
Abstract
Background
Animal models of nephrotic syndrome (NS) revealed that tight junction (TJ)-like structures are generated together with a concomitant decrease in slit diaphragms (SDs). Claudins (CLDNs) are capable of forming TJ strands and thereby the backbone of TJs. We showed the ectopic expression of CLDN2 in podocytes in pediatric NS, and detected its localization.
Methods
Renal frozen specimens were obtained by biopsy from 49 pediatric patients: 21 subjects with MCD, 18 with FSGS, and 10 with IgA nephritis (IgA-N). CLDN2 expression was observed by immunohistochemistry and the CLDN2-positive area was calculated. Moreover, its localization was detected using immunoelectron microscopy.
Results
CLDN2 is ectopically detected in cases with MCD and FSGS before remission. The CLDN2-stained region in MCD and FSGS glomeruli before remission was significantly greater than that after remission as well as in IgA-N patients. Immunoelectron microscopy revealed that CLDN2 was concentrated along newly formed TJs in podocytes.
Conclusion
The same pathological findings in terms of ectopic CLDN2 expression in podocytes were shown in cases with MCD and FSGS before remission. Immunofluorescence and immunoelectron studies of CLDN2 appear to afford a powerful tool for the diagnosis of primary NS. In addition, CLDN2 expression level may be related to disease status.
Similar content being viewed by others

Introduction
Nephrotic syndrome (NS) is a complex disorder characterized by severe proteinuria along with hypoalbuminemia, edema and hyperlipidemia. Primary NS in children is most frequently caused by minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS). In both diseases, podocyte injury initiates foot process effacement, whereas the change in podocyte morphology and the resulting proteinuria are usually reversible and irreversible in MCD and FSGS, respectively. However, the pathogenesis of these diseases remains obscure, and the majority of cases cannot be explained by mutations in various podocyte genes.1,2 In addition, whether MCD and FSGS are distinct types of one disease or two different diseases remains to be resolved.3
During early stage of glomerulogenesis, immature podocytes present as columnar epithelia with tight junctions (TJs).4,5,6 On the other hand, mature podocytes lack TJs and form slit diaphragms (SDs) between opposing foot processes, establishing the final barrier to urinary protein loss. Interestingly, in several animal models of NS, TJ-like structures are generated with a concomitant decrease in or disappearance of SDs.7,8,9,10 This SD−TJ transition is also observed in human MCD cases.11 Nevertheless, the mechanism by which the SD−TJ transition occurs in both MCD and FSGS remains unclear.
Claudins (CLDNs) are capable of forming TJ strands12 and thereby the backbone of TJs. The CLDN family consists of 27 members in mammals, and shows distinct tissue- and cell-type-specific expression patterns.13,14,15,16 Among the CLDNs expressed in normal renal corpuscles, CLDN1 and CLDN2 are known to be observed in parietal epithelial cells (PECs), which cover the inner surface of Bowman’s capsules, but not in podocytes.17,18,19 During the SD−TJ transition in MCD and FSGS, however, it is unknown which CLDN subtype is responsible for the newly formed TJs in the injured podocytes.
On the other hand, CLDN2 is also detected in the epithelial cells of the proximal tubules and the thin descending limbs of Henle along the normal renal tubules.17 CLDN2 is a pore-forming CLDN, with a role in the bulk reabsorption of salt and water in the proximal tubules.20 Therefore, we focused on CLDN2 and found that ectopic expression of CLDN2 existed in the glomeruli of primary NS. In the present study, we show the ectopic expression of CLDN2 in podocytes in cases of pediatric MCD and FSGS. We also demonstrate that CLDN2 is associated with their pathogenesis, suggesting that both diseases are “CLDN2-related podocytopathies”. Moreover, we discuss the possible mechanism by which CLDN2 expression in podocytes leads to their dysfunction.
Methods
Patients
Renal frozen specimens were obtained by needle biopsy from 49 pediatric patients: 21 subjects (8 subjects before remission (BR) and 13 after remission (AR)) with MCD, 18 (8 BR and 10 AR) with FSGS, and 10 with IgA nephritis (IgA-N) as disease controls. This study was approved by the Ethical Committee of Fukushima Medical University (Approval number: 1809).
Clinical data for the subjects were documented at the time of biopsy, and are summarized in Tables 1 and 2. Proteinuria and urinary occult blood were semi-quantitatively scored as follows: (−) = 0, (±) = 0.5, (1+) = 1, (2+) = 2, (3+) = 3, and (4+) = 4.
Antibodies
Rabbit polyclonal antibodies (pAbs) against CLDN2 and CLDN1 were purchased from IBL (Gunma, Japan). Rabbit pAb against podocin was purchased from Sigma Aldrich (St. Louis, MO, USA). Mouse monoclonal antibodies (mAbs) against CD34 (clone NU-4A1), β-Actin (clone AC-15), podocalyxin (clone #222328) and synaptopodin (clone G1D4) were obtained from Nichirei Bioscience (Tokyo, Japan), Sigma Aldrich, R&D Systems (Minneapolis, MN, USA) and Progen Biotechnik (Heidelberg Germany), respectively. A rat anti-Heparan Sulfate Proteoglycan (HSPG) (Perlecan) mAb (clone A7L6) was purchased from Merck Millipore (Temecula, CA, USA). The following secondary antibodies were used: AlexaFluor488-labeled donkey anti-rabbit IgG (H+L) (Invitrogen, Waltham, MA, USA), Cy3-conjugated AffiniPure donkey anti-mouse IgG (H+L) (Jackson ImmunoResearch, West Grove, PA, USA), AlexaFluor647-labeled AffiniPure donkey anti-rat IgG (H+L) (Jackson ImmunoResearch) and immunogold conjugate EM goat anti-rabbit IgG (BBI Solutions, Cardiff, UK).
Immunohistochemistry
Renal biopsy specimens were frozen on dry ice and kept at −80 °C until use. They were sectioned at a thickness of 5 µm and fixed in ice-cold methanol for 15 min at −20 °C. After washing with phosphate-buffered saline (PBS), sections were blocked in 2% bovine serum albumin (BSA) for 1 h at room temperature. After washing, they were subsequently incubated with primary antibodies overnight at 4 °C and rinsed again with PBS followed by reaction with the appropriate secondary antibodies for 1 h at room temperature. They were then mounted after washing with PBS. All samples were examined using a laser-scanning confocal microscope (FV1000, OLYMPUS, Tokyo, Japan).
Calculations
The CLDN2-positive area was calculated using image processing software (ImageJ, Java). The thresholds for images stained with CLDN2 and HSPG (Perlecan) were set from 100 to 255 in order to exclude background signals. A circle was drawn free hand along the inside of the PECs, and the total area inside the circle (A) and the CLDN2-expression area inside the circle (B) were determined. The CLDN2-positive area was defined as B/A × 100 (%) and represented using box-and-whisker plots.
Cell culture, plasmids and western blot analysis
HEK293T cells were cultured at 37 °C under 5% CO2 in Dulbecco’s modified Eagle’s medium high glucose (Sigma Aldrich). The medium was supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were transiently transfected with the pCSII-IRES-Venus expression vectors coding GFP and human CLDNs using PEI-Max solution (Sigma Aldrich). After 48 h of transfection, the cells were lysed in CelLytic MT (Sigma Aldrich) containing a protease inhibitor (Complete mini EDTA-free; Roche Diagnostics, Mannheim, Germany). After centrifugation for 30 min at 4 °C at 15,000 × g, the lysates were resolved by SDS-PAGE and electrophoreticallly transferred onto a polyvinylidene difluoride membrane (Immobilon; Merck Millipore). The membrane was incubated overnight at 4 °C with primary antibodies against CLDN2 or Actin. After washing, they were incubated for 1 h at room temperature with horseradish peroxidase-labeled, anti-rabbit IgG (GE Healthcare Bio-Sciences, Buckinghamshire, UK), or anti-mouse IgG (GE Healthcare Bio-Sciences).
Immunoelectron microscopy
Renal biopsy tissues were fixed with periodate-lysine-paraformaldehyde for 2 h at 4 °C and, after washing with PBS, incubated with polyvinylpyrrolidone-sucrose overnight at 4 °C. They were then frozen by liquid nitrogen and ultrathin cryosections were prepared using a Leica Ultracut UCT microtome equipped with the FCS cryoattachment (Wien, Austria) at −20 °C. They were transferred to nickel grids (150 mesh) coated with formvar and carbon, and the subsequent incubation steps were carried out by floating the grids on droplets of the filtered solutions. After quenching free aldehyde groups with PBS/0.01 M glycine, sections were incubated with rabbit anti-CLDN2 pAb overnight at 4 °C, and reacted with 10 nm gold-labeled goat anti-rabbit IgG for 1 h at room temperature followed by fixation with 2.5% glutaraldehyde buffered with 0.1 M PBS (pH 7.4). They were subsequently contrasted with 3% uranyl acetate solution for 40 min, and absorption-stained with 3% polyvinyl alcohol containing 0.2% acidic uranyl acetate for 40 min. Micrographs were captured using an electron microscope (JEM1230, JOEL).
Statistical analysis
All values are shown as the mean ± standard deviation (SD) except for those of the CLDN2-positive area. Statistical analyses were performed using IBM SPSS statistics 23 software (Chicago, IL, USA). Results were analyzed using two-sample t test and one-way analysis of variance (ANOVA).
Results
CLDN2 is ectopically detected in MCD and FSGS glomeruli
We first examined, by immunohistochemistry, the expression of CLDN2 in glomeruli obtained from pediatric MCD and FSGS patients, as well as in those from IgA-N subjects as disease controls. To distinguish the overall structure of the glomeruli, the endothelial marker CD34 and the basement membrane marker HSPG (Perlecan) were co-immunostained with CLDN2. As shown in Fig. 1, strong filamentous signals for CLDN2 appeared to be detected in the cases with MCD and FSGS before remission, but not in subjects with IgA-N. CLDN2 was also occasionally observed within the whole cell bodies. These CLDN2 signals were generally distributed close to the basement membrane and were separated from endothelial cells, implying that the CLDN2-expressing cells corresponded to the podocytes. In contrast, in the cases with MCD and FSGS after remission, the CLDN2 expression was strikingly decreased, and the filamentous and cytoplasmic staining disappeared.

CLDN2 is ectopically expressed in MCD and FSGS glomeruli. Renal biopsy sections were subjected to immunostaining with the corresponding antibodies. Typical micrographs are shown for cases with MCD and FSGS before remission (BR) and after remission (AR), as well as for cases with IgA-N. In the merged panels, CLDN2 is stained green, CD34 is red and HSPG (perlecan) is blue. Bar, 50 µm. MCD minimal change disease, FSGS focal segmental glomerulosclerosis, IgA-N IgA nephritis
We also quantitatively evaluated CLDN2 expression by calculating the CLDN2-positive area in glomeruli (Fig. 2). The CLDN2-stained region in MCD and FSGS glomeruli before remission was significantly greater than that after remission as well as in IgA-N patients. In addition, the level of CLDN2 expression was well correlated with the degree of proteinuria in each group at the time of biopsy (Tables 1 and 2). Interestingly, among the subjects before remission, CLDN2 was expressed at higher levels in MCD than in FSGS glomeruli.

The CLDN2-positive area is increased in MCD and FSGS glomeruli. Data are represented using box-and-whisker plots for cases with MCD before remission (BR; n = 8) and after remission (AR; n = 13), cases with FSGS BR (n = 8) and AR (n = 10), and IgA-N cases (n = 10). Results are analyzed using one-way ANOVA. *p < 0.05, and **p < 0.01. MCD minimal change disease, FSGS focal segmental glomerulosclerosis, IgA-N IgA nephritis
The anti-CLDN2 antibody has high specificity
To evaluate antibody specificity against CLDN2, we performed the western blot analysis. The anti-CLDN2 antibody did not react against the proteins derived from HEK293T cells expressing CLDNs and GFP, except for CLDN2 (Supplemental Fig. S1). Furthermore, to exclude nonspecific binding, we examined immunostaining with the control rabbit IgG as the primary antibody in the cases with MCD and FSGS before remission. This IgG was not incorporated with the protein existing in the glomeruli (Supplemental Fig. S2).
CLDN2 is expressed in MCD and FSGS podocytes
To determine whether CLDN2-expressing cells represent podocytes, we next performed multiple immunostaining using the podocyte markers synaptopodin (SYNPO) and podocalyxin (PODXL)21,22 (Fig. 3). In both MCD and FSGS glomeruli before remission, CLDN2 was at least in part colocalized with SYNPO and PODXL, suggesting that the observed CLDN2 expression occurred in podocytes.

CLDN2 is at least partially colocalized with the podocyte markers synaptopodin (SYNPO) and podocalyxin (PODXL) in MCD and FSGS glomeruli before remission. Renal biopsy sections were subjected to immunostaining with the corresponding antibodies. CLDN2 is stained green, SYNPO and PODXL are red, and HSPG (perlecan) is blue. Arrowheads indicate colocalization of CLDN2 with the podocyte markers. Bars, 50 µm in the left panel, 10 µm in the middle and right panels. MCD minimal change disease, FSGS focal segmental glomerulosclerosis
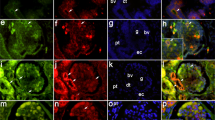
We subsequently verified, by immunogold immunoelectron microscopy, the nature of the CLDN2-positve cells in MCD glomeruli before remission, as well as the detailed subcellular localization of CLDN2 (Fig. 4). CLDN2 labeling was detected not only in the residual foot processes of podocytes (Fig. 4a) but also in the fused foot processes (Fig. 4b). Importantly, CLDN2 was also concentrated along newly formed TJs in the podocytes (Fig. 4c).

Immunogold electron micrographs showing the presence of CLDN2 in podocytes from MCD before remission. White and black arrowheads indicate SDs and newly formed TJs, respectively. Arrows reveal CLDN2 labeling in residual (a) and fused (b) foot processes, as well as at TJs (c). Bars, 200 nm. MCD minimal change disease, SD slit diaphragm, TJ tight junction
CLDN1 is not expressed in MCD glomeruli but is segmentally observed in FSGS
We also evaluated CLDN1 expression in the renal corpuscles of MCD and FSGS cases before remission (Fig. 5). As expected, PECs were positive for CLDN1 in both diseases. By contrast, no CLDN1 signals were apparent in the glomeruli of MCD subjects. In addition, CLDN1 was only focally and segmentally expressed in FSGS glomeruli with a trabecular pattern.

CLDN1 is segmentally observed in FSGS glomeruli but not in MCD glomeruli before remission. Renal biopsy sections were subjected to immunostaining with the corresponding antibodies. CLDN1 is stained green and HSPG (perlecan) is blue. Bar, 25 µm. FSGS focal segmental glomerulosclerosis, MCD minimal change disease
Newly formed TJs constructed by CLDN2 are generated together with a decrease in SDs in MCD and FSGS glomeruli before remission
To confirm the SD−TJ transition, we performed multiple immunostaining using the SD marker podocin,23 and compared CLDN2 and podocin expression in MCD and FSGS glomeruli (Fig. 6). In the cases with MCD and FSGS before remission, the filamentous signals for podocin were decreased and changed to the granulated signals together with the expression of strong filamentous signals for CLDN2. By contrast, in the cases with MCD and FSGS after remission, the filamentous signals for podocin were recovered together with decreased expression for CLDN2.

Strong filamentous signals for CLDN2 appeared together with a decrease in filamentous signals and changes to the granulated pattern for podocin in MCD and FSGS glomeruli before remission. Renal biopsy sections were subjected to immunostaining with the corresponding antibodies. Typical micrographs are shown for cases of MCD and FSGS before remission (BR) and after remission (AR). In the merged panels, CLDN2 and podocin are stained green, CD34 is red and HSPG (perlecan) is blue. Bar, 25 µm. MCD minimal change disease, FSGS focal segmental glomerulosclerosis
Discussion
In the present study, we found that CLDN2, which is detected in the epithelial cells of Bowman’s capsules, the proximal tubules and the thin descending limbs of Henle along the normal nephron,17 was ectopically expressed in injured podocytes in pediatric MCD and FSGS. In both diseases, CLDN2 was distributed along the glomerular basement membrane maker HSPG (Perlecan), and separated from the vascular endothelial marker CD34. In addition, CLDN2 was at least in part colocalized with the podocyte markers SYNPO and PODXL in MCD and FSGS cases. Moreover, CLDN2-immunogold signals were observed in podocytes, especially in the residual and fused foot processes as well as at the TJs, definitively indicating that the CLDN2-expressing cells were podocytes. Hence, immunofluorescence and immunoelectron studies for CLDN2 appear to afford a powerful tool for the diagnosis of primary NS. As no substantial abnormalities in glomerular structure are detected in MCD by light microcopy, CLDN2 could be a novel diagnostic marker, especially for patients who are resistant to steroid therapy and underwent renal biopsy.24
Although both MCD and FSGS are typical podocyte diseases,25,26 information on the pathophysiological basis of these diseases is still incomplete. In this regard, it is noteworthy that ectopic expression of CLDN2 in podocytes was observed not only in MCD but also in FSGS. Thus, both MCD and FSGS could be regarded as “CLDN2-related podocytopathies”. Lower expression levels of CLDN2 in FSGS before remission compared with those in MCD most probably reflect podocyte loss in FSGS. Even in the cases with FSGS before remission, there was a difference in CLDN2 staining. In these cases, only one patient did not achieve remission despite treatment. The CLDN2 expression in this case showed a more obvious segmental pattern compared to the other cases in this group. This finding also reflects podocyte loss. The CLDN2-immunoreactive area in the podocytes of both diseases after remission was significantly decreased to levels similar to that of the disease control group, further supporting the idea that CLDN2 expression is involved in their pathogeneses. As circulating glomerular permeability factors, including angiopoietin-like-4 and urokinase plasminogen activator receptor in MCD and FSGS, respectively, are expected to result in the onset of these diseases,3,27,28,29 it is intriguing to elucidate whether and how these factors are associated with CLDN2 expression in damaged podocytes.
CLDN1 expression, which is restricted to the PECs along the healthy nephron,17,18 is induced in the glomeruli from humans and animals with diabetic nephropathy.30 On the other hand, we demonstrated that CLDN1 was not principally observed in the glomerular tufts of pediatric MCD. In FSGS glomeruli before remission, the CLDN1 signals displayed a cord-like array with focal and segmental patterns, which differed completely from those of CLDN2. In FSGS, PECs are activated on Bowman’s capsules and migrate onto the glomerular capillary to substitute for or dislocate podocytes.31 Activated PECs in glomerulosclerotic lesions are also known to be positive for CLDN1.32 Taken collectively, CLDN1-expressing cells in the glomeruli of our FSCG cases appeared to correspond to activated PECs.
Podocin is one of the proteins forming SDs, and its mutations (NPHS2 gene) are responsible for the autosomal recessive form of steroid-resistant NS.23 The strong filamentous signals for CLDN2 appeared together with a decrease in the filamentous signals and changes to a granulated pattern for podocin in the cases with MCD and FSGS before remission. These changes suggest that SDs are displaced by TJs constructed by CLDN2 in the cases of MCD and FSGS before remission. In the cases with MCD and FSGS after remission, the filamentous signals for podocin were recovered. It appears that the SD-related molecules containing podocin accumulate to reform SDs.
Several TJ proteins, such as junctional adhesion molecule-A, coxsackie and adenovirus receptor, ZO-1 and cingulin, are concentrated at the SDs in mature podocytes.33,34,35 Among them, ZO-1 is indispensable for the interdigitation of foot processes and the formation of SDs,36 even though the precise roles of the other TJ components in the glomerular filtration barrier remain elusive. Therefore, we speculate that the ectopically expressed CLDN2 could recruit these TJ constituents from the SD pool and disrupt the architecture of the foot processes and SDs, resulting in their dedifferentiation into immature podocytes with glomerular dysfunction.
There were some limitations to the present study. First, the cases before and after remission in the MCD and FSGS groups were different patients, respectively. Therefore, we could not compare changes before and after treatment in the same patient. Second, other histologic types that present nephrotic conditions exist in NS patients. Therefore, we also examined the expression of CLDN2 in a few cases with membranous nephropathy (MN) and membranoproliferative glomerulonephritis (MPGN). However, our cases with MN and MPGN presented chronic glomerulonephritis, not a nephrotic condition. The expression of CLDN2 in such cases did not differ significantly from the IgA-N cases.
In conclusion, we showed that both MCD and FSGS in children possessed the same pathological findings in terms of the ectopic CLDN2 expression in podocytes. We also demonstrated that the level of CLDN2 was diminished after remission, indicating that the levels of CLDN2 expression are related to disease status. Further studies are required to clarify the functional relevance of CLDN2 expression in the pathogenesis of these diseases.
References
Eddy, Allison A. & Symons, Jordan M. Nephrotic syndrome in childhood. Lancet 362, 629–639 (2003).
D’Agati, V. D., Kaskel, F. J. & Falk, R. J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 365, 2398–2411 (2011).
Floege, J. & Amann, K. Primary glomerulonephritides. Lancet 387, 2036–2048 (2016).
Reeves, W., Caulfield, J. P. & Farquhar, M. G. Differentiation of epithelial foot processes and filtration slits: sequential appearance of occluding junctions, epithelial polyanion, and slit membranes in developing glomeruli. Lab Invest. 39, 90–100 (1978).
Grahammer, F., Schell, C. & Huber, T. B. The podocyte slit diaphragm—from a thin grey line to a complex signalling hub. Nat. Rev. Nephrol. 9, 587–598 (2013).
Schell, C., Wanner, N. & Huber, T. B. Glomerular development—shaping the multi-cellular filtration unit. Semin Cell Dev. Biol. 36, 39–49 (2014).
Pricam, C. et al. Intercellular junctions in podocytes of the nephrotic glomerulus as seen with freeze-fracture. Lab Invest. 33, 209–218 (1975).
Ryan, G. B., Leventhal, M. & Karnovsky, M. J. A freeze-fracture study of the junctions between glomerular epithelial cells in aminonucleoside nephrosis. Lab Invest. 32, 397–403 (1975).
Caulfield, J. P., Reid, J. J. & Farquhar, M. G. Alterations of the glomerular epithelium in acute aminonucleoside nephrosis. Evidence for formation of occluding junctions and epithelial cell detachment. Lab Invest. 34, 43–59 (1976).
Kurihara, H. et al. The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am. J. Pathol. 141, 805–816 (1992).
Lahdenkari, A. T. et al. Podocytes are firmly attached to glomerular basement membrane in kidneys with heavy proteinuria. J. Am. Soc. Nephrol. 15, 2611–2618 (2004).
Furuse, M. et al. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 143, 391–401 (1998).
Tsukita, S., Furuse, M. & Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2, 285–293 (2001).
Van Itallie, C. M. & Anderson, J. M. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 68, 403–429 (2006).
Chiba, H. et al. Transmembrane proteins of tight junctions. Biochim. Biophys. Acta 1778, 588–600 (2008).
Günzel, D. & Yu, A. S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 93, 525–569 (2013).
Kiuchi-Saishin, Y. et al. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J. Am. Soc. Nephrol. 13, 875–886 (2002).
Ohse, T. et al. Establishment of conditionally immortalized mouse glomerular parietal epithelial cells in culture. J. Am. Soc. Nephrol. 19, 1879–1890 (2008).
Ohse, T. et al. The enigmatic parietal epithelial cell is finally getting noticed: a review. Kidney Int. 76, 1225–1238 (2009).
Yu, A. S. Claudins and the kidney. J. Am. Soc. Nephrol. 26, 11–19 (2015).
Kerjaschki, D., Sharkey, D. J. & Farquhar, M. G. Identification and characterization of podocalyxin—the major sialoprotein of the renal glomerular epithelial cell. J. Cell Biol. 98, 1591–1596 (1984).
Mundel, P. et al. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J. Cell Biol. 139, 193–204 (1997).
Caridi, G. et al. Prevalence, genetics, and clinical features of patients carrying podocin mutations in steroid-resistant nonfamilial focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 12, 2742–2746 (2001).
Gulati, S. et al. Do current recommendations for kidney biopsy in nephrotic syndrome need modifications? Pedia. Nephrol. 17, 404–408 (2002).
Wiggins, R. C. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 71, 1205–1214 (2007).
D’Agati, V. D. The spectrum of focal segmental glomerulosclerosis: new insights. Curr. Opin. Nephrol. Hypertens. 17, 271–281 (2008).
Clement, L. C. et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat. Med. 17, 117–122 (2011).
Wei, C. et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 17, 952–960 (2011).
Chugh, S. S., Clement, L. C. & Macé, C. New insights into human minimal change disease: lessons from animal models. Am. J. Kidney Dis. 59, 284–292 (2012).
Hasegawa, K. et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 19, 1496–1504 (2013).
Shankland, S. J. et al. The emergence of the glomerular parietal epithelial cell. Nat. Rev. Nephrol. 10, 158–173 (2014).
Smeets, B. et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 22, 1262–1274 (2011).
Schnabel, E., Anderson, J. M. & Farquhar, M. G. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J. Cell Biol. 111, 1255–1263 (1990).
Nagai, M. et al. Coxsackievirus and adenovirus receptor, a tight junction membrane protein, is expressed in glomerular podocytes in the kidney. Lab Invest. 83, 901–911 (2003).
Fukasawa, H. et al. Slit diaphragms contain tight junction proteins. J. Am. Soc. Nephrol. 20, 1491–1503 (2009).
Itoh, M. et al. The structural and functional organization of the podocyte filtration slits is regulated by Tjp1/ZO-1. PLoS ONE 9, e106621 (2014).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kanno, S., Kume, Y., Maeda, R. et al. Ectopic expression of CLDN2 in podocytes is associated with childhood onset nephrotic syndrome. Pediatr Res 86, 485–491 (2019). https://doi.org/10.1038/s41390-019-0423-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0423-7
