of the University of Barcelona and were in accordance with the Catalan Government guideline 214/97. First-time-pregnant Wistar rats (Janvier, Le Genest Saint Isle, France) were housed in a facility with constant temperature (21 ± 1 °C) and humidity (50–60%) and a controlled 12:12-h light-dark cycle. The first day of gestation was determined by observation of spermatozoa in vaginal smears. From day 1 of pregnancy to term, control dams (C) were provided an ad libitum diet consisting of a standard laboratory chow (Teklad Global Rodent Maintenance Diet, Harlan Laboratories Inc.; 14% protein, 4% fat, 2900 kcal/kg) and restricted dams (R) were fed a 50% less of diet used in control dams (Supplementary Figure S1). Rat dams gave birth normally and pups were limited to ten per litter at day 1 after birth to normalize pups feeding. Throughout the 21-day lactation period, different control or restricted diets were provided in order to create 3 experimental groups: control dams were fed an ad libitum diet (C/C) and restricted dams were fed either an ad libitum diet (R/C) or a restricted diet (R/R) to create catch-up or non-catch-up growth groups, respectively (Supplementary Figure S1). At 22-days of age, after the lactation period, offspring in all groups were weaned to ad libitum feed. All pups were weighed twice a week from postnatal day 1 (PD1) until PD42. At PD42 all pups (N = 14 per group; 7 females and 7 males) fasted overnight were anesthetized with 3% isoflurane and euthanized by cardiac exsanguination. Blood was collected via cardiac puncture in heparinized syringes and centrifuged at 3500 rpm for 15 min at 4 °C to obtain plasma samples which were stored at −80 °C. Adipose tissue from different locations in the abdomen and skeletal gastrocnemius muscle samples were immediately frozen in liquid nitrogen. Additionally, an approximation to body composition was obtained by calculating visceral fat tissue mass (sum of weights of perigonadal, retroperitoneal and mesenteric adipose tissues). For gene and protein expression studies, rat retroperitoneal adipose tissue was selected as being equivalent to human omental adipose tissue.
Plasma metabolic profile
Plasma adiponectin and insulin concentrations were determined by ELISA using a commercial kit (Rat Adiponectin ELISA Kit, and Ultrasensitive Rat Insulin ELISA kit; Crystal Chem Inc.). For adiponectin, the sensitivity of the kit was 0.25 mg/l and intra-assay CVs were <10% and for insulin, the sensitivity was 0.1 ng/ml and intra-assay CVs were less than 10%. Plasma glucose, total cholesterol, HDL-cholesterol and triglycerides were determined by dry chemistry technique (Spotchem II, Spotchem EZ SP-4410, Menarini Diagnostics-Akray, Japan). Homeostatic model assessment of insulin resistance (HOMA-IR) was assessed using the following formula: HOMA-IR index = glucose×insulin/2430.11
Gene and protein expression
For gene expression, RNA (RNAeasy mini kit, Qiagen) was extracted and 2 µg of total RNA was retro-transcribed to cDNA (High-capacity cDNA Reverse Transcription Kit, Applied Biosystems, Thermo Fisher Scientific Inc.). cDNA samples were used to quantify the relative expression by real-time PCR (Taqman Assays 20 × [Dlk1: Rn00587011_m1 and endogenous control GAPDH: Rn01775763_g1], Applied Biosystems, Thermo Fisher Scientific Inc.). For protein quantification, total protein was extracted from tissue (DC protein assay, Bio-Rad Laboratories Inc.). Total protein concentration was quantified by the Lowry method and Dlk1 protein concentrations by an ELISA commercial kit (Rat Delta Like 1 Homolog Dlk1 ELISA kit, MyBioSource, Inc.). The sensitivity of the kit was 0.27 ng/ml and intra-assay CVs were <9%.
Tissue morphology
For morphometric studies, retroperitoneal adipose tissue from the abdominal cavity and skeletal gastrocnemius muscle from a set of 12 animals (N = 4 animals per each treatment, 2 males and 2 females) were used. Tissues were paraffin-embedded, tissue sections were deparaffinized by the xylene-ethanol sequence and processed using the hematoxilin-eosin dye method. Images were taken at ×200 (Olympus BX51, XC50 CellVe). One histological section of each animal was randomly selected from the histological slides. Twelve randomly chosen fields per section were considered for the measurement of both adipocyte area and density (>250 adipocytes were analyzed per group) and skeletal muscle cross-sectional myofiber density and cross-sectional myofiber area. The measurements were analyzed in a blinded manner and both manual and automatic tools of the Image J software (NIH, public domain) were used.
Statistical analysis
All statistics were performed with SPSS 22.0 (SPSS software, IBM, Inc.) and graphs were done with Prism 6.0 (GraphPad Software, Inc.). Differences between experimental groups were compared using ANOVA test and least significance difference (LSD) post hoc tests. Spearman correlation was used to analyze associations between variables. Combined data for males and females are shown, since no sex differences were evident. Data are expressed as mean ± standard deviation (SD) and p value was set at 0.05.
Results
Catch-up growth Wistar rats following prenatal growth restriction exhibited increased fat mass and higher HOMA-IR
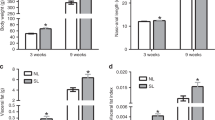
At birth, R/R and R/C pups showed less body weight (p < 0.0001, Table 1) relative to C/C pups: both R/R and R/C pups showed a 22% lower value in body weight at birth in relation to C/C pups. At weaning (PD22), R/C pups showed higher body weight (p < 0.0001; Table 1) and at PD42 these pups experienced further postnatal weight gain (p < 0.0001; Table 1), together with higher retroperitoneal adipose tissue weight and higher visceral fat (from p < 0.05 to p < 0.0001; Table 1), as compared to other pup groups. At PD42, R/C pups showed also lower total adiponectin, higher plasma triglycerides (both p < 0.01; Table 1) and higher insulin concentration and HOMA-IR index (both p < 0.0001; Table 1), as compared to R/R pups.
Decreased Dlk1 expression in adipose tissue and skeletal muscle relates to visceral fat and HOMA-IR in catch-up growth Wistar rats following prenatal growth restriction
Relative Dlk1 expression in R/C pups was decreased in both retroperitoneal adipose tissue and skeletal muscle compared to R/R pups (p < 0.0001; Table 1). Likewise, Dlk1 protein concentrations in retroperitoneal adipose tissue were lower in R/C pups (p < 0.001; Table 1) compared to other pup groups. In adipose tissue, negative correlations between Dlk1 relative expression and body weight, postnatal body weight gain and visceral fat at PD42 were observed (all between r = −0.820 and r = −0.895, p < 0.0001; Fig. 1 and Supplementary Table S1). In muscle, negative correlations between Dlk1 relative expression and plasma insulin and HOMA-IR index were observed (r = −0.701, p = 0.005 and r = −0.745, p = 0.002; respectively; Fig. 1 and Supplementary Table S1).

Correlation graphs between Dlk1 expression in adipose and skeletal muscle tissues and studied parameters in a Wistar rat catch-up growth model. Correlation graphs of different R/R, C/C and R/C groups between Dlk1 relative expression in retroperitoneal adipose tissue (AT) and a body weight at PD42, b postnatal body weight gain and c visceral fat; as well as Dlk1 relative expression in skeletal muscle and d insulin and e HOMA-IR values. Spearman r and p values are shown, P value was set at p < 0.05 and significant correlations are marked in bold style. Gene expression variables have been logarithmically transformed (log10). R/R: restricted, C/C: control, R/C: catch-up growth
Catch-up growth Wistar rats following prenatal growth restriction showed hypertrophied adipocytes and enlarged skeletal muscle myofibers
The histological studies showed higher adipocyte area (4933 μm2 ± 1336; 3277 μm2 ± 900; 2144 μm2 ± 533; p < 0.0001) and lower adipocyte density (242N/mm2 ± 85; 340N/mm2 ± 83; 531N/mm2 ± 153; p < 0.0001) in retroperitoneal adipose tissue and lower density or number of myofibers per area (491N/mm2 ± 90; 474N/mm2 ± 45; 726N/mm2 ± 95; p = 0.003) in skeletal muscle of R/C pups, as compared to C/C and R/R groups, respectively (Fig. 2).

Adipose tissue and skeletal muscle morphometric parameters in a Wistar rat catch-up growth model. Representative images of a retroperitoneal adipose tissue and c skeletal muscle at PD42 are shown (scale bar: 50 µm). Bar graphs showing b adipocyte area and adipocyte density (>250 adipocytes per group were measured), d myofibers area and myofiber density (>20 fields per group were measured) are shown (*p < 0.05 compared to C/C and #p < 0.05 compared to R/R)
Discussion
In the present model of catch-up growth following fetal growth restriction, we observed decreased Dlk1 in both visceral fat and skeletal muscle in pups experiencing catch-up growth. Moreover, in these pups, reduced Dlk1 gene expression in retroperitoneal adipose tissue was related to visceral fat mass accumulation, and reduced Dlk1 gene expression in skeletal muscle was associated with insulin resistance.
As expected, in our experimental model of catch-up growth following fetal weight restriction, R/C pups successfully presented catch-up growth, as judged by higher body weight gain and visceral adiposity at P42. Previous studies have shown excessive visceral fat accumulation during catch-up growth.12 It has also been suggested that permanent changes in the expression of regulatory proteins in liver, muscle and adipose during catch-up growth relate to insulin resistance and metabolic disorders later in life.4 In our study, R/C pups showed not only increased visceral fat but also decreased circulating adiponectin and increased plasma triglycerides. It is well known that circulating adiponectin concentrations decrease along with visceral fat accumulation.13 Consequently, the observed visceral fat mass accumulation, low adiponectin levels and high triglycerides during catch-up growth may reflect dysregulation of adipose tissue function, which may plausibly contribute to the development of insulin resistance in these animals. This is substantiated by previous studies relating catch-up growth and insulin resistance in Wistar rats14 and by our results showing higher HOMA-IR values in R/C pups. Catch-up growth in these pups caused both visceral fat accumulation and insulin resistance as early as at postnatal period PD42. On the contrary, R/R pups kept postnatally under restricted conditions, in order to prevent the mismatch between fetal and postnatal nutrition, exhibited rather insulin sensitivity. Metabolic programming has been described to present both altered fat expansion and disrupted insulin signaling pathway leading to insulin resistance, although the molecular mechanisms implicated in these processes are not fully understood.
Dlk1 is a regulator of adipocyte differentiation and adipogenesis.6,15 The decrease in Dlk1 relative gene expression and Dlk1 protein concentrations in retroperitoneal adipose tissue at P42 together with the negative correlations between Dlk1 expression and body weight, body weight gain and visceral fat mass in R/C pups point to a relevant role of Dlk1 in the control of adipocyte growth and differentiation during catch-up growth. It was shown that mice with Dlk1 deficiency presented higher amounts of visceral fat, circulating lipids and adipocyte cell size at weaning and developed obesity and fatty liver disease.6,8,16 Histologically, hypertrophied adipocytes have been consistently found in adipose tissue of individuals with obesity,17 as well as in restricted pups at weaning.12,18,19 This observation has been related to a lower number of progenitor adipocytes capable of differentiating into healthy adipocytes.20 Accordingly, our results showed hypertrophy of the retroperitoneal adipose tissue in R/C pups at P42, which was characterized by higher adipocyte area and lower adipocyte density. The low Dlk1 expression observed in R/C pups may thus predispose these pups to postnatal adipose tissue expansion, especially of visceral fat, with a low number of preadipocytes being capable of differentiating into mature adipocytes, in turn contributing to adipocyte dysfunction. Indeed, low Dlk1 gene expression in adipose tissue is likely to cause excessive adipocyte lipid storage, inflammation and altered adipokine secretion, all known to be involved in the development of insulin resistance and obesity.18 As it has been described that cell size and/or lipid droplet locularity influence cortical actin structures that mediate GLUT4 translocation,21 we hypothesize that low Dlk1 and related adipocyte hypertrophy could directly influence the development of insulin resistance.
Dlk1 is known to be a crucial regulator of the myogenic program in skeletal muscle, and it has been shown that Dlk1 has a dual biological function in this tissue, both as an enhancer of muscle development and as an inhibitor of adult muscle regeneration.22 The observed low Dlk1 expression in skeletal muscle could be involved in restraining of muscle development as we observed histologically lower number of myofibers per area (myofiber density) in skeletal muscle of R/C pups at P42. In this sense, catch-up growth towards fat mass accumulation in detriment of muscular mass has been described in humans.23 Low birth weight following free access to food during the growing-finishing period in pigs also showed enlarged cross-sectional myofiber areas due to enhanced fat deposition.24 Moreover, fat mass accumulation in detriment of muscular mass has been related to insulin resistance and overexpression of insulin receptor in skeletal muscle in experimental animals.25 In our model we observed decreased Dlk1 expression in skeletal muscle in R/C pups that was related to increased HOMA-IR values. We therefore suggest that Dlk1 expression in skeletal muscle is contributing to the development of insulin resistance during catch-up growth. It is tempting to speculate that lower Dlk1 gene expression could enhance C/EBPα, a known downstream Dlk1 target molecule, which in turn would regulate GLUT4 and affect glucose homeostasis.26 Then, Dlk1 dysregulation would be among the mechanisms explaining why skeletal muscle presents lower insulin-stimulated glucose utilization and develops insulin resistance during catch-up growth.
We acknowledge some limitations in our study. Major differences were observed between R/C and R/R pups, with fewer differences being evident between R/C and C/C pups. It should be highlighted, however, that there were strong linear differences between the 3 groups of pups in most of the examined parameters, which give relevance to our results. In future studies, our goal will be to examine more severe fat mass accumulation or obesity phenotypes during catch-up growth as well as to extend the growth period since P42 may have been rather a short juvenile time-point to examine the association between Dlk1 and metabolic risk markers. Interestingly though, our findings indicate that these metabolic abnormalities take place at an early age following catch-up growth. Future studies assessing these parameters for a longer period should take into account the age-related sex dimorphism in insulin sensitivity that is evident starting at PD90 (3 months), and which has not been described in early stages, such as at PD26 or PD40, when the juvenile rats are still not sexually mature.27,28,29 Finally, we could not analyze the direction of causality of the molecular changes observed at PD42 in pups experiencing catch-up growth since the Wistar rat model of intrauterine growth restriction induced by calorie restriction throughout gestation used in the present study did not include tissues at birth: a few pups were removed to equal litter sizes within groups but they were insufficient in number to provide tissue samples at birth. Despite these limitations, we have observed novel molecular changes in Dlk1 gene expression during catch-up growth.
In conclusion, decreased gene expression of Dlk1 in adipose tissue and skeletal muscle may contribute to visceral fat expansion and development of insulin resistance during catch-up growth following fetal growth restriction. It is tempting to speculate that future intervention strategies based on Dlk1 activation may improve the outcome of catch-up growth following prenatal growth restriction.
Change history
15 June 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41390-023-02691-2
References
Barker, D. J., Osmond, C., Golding, J., Kuh, D. & Wadsworth, M. E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 298, 564–567 (1989).
Ravelli, A. C. et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet 351, 173–177 (1998).
Bol, V. V., Delattre, A.-I., Reusens, B., Raes, M. & Remacle, C. Forced catch-up growth after fetal protein restriction alters the adipose tissue gene expression program leading to obesity in adult mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R291–R299 (2009).
Hales, C. N. & Ozanne, S. E. The dangerous road of catch-up growth. J. Physiol. 547(Pt 1), 5–10 (2003).
Barker, D. J. et al. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36, 62–67 (1993).
Sul, H. S. Minireview: Pref-1: role in adipogenesis and mesenchymal cell fate. Mol. Endocrinol. 23, 1717–1725 (2009).
Mortensen, S. B. et al. Membrane-tethered delta-like 1 homolog (DLK1) restricts adipose tissue size by inhibiting preadipocyte proliferation. Diabetes 61, 2814–2822 (2012).
Falix, F. A., Aronson, D. C., Lamers, W. H. & Gaemers, I. C. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim Biophys. Acta 1822, 988–995 (2012).
Prats-Puig, A. et al. The placental imprinted DLK1-DIO3 domain: a new link to prenatal and postnatal growth in humans. Am. J. Obstet. Gynecol. 217, 350.e1–350.e13 (2017).
Geach, T. DLK1 levels predict fetal growth restriction. Nat. Rev. Endocrinol. 13, 4–4 (2017).
Cacho, J., Sevillano, J., de Castro, J., Herrera, E. & Ramos, M. P. Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague-Dawley rats. Am. J. Physiol. Endocrinol. Metab. 295, E1269–E1276 (2008).
De Toro-Martín, J. et al. Predominant role of GIP in the development of a metabolic syndrome-like phenotype in female Wistar rats submitted to forced catch-up growth. Endocrinology 155, 3769–3780 (2014).
Turer, A. T. et al. Adipose tissue mass and location affect circulating adiponectin levels. Diabetologia 54, 2515–2524 (2011).
Berends, L. M., Fernandez-Twinn, D. S., Martin-Gronert, M. S., Cripps, R. L. & Ozanne, S. E. Catch-up growth following intra-uterine growth-restriction programmes an insulin-resistant phenotype in adipose tissue. Int J. Obes. 37, 1051–1057 (2012).
Lee, K. et al. Inhibition of adipogenesis and development of glucose intolerance by soluble preadipocyte factor-1 (Pref-1). J. Clin. Invest. 111, 453–461 (2003).
Villena, J. A., Kim, K. H. & Sul, H. S. Pref-1 and ADSF/resistin: two secreted factors inhibiting adipose tissue development. Horm. Metab. Res. 34, 664–670 (2002).
Bieswal, F. et al. The importance of catch-up growth after early malnutrition for the programming of obesity in male rat. Obesity 14, 1330–1343 (2006).
Duque-Guimarães, D. E. & Ozanne, S. E. Nutritional programming of insulin resistance: causes and consequences. Trends Endocrinol. Metab. 24, 525–535 (2013).
Somm, E. et al. Early metabolic defects in dexamethasone-exposed and undernourished intrauterine growth restricted rats. PLoS ONE 7, e50131 (2012).
Lafontan, M. Adipose tissue and adipocyte dysregulation. Diabetes Metab. 40, 16–28 (2014).
Kim, J. I. et al. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol. Cell Biol. 35, 1686–1699 (2015).
Andersen, D. C. et al. Dual role of delta-like 1 homolog (DLK1) in skeletal muscle development and adult muscle regeneration. Development 140, 3743–3753 (2013).
Mullis, P.-E. & Tonella, P. Regulation of fetal growth: consequences and impact of being born small. Best. Pr. Res Clin. Endocrinol. Metab. 22, 173–190 (2008).
Gondret, F., Lefaucheur, L., Juin, H., Louveau, I. & Lebret, B. Low birth weight is associated with enlarged muscle fiber area and impaired meat tenderness of the longissimus muscle in pigs. J. Anim. Sci. 84, 93–103 (2006).
Ozanne, S. E. Metabolic programming in animals. Br. Med. Bull. 60, 143–152 (2001).
Cha, H. C. et al. Phosphorylation of CCAAT/enhancer-binding protein α regulates GLUT4 expression and glucose transport in adipocytes. J. Biol. Chem. 283, 18002–18011 (2008).
Amengual-Cladera, E., Lladó, I., Proenza, A. M. & Gianotti, M. Sex dimorphism in the onset of the white adipose tissue insulin sensitivity impairment associated with age. Biochimie 106, 75–80 (2014).
Guarda, D. S. et al. Maternal flaxseed oil intake during lactation changes body fat, inflammatory markers and glucose homeostasis in the adult progeny: role of gender dimorphism. J. Nutr. Biochem. 35, 74–80 (2016).
Garcia-Carrizo, F., Priego, T., Szostaczuk, N., Palou, A. & Picó, C. Sexual dimorphism in the age-induced insulin resistance, liver steatosis, and adipose tissue function in rats. Front Physiol. 8, 445 (2017).
Acknowledgements
We would like to thank Dr. Glòria Oliveras Serrat and Ms. Yaiza Martin Gonzalez for the preparation of the tissue histological sections. The study was supported by grant no. PI13/01257 and PI16/01335 (to A.L.-B.) from the National Institute of Health Carlos III (Fund for Health Research FIS, Spain), project co-financed by FEDER.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. G.C.-B., X.R., J.B. and A.L.-B. designed research; G.C.-B. and A.L.-B. wrote the first draft of the manuscript; G.C.-B., X.R. and A.P.-P. conducted research; G.C.-B., S.X.-T. and E.L.-M. analyzed data; and F.d.Z. and L.I. contributed to review the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Acknowledgment was revised.
Supplementary information
Rights and permissions
About this article

Cite this article
Carreras-Badosa, G., Remesar, X., Prats-Puig, A. et al. Dlk1 expression relates to visceral fat expansion and insulin resistance in male and female rats with postnatal catch-up growth. Pediatr Res 86, 195–201 (2019). https://doi.org/10.1038/s41390-019-0428-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0428-2
This article is cited by
-
Catch-up growth in juvenile rats, fat expansion, and dysregulation of visceral adipose tissue
Pediatric Research (2022)
