
Abstract
The placenta is vital for fetal growth, and compromised function is associated with abnormal development, especially of the brain. Linking placental function to brain development is a new field we have dubbed neuroplacentology. Approximately 380,000 infants in the United States each year abruptly lose placental support upon premature birth, and more than 10% of pregnancies are affected by more insidious placental dysfunction such as preeclampsia or infection. Abnormal fetal brain development or injury can lead to life-long neurological impairments, including psychiatric disorders. The majority of research connecting placental compromise to fetal brain injury has focused on gas exchange or nutritional programming, neglecting the placenta’s essential neuroendocrine role. We will review the current evidence that placental dysfunction, particularly endocrine dysfunction, secretion of pro-inflammatory cytokines, or barrier breakdown may place many thousands of fetuses at risk for life-long neurodevelopmental impairments each year. Understanding how specific placental factors shape brain development and increase the risk for later psychiatric disorders, including autism, attention deficit disorder, and schizophrenia, paves the way for novel treatment strategies to maintain the normal developmental milieu and protect from further injury.
Similar content being viewed by others

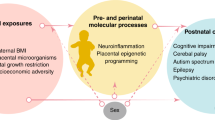
The placenta is a vital component of healthy pregnancy; compromised placental function has been linked to diverse developmental brain disorders. Some neuropsychiatric disorders present in early childhood, such as autism spectrum disorders (ASDs) or attention deficit hyperactivity disorder (ADHD), while some, such as schizophrenia or depression, usually present later.1,2 Early insults that alter in utero brain development have been linked to these disorders (Fig. 1). During developmental critical periods, the brain may be “programmed” for conditions that will occur later in life. The placenta, a fetal organ with the same genetic makeup as the fetus and the organ controlling the intrauterine environment, can play a key role in this programming.

Critical periods of development and corresponding symptomatology of neuropsychiatric disorders
The placenta’s role in protecting and shaping the fetal brain, what we call “neuroplacentology,” has only recently started to be investigated. As the interface between maternal and fetal circulation, the placenta monitors maternal well-being and aims to modify this environment to optimize fetal development through changes in placental function. Placental substrate transport, metabolism, and placental hormone production are all important to fetal development. The life-long developmental potential of the fetus can be compromised when the placenta fails to function properly. Many events, such as infection, malnutrition, and genetic abnormalities, can disrupt placental function or, as in preterm birth, can abruptly change the fetal brain’s environment. More than 10% of all pregnancies are affected by some degree of placental failure that can lead to growth restriction, preterm delivery, or still birth.3 Placental failure can directly damage the developing brain or increase its susceptibility to injury, leading to permanent neurological disabilities. We will review the current evidence for the hypothesis that placental dysfunction or loss may “program” or predispose an individual to psychiatric disorders not seen until childhood or adulthood. Understanding how the placenta integrates maternal and fetal information that lead to long-term programming may suggest new ways to manage pregnancy complications and prevent abnormal neurodevelopment or injury.
Placental function and fetal programming
Epidemiological evidence first suggested that interactions between maternal and fetal systems promote fetal growth and normal pregnancy outcomes and alter the in utero environment in ways that lead to long-term changes, particularly cardiovascular and metabolic disease.4 The links between the in utero nutritional environment and later cardiovascular morbidities were recognized by Barker et al.5 in the late 1980s. Since then, a new field of “developmental origins of health and disease” has emerged.6 Nutrition is a well-described mechanism of fetal programming; maternal undernutrition can elicit placental and fetal adaptive responses that lead to metabolic, hormonal, and immune reprogramming, resulting in intrauterine growth-restricted (IUGR) fetuses with increased risk of metabolic and cardiovascular disease in adulthood. Adult diseases such as coronary heart disorders, hypertension, atherosclerosis, type 2 diabetes, insulin resistance, altered cell-mediated immunity, and cancer have all been linked to fetal programming.7
Maternal health, dietary status, and exposure to environmental factors, uteroplacental blood flow, placental transfer, and fetal genetic and epigenetic responses likely all contribute this programming. Cytokines, hormones, growth factors, and the intrauterine immune milieu can also contribute to in utero programming. In addition to these chemical signals, a role for extracellular vesicles and shedding of micro-particles (trophoblast “debris”) has been recently identified as a mechanism of communication between mother and fetus. These particles comprise heterogeneous collection of membrane-bound carriers with complex cargoes, including proteins, lipids, and nucleic acids.8 How these signaling pathways alter maternal response to pregnancy and fetal development is an active area of current investigation.9,10
Placental factors that predispose to mental illness
Both acute and chronic placental dysfunction is linked to adverse life-long neurological injury11 (Table 1). Evidence is now emerging that links the placenta to changes in the developing brain that may increase the risk of, or even cause, psychiatric disorders. Acute interruption of placental circulation (umbilical cord occlusion or placental abruption) may result in severe neurological hypoxic–ischemic damage before or during delivery.3,11,12,13,14 Chronic placental vascular lesions (chronic villitis, fetal thrombotic vasculopathy, or infection-associated fetal vasculitis) are correlated with neurological injury.3 Almost 90% of term infants with neonatal encephalopathy and brain injury have placental lesions noted post delivery, although the high frequency of placental lesions in apparently neurologically intact newborns makes interpretation complex.3,13,14
There are important time-points in which the embryo and fetus is more vulnerable to stressful events, and the impact of such events will vary depending on when they occur during gestation. In the embryonic period, the first 8 weeks post conception in humans, the extent of developmental programming via the placenta is unclear. Early placental transport functions for nutrients, such as folate, may substantially impact embryonic organogenesis. However, with the transition to the fetal period, placental blood flow in the intravillous spaces allows a significant increase in maternal–fetal information exchange, with molecular and chemical signals transmitted to the fetus about the maternal environment and vice versa (Table 2). It is primarily during fetal developmental windows or “critical periods” that the nervous system may be programmed for conditions that will occur later in life. Altered brain development and subsequent brain disorders maybe impacted by placental events, including:
-
1.
Prematurity: The loss of placental support due to preterm birth is an independent risk factor for neurological injury. Preterm birth has profound impacts on postnatal neurodevelopment,2 although the extent to which these changes are related to postnatal insults remains controversial.15 Additional complications, such as infection, may exacerbate issues arising from prematurity.14,16,17,18,19 Events preceding premature delivery, particularly IUGR due to impaired placental function, may also enhance the risk of neurodevelopmental impairment in premature infants.18,19,20,21,22
-
2.
Infection: The role of placental immune activation in poor neonatal outcome, particularly in neurological complications, has become an area of intense study in the past decade.18,23,24 Maternal infection has been implicated in the etiology of neuropsychiatric disorders, including ASD, schizophrenia, and generalized cognitive impairment.24 Direct infection of the placenta is often associated with devastating outcomes in the embryo and fetus.25 Additionally, the placenta itself can sense and respond to pathogen molecular patterns or immune cytokines released into the blood stream from distant sites of infection.26,27 Animal studies have established that activation of the maternal immune system during pregnancy is alone sufficient to cause abnormalities in social behavior, complex learning tasks and sensorimotor gating in offspring.28,29,30 Evaluation of the postnatal or adult brains of offspring show diffuse anatomical alterations following the prenatal challenge.31,32,33,34 Viral infections such as influenza may also modulate genetic predisposition associated with schizophrenia or autism.31,35,36 The placenta is likely the key mediator of these injuries.
-
3.
Genetic factors: The placenta maintains a fine equilibrium between the genomic signature of the mother and the fetus. The placental genetic program—remarkable for an abrupt shift in overall gene expression pattern in mid-gestation—is strikingly similar between mouse and human37,38,39 with high conservation of genes and proteins related to placental dysfunction.40 Interestingly, gene expression in human placentas follows a different pattern after in vitro fertilization or in intracytoplasmic sperm injection with particular changes in imprinted genes, potentially leading to expression changes that could alter placental function, and thus brain development, across pregnancy.41 It is intriguing that many genes highly expressed in placenta are also expressed in brain, particularly those that may be linked to neuropsychiatric disease.42
-
4.
Epigenetic factors: Epigenetic mechanisms used by the placenta are unique, but are most similar to those seen in cancer, reflecting their invasive properties. Large-scale patterns of hypomethylation, as well as site-specific hypermethylation of tumor suppressor genes, occurs in placenta.43 In addition, microRNAs (miRNAs) that have been implicated in regulating uterine invasion are associated with malignancies when reactivated outside of the placental context.13 Given the limited invasive potential of the placenta under normal circumstances, there is likely to be an additional layer of regulation controlling the placental epigenetic response to both intrinsic and environmental cues. Massive epigenetic reprogramming with loss of DNA methylation occurs just after fertilization and prior to implantation.44 The placenta and other extra-embryonic tissue remain hypomethylated, even though methylation increases with cellular differentiation, so that even at term, the placenta is the most hypomethylated human tissue.44 Within the placenta, the various cell types have distinct methylation profiles. Nutritional components, such as folate, can alter placental and fetal methylation patterns.45 Placental methylation changes have been linked to preeclampsia, IUGR, and preterm birth, although whether these changes are causal or result from disease is undetermined.44,45,46
-
5.
Barrier dysfunction: As an interface, the placenta is responsible for protection of the developing fetus from maternal insults. For example, in the human placenta, fetal cortisol exposure is controlled by expression of placental 11-β-hydroxysteroid dehydrogenase (11βHSD), with maternal cortisol metabolized to inactive cortisone primarily by the enzyme isoform 11βHSD247,48 A recent study in an ex vivo placental perfusion model also demonstrated that there can be de novo synthesis of glucocorticoids in the placenta on both fetal and maternal sides.49 Cortisol is the common mediator of many programming events. High levels of maternal cortisol due to stress, or impaired activity of placental 11βHSD2 isoform due to stressors (e.g., hypoxia or other insults) may lead to fetal tissues being exposed to higher than normal glucocorticoid levels, suppressing cell proliferation and inducing epigenetic changes.47,50,51
-
6.
Maternal stressors: Stressors, such hypoxia or malnutrition, may directly impact the developing fetus or alter gene expression, thus altering offspring phenotype.52,53 Changes in fetal cortisol exposure and inflammatory processes have both been implicated. For example, in preeclampsia, oxidative stress of the placenta caused by malperfusion can elicit a sterile inflammatory response, releasing pro-inflammatory cytokines into both the maternal and fetal circulations.44,54 Furthermore, the effect of maternal stress on the placenta is potentiated by the presence of comorbidities, such as severe maternal obesity, which alters the maternal inflammatory status and increases fetal glucocorticoid exposure.55 Placental exposure to cortisol also increases placental corticotropin-releasing hormone (CRH), resulting in a positive feedback loop, rather than the negative feedback loop seen in the hypothalamus. Placental CRH has been postulated to play a role in timing of parturition, as well as in fetal HPA axis programming.56 As maternal cortisol increases, placental CRH increases further increasing glucocorticoid production, the placental enzymatic barrier is overwhelmed and the fetal brain may be exposed to high glucocorticoid levels that alter the trajectory of multiple neurodevelopmental processes.56
The role of placenta in specific neuropsychiatric disorders
Autism
There is emerging evidence that the placenta plays a major role in ASD pathogenesis.57 Human studies have focused on anatomical placental differences and epigenetic risk, while animal studies have begun to reveal potential mechanisms linking placental dysfunction to later ASD-like behaviors.
Gross pathology of the placenta from ASD patients or patients at increased risk of ASD suggests a correlation between placental architecture and later ASD. Anatomical variations in the placental chorionic surface vascular network may reflect both genetic and environmental influences on vascular branching morphogenesis and have therefore been explored in many pregnancy conditions.58 In a cohort with an elevated risk of ASD, placental evaluation revealed fewer branch points, thicker and less tortuous arteries, better extension to the surface boundary, and smaller branch angles than population-based counterparts.58 In another histological study of placentas from the Early Autism Risk Longitudinal Investigation network and National Children’s Study, placentas from mothers who had a prior child with ASD were thicker and rounder with a more regular perimeter compared to placentas from the general population, suggesting restricted placental responses to the in utero environment.59 In addition to anatomical variations, histological evidence of placental inflammation is increased in ASD patients. For example, a recent retrospective review of placentas from individuals with ASD showed significantly higher rates of acute generalized inflammation, fetal inflammatory response, and maternal vascular malperfusion, especially in males.57 Currently, the relationship between placental histology and placental function is not well defined, but the correlations with ASD suggest that placentas may reflect or even help to create an adverse environment for fetal brain development.
Beyond histological studies, placental epigenetic modifications suggest a link to ASD. The MARBLES (Markers of Autism Risk in Babies: Learning Early Signs) prospective pregnancy study follows high-risk younger siblings of children with ASD and has examined placental DNA methylation. Clinical pregnancy features and environmental exposures were correlated with methylation changes. While the placenta is a hypomethylated tissue, it does have partially methylated domains (PMDs), a specific embryonic feature of the placental methylomes.60 The strongest, most robust associations reported from MARBLES suggested that pesticide exposures could alter placental DNA methylation more than other factors, with professional pesticide application correlating with higher than average genomic methylation and decreased genomic PMDs.60 Whole-genome bisulfite sequencing of term placentas from MARBLES revealed that human placental methylomes have highly reproducible PMD and highly methylated domain (HMD) locations, but that there was more variation between PMD regions than HMD regions in individuals, making interpretation of placental methylation data complex.60 However, placentas from ASD diagnosed individuals showed genome-wide significance of higher methylation in an HMD containing a putative fetal brain enhancer near Delta-like protein 1 (DLL1).61
Preclinical animal models have also supported a link between placenta and ASD-like behavioral features. When pregnant rodents are treated with immunogenic agents [viral polyriboinosinic-polyribocytidylic acid (polyI:C) or bacterial lipopolysaccharide (LPS)], acute viral infection or stress induction in mid-pregnancy, the offspring frequently show ASD-like behavioral features. Maternal inflammation and cytokine production, especially interleukin-6, -2, and -17a (IL-6, IL-2, and IL-17a), is particularly strongly correlated with neurodevelopmental impairment in offspring.62,63,64,65 In another elegant study, IL-6 receptor was specifically deleted only from placenta in pregnant dams exposed to an inflammatory stimulus, leading to the blockade of brain inflammation in the offspring.64
Additional environmental factors, such as vitamin D deficiency (DVD), can contribute to placental cytokine response and neurobehavioral outcomes. Vitamin D is an essential regulator of immune function. DVD was found to have no effect on the baseline inflammatory cytokines expressed in the placenta. However, when DVD rats received an inflammatory challenge, IL-6 and IL-1β placental production increased after polyI:C exposure compared to control placentas, especially in males.66 LPS exposure did not alter these cytokines in DVD rats. These results highlight differences in placental response depending on both fetal sex and immunological agent. In mice exposed to polyI: C, vitamin D administration during gestation prevented ASD-like behaviors in male offspring.67 These preclinical results suggesting that the male fetus may be particularly vulnerable to maternal viral exposures, a finding that reflects the human epidemiological data.68 In addition, there is some epidemiological evidence that low vitamin D, without obvious exposure to inflammatory agents, may be a risk factor for ASD.69,70,71,72,73 The placenta can produce vitamin D precursors that may protect the fetal brain against glutamate excitotoxicity and enhance serotonin production.68,74,75 A recent meta-analysis suggested that higher concentrations of 25(OH)D in the prenatal period were associated with decreased probability of autistic phenotypes.75
ASD is strikingly sex linked, with a 4× greater prevalence in males than females.76,77 Whether this sex difference is linked directly to placental sex is still unclear, but there is growing evidence that changes in placental function may program later sex-linked neurodevelopmental outcomes.53,78,79,80 A series of studies by Bale and colleagues has provided strong evidence that early pregnancy stress programs not only hypothalamic development and feeding behaviors, but also long-term ASD-like behaviors in mice in a sex-linked manner.80,81 Offspring of pregnant mice exposed to stress during the first week of pregnancy have increased placental insulin-like growth factor 2 expression and sex-specific changes in 11βHSD2.79 Placental insulin receptor was also altered in this paradigm, with sex-specific neurodevelopment consequences.78,82
Whether or not there is a female “protective effect” against ASD or whether the female ASD phenotype is somewhat different remains unclear, but X-linked gene expression suggests one potential mechanism of protection and programming.77,83,84 Other core features of ASD, such as anxiety, can be produced more readily in female offspring, an outcome potentially linked to placental endocrine function or differences in fetal brain programming.85,86
Schizophrenia
Several theories regarding the causative mechanism of schizophrenia have been proposed that link the in utero environment to later disease development.32,35,87,88,89,90 Epidemiological data strongly supports an association between maternal viral illnesses, particularly influenza infection, in early gestation with later development of schizophrenia in offspring. Viral infections during pregnancy often result in overexpression of IL-1β and IL-6, and tumor necrosis factor-α by the placenta and the fetus. Maternal infection may permanently affect the placenta and fetus either through altered gene expression or epigenetic modification.36,88
Schizophrenia has been linked to insults that may occur early in life and might be related to pregnancy (hemorrhage, preeclampsia, or hyperglycemic conditions), or labor (birth asphyxia, uterine rupture), or to fetal conditions such as IUGR or genetic anomalies.91 Recent work by Ursini et al. has linked a multitude of insults, defined broadly as early life complications (ELCs), with placental genomic signatures that may increase the risk of schizophrenia.42 Genetic risk for schizophrenia was defined as polygenic risk scores derived from genome-wide association studies done in independent populations from the United States, Europe, and Japan. Report of ELCs greatly increased the association of PRS with schizophrenia.42 The gene loci linked to schizophrenia risk were highly expressed in placenta, were increased in male placenta compared to female, and were differentially expressed in placentas from complicated pregnancies compared to normal. To definitively link these placental genetic patterns with schizophrenia, there needs to be a collection of placentas from a cohort enriched for later schizophrenia (as is being done in the MARBLES study for ASD), but these new findings support the need for such investigations. Intriguingly, gene pathway analysis suggests that the interaction of ELC and PRS is mainly driven by genes in the cellular stress response pathways in the brain. Thus, a subset of the most significant genetic variants associated with schizophrenia in conjunction with the ability of the brain cells to respond to stress may determine the final phenotype.42
In addition to a fetal genetic contribution to schizophrenia risk via placental gene expression, there is likely to be a maternal contribution. Abnormal brain levels of kynurenic acid (KYNA), an endogenous antagonist of N-methyl-d-aspartate and α7 nicotinic acetylcholine receptors, have been associated with schizophrenia-like phenotypes in animal models.92 It was recently reported that genetic alteration of maternal kynurenine 3-monooxygenase, and thus fetal KYNA exposure, may lead to increased placenta and brain KYNA, suggesting that maternal placental genotype may also contribute to schizophrenic pathology.93 While studies on schizophrenia risk related to placental function or gene expression remain limited, they hint that the placenta is a key player in controlling the impact of in utero genetic and environmental risks that can later lead to schizophrenia.
Mood disorders
Both human and rodent studies have investigated the association between maternal mood disorders or stress, respectively, in terms of offspring resilience to stress and the later development of mood disorders. The developing brain is highly sensitive to glucocorticoids, which play a role in neuronal maturation as well as programmed cell death. Many maternal stressors—depression, trauma, and malnutrition—can alter maternal glucocorticoid levels and affect the placenta. In addition, poor placental function that is associated with IUGR can alter fetal glucocorticoid exposure. Placental enzymes that regulate maternal–fetal glucocorticoid and serotonin transfer appear to be key modulators of fetal brain programming linked to maternal depression.94 11βHSD2 is highly expressed in placenta, protecting the fetus from the normal increase of maternal cortisol that occurs across gestation. Maternal mood disorders are associated with disruption of this placental barrier, in part by suppressing 11βHSD2 expression, leading to abnormal neurodevelopment and potentially to mood disorders in the offspring as well.95
Most studies have focused on alterations in cortisol and serotonin because of their strong link to mood disorders.94 One study aimed to link maternal mood with fetal distress and epigenetic changes in placental genes. While perceived stress (Perceived Stress Scale) was associated with altered methylation patterns in the placenta, cortisol levels were not.96 Of note, 11βHSD2 methylation was significantly modified. Another study prospectively assessed maternal depressive symptoms during pregnancy and then explored both placental gene expression for genes regulating glucocorticoid and serotonin levels in term placentas from these pregnancies, as well as association of depression with later infant regulatory behaviors.97 Higher placental expression of the glucocorticoid receptor, encoded by nuclear receptor subfamily 3 group C member 1, partly mediated the association between maternal depressive symptoms during pregnancy and infant regulatory behaviors. Such studies suggest that maternal depression acts, at least partly, by altering glucocorticoid action in the placenta leading to changes in fetal cortisol exposure that program the neonatal neurobehavioral phenotype.97
Genetically modified mouse models have been particularly useful in beginning to provide a mechanistic link between glucocorticoid exposure, placental alterations, and neurodevelopmental changes in the offspring. Mice in which 11βHSD2 is globally knocked out show increased anxiety and other abnormal behavioral phenotypes.98 Placental-specific deletion of 11βHSD2 has not yet been reported, but results of such an experiment could directly link placental enzyme loss with neurodevelopmental reprogramming.
In addition to changing fetal glucocorticoid exposure, genetic loss of 11βHSD2 changes placental nutrient transport as well as neurotransmitter pathways in the brain, including serotonin.99 Serotonin, a neurotransmitter highly associated to depression and its treatment, is produced by the placenta in early gestation, and regulates wiring patterns in the developing mouse forebrain. In a series of elegant experiments, Bonnin et al.100 showed that the source of serotonin guiding axon trajectories is not maternal, but rather generated by a placental serotonin synthetic pathway. This work provided early evidence for a new direct role of placental metabolic pathways in modulating early fetal brain development. Both rodent and human studies suggest that the placenta can signal adverse maternal conditions, such as depression and stress, to the fetus and that these signals may produce long-lasting mental health disorders, including mood disorders, in offspring. Better understanding of placental signaling mechanisms and how these pathways may contribute to later outcomes, including the intersection of the glucocorticoid and serotonin pathways, may provide new placental biomarkers for later risk of mood disorders, as well as new opportunities for early interventions to increase resilience in offspring at risk.
Additional placental alterations have also been noted to occur with maternal depression. One study showed aberrant placental expression of several imprinted genes, including paternally expressed gene 3 in male placentas. Human placental lactogen expression was also significantly decreased in placentas from women with clinically diagnosed depression and in those with high depression scores.101 Whether these gene expression changes are related to maternal cortisol levels or other factors is unclear. Even more importantly, the extent to which placental changes directly alter neurodevelopment and contribute to later depressive disorders in offspring, particularly compared to the significant contribution of genetic factors, is not yet clear.
Disorders of executive functioning
Executive functioning (EF) is the ability to function and utilize working memory, inhibition, cognitive flexibility, goal selection, organization, and planning. Recent research shows that learning difficulties and behavioral problems are significantly associated with EF deficits that are thought to stem primarily from prefrontal lobe dysfunction. EF plays a critical role in cognitive development and is vital to individual social and intellectual success.16 It has recently been recognized that premature neonates exhibit EF deficits later in childhood or adult life.102 While there is not yet any data directly linking placental loss due to preterm birth directly to this impaired EF, there is circumstantial evidence accumulating for placental involvement. In a study from 2016, children born prematurely were assessed up to 10 years of age. More than half of the extremely preterm cohort exhibited moderate or severe neurocognitive deficits at age 10 years, with the most extensive impairments found among those born at the lowest gestational age, the population that loses placental function at the earliest time.17 A prospective study by Baron et al.103 showed that late-premature neonates are also at higher risk for cognitive impairment, although to a lesser degree, and that male gender is an additive risk factor.103 In a separate study, late premature neonates were divided into two categories based on the reason for the premature birth—medically indicated premature delivery versus spontaneous birth. Those delivered for medical indications had higher levels of childhood attention problems, suggesting that pregnancy complications motivating medical intervention during the late-preterm period, complications that are primarily due to placental dysfunction with preeclampsia and fetal growth restriction, can increase the risk of EF.104 The lack of representative animal models for EF deficits make mechanistic links difficult to establish, but preclinical models of prematurity provide opportunities to define the key placental factors that may be lost; human studies can then determine these factors are deficient in preterm infants and whether replacement improves EF and overall cognitive development.105
Conclusion
Investigating placental changes associated with human neuropsychiatric outcomes has suggested a significant role for placental programming, but mechanistic studies in preclinical models are needed to demonstrate that some, or any, of these placental changes are directly causal. The NICHD Human Placenta Project is beginning to provide new tools to measure early human placental function.106,107 Short- and long-term tracking of offspring from studied pregnancies will require coordination between researchers, obstetricians, neonatologists, and other pediatrician, but such studies would provide more precise correlations between placental function and newborn reactivity and later psychological development. Causal relationships are more difficult to demonstrate, but genetic manipulations of rodent models are beginning to reveal mechanisms that may underlie these associations. Many questions remain to be definitively answered: what is the relative importance of placental genetic versus epigenetic change in fetal brain programming? To what extent will these associations provide actionable information from which biomarkers can be developed or even new interventions to reduce risk?
The complex maternal–fetal genetic and epigenetic milieu maybe altered by a myriad of insults during gestation, subtly changing placental function and only later presenting as neuropsychiatric disorders. In utero insults, such as viral infection or stress, may lead to very different neurodevelopmental outcomes depending on both the insult and the timing of exposure. However, common themes are emerging that suggest that the placenta is a critical hub that combines information about the in utero environment with signals that shape the development of the fetal brain. There are emerging roles for both genetic and epigenetic changes in placenta and brain, changes that may be linked by inheritance as well as by environmental modifiers that alter later risk. In addition, significant placental sex-linked differences are becoming apparent across multiple disorders and imply that interventions to reduce long-term risk will require sex-specific approaches. The consequences of impaired placental function may have life-long impact not only on the mental health of individual offspring, but also on multiple generations affected by both maternal conditions and epigenetic placental changes.
References
Nosarti, C. et al. Preterm birth and psychiatric disorders in young adult life. Arch. Gen. Psychiatry 69, E1–E8 (2012).
Marin, O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 22, 1229–1238 (2016).
Redline, R. W. Severe fetal placental vascular lesions in term infants with neurologic impairment. Am. J. Obstet. Gynecol. 192, 452–457 (2005).
Barker, D. J. & Osmond, C. Childhood respiratory infection and adult chronic bronchitis in England and Wales. BMJ (Clin. Res. Ed.) 293, 1271–1275 (1986).
Barker, D. J. et al. Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580 (1989).
Burton, G. J., Fowden, A. L. & Thornburg, K. L. Placental origins of chronic disease. Physiol. Rev. 96, 1509–1565 (2016).
Sallout, B. & Walker, M. The fetal origin of adult diseases. J. Obstet. Gynaecol. 23, 555–560 (2003).
Nair, S. & Salomon, C. Extracellular vesicles and their immunomodulatory functions in pregnancy. Semin. Immunopathol. 40, 425–437 (2018).
Fallen, S. et al. Extracellular vesicle RNAs reflect placenta dysfunction and are a biomarker source for preterm labour. J. Cell Mol. Med. 22, 2760–2773 (2018).
Liu, H. et al. Estimation of the burden of human placental micro- and nano-vesicles extruded into the maternal blood from 8 to 12 weeks of gestation. Placenta 72–73, 41–47 (2018).
Nelson, K. B. & Blair, E. The placenta and neurologic and psychiatric outcomes in the child: study design matters. Placenta 32, 623–625 (2011).
VanWijk, M. J. et al. Vascular function in preeclampsia. Cardiovasc. Res. 47, 38–48 (2000).
Wintermark, P. et al. Placental pathology in asphyxiated newborns meeting the criteria for therapeutic hypothermia. Am. J. Obstet. Gynecol. 203, 579 e571–e579 (2010).
Lachapelle J. et al. Placental pathology in asphyxiated newborns treated with therapeutic hypothermia. J. Neonatal. Perinatal. Med. 8, 33–40 (2015).
Bonifacio, S. L. et al. Extreme premature birth is not associated with impaired development of brain microstructure. J. Pediatr. 157, 726–732 e721 (2010).
Taylor, H. G. & Clark, C. A. Executive function in children born preterm: risk factors and implications for outcome. Semin. Perinatol. 40, 520–529 (2016).
Joseph, R. M. et al. Investigators ES. Neurocognitive and academic outcomes at agosartie 10 years of extremely preterm newborns. Pediatrics 137 (2016). https://doi.org/10.1542/peds.2015-4343e20154343.
Kim, C. J. et al. Acute chorioamnionitis and funisitis: definition, pathologic features, and clinical significance. Am. J. Obstet. Gynecol. 213, S29–S52 (2015).
Romero, R., Dey, S. K. & Fisher, S. J. Preterm labor: one syndrome, many causes. Science 345, 760–765 (2014).
Apel-Sarid, L. et al. Term and preterm (<34 and <37 weeks gestation) placental pathologies associated with fetal growth restriction. Arch. Gynecol. Obstet. 282, 487–492 (2010).
Redline, R. W. et al. Placental lesions as predictors of cerebral palsy and abnormal neurocognitive function at school age in extremely low birth weight infants (<1 kg). Pediatr. Dev. Pathol. 10, 282–292 (2007).
Leviton, A. et al. Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Developmental Epidemiology Network Investigators. Pediatr. Res 46, 566–575 (1999).
Kim, C. J. et al. Chronic inflammation of the placenta: definition, classification, pathogenesis, and clinical significance. Am. J. Obstet. Gynecol. 213, S53–S69 (2015).
Faa, G. et al. Fetal programming of neuropsychiatric disorders. Birth Defects Res. C 108, 207–223 (2016).
Elovitz, M. A. et al. Intrauterine inflammation, insufficient to induce parturition, still evokes fetal and neonatal brain injury. Int. J. Dev. Neurosci. 29, 663–671 (2011).
Ellman, L. M. et al. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr. Res. 121, 46–54 (2010).
Curtis, D. J. et al. Secretions from placenta, after hypoxia/reoxygenation, can damage developing neurones of brain under experimental conditions. Exp. Neurol. 261, 386–395 (2014).
Patterson, P. H. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav. Brain Res. 204, 313–321 (2009).
Wolff, A. R. & Bilkey, D. K. The maternal immune activation (MIA) model of schizophrenia produces pre-pulse inhibition (PPI) deficits in both juvenile and adult rats but these effects are not associated with maternal weight loss. Behav. Brain Res. 213, 323–327 (2010).
Ozawa, K. et al. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol. Psychiatry 59, 546–554 (2006).
Buehler, M. R. A proposed mechanism for autism: an aberrant neuroimmune response manifested as a psychiatric disorder. Med. Hypotheses 76, 863–870 (2011).
Brown, A. S. & Derkits, E. J. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am. J. Psychiatry 167, 261–280 (2010).
Ashdown, H. et al. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol. Psychiatry 11, 47–55 (2006).
Hsiao, E. Y. & Patterson, P. H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 25, 604–615 (2011).
Brown, A. S. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev. Neurobiol. 72, 1272–1276 (2012).
Careaga, M., Murai, T. & Bauman, M. D. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol. Psychiatry 81, 391–401 (2017).
Knox, K. & Baker, J. C. Genomic evolution of the placenta using co-option and duplication and divergence. Genome Res. 18, 695–705 (2008).
Winn, V. D. et al. Gene expression profiling of the human maternal-fetal interface reveals dramatic changes between midgestation and term. Endocrinology 148, 1059–1079 (2007).
Tanaka, T. S. et al. Genome-wide expression profiling of mid-gestation placenta and embryo using a 15,000 mouse developmental cDNA microarray. Proc. Natl. Acad. Sci. USA 97, 9127–9132 (2000).
Cox, B. et al. Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology. Mol. Syst. Biol. 5, 279 (2009).
Nelissen, E. C. et al. Altered gene expression in human placentas after IVF/ICSI. Hum. Reprod. 29, 2821–2831 (2014).
Ursini, G. et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat. Med. 24, 792–801 (2018).
Mouillet, J. F. et al. MicroRNAs in placental health and disease. Am. J. Obstet. Gynecol. 213, S163–S172 (2015).
Bianco-Miotto, T. et al. IFPA meeting 2015 workshop report I: placental mitochondrial function, transport systems and epigenetics. Placenta 48(Suppl. 1), S3–S6 (2016).
Jansson, T. & Powell, T. L. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin. Sci. (Lond.) 113, 1–13 (2007).
Bianco-Miotto, T. et al. Recent progress towards understanding the role of DNA methylation in human placental development. Reproduction 152, R23–R30 (2016).
McMullen, S. et al. Alterations in placental 11 beta-hydroxysteroid dehydrogenase (11 betaHSD) activities and fetal cortisol:cortisone ratios induced by nutritional restriction prior to conception and at defined stages of gestation in ewes. Reproduction 127, 717–725 (2004).
Murphy, V. E. & Clifton, V. L. Alterations in human placental 11beta-hydroxysteroid dehydrogenase type 1 and 2 with gestational age and labour. Placenta 24, 739–744 (2003).
Stirrat, L. I. et al. Transfer and metabolism of cortisol by the isolated perfused human placenta. J. Clin. Endocrinol. Metab. 103, 640–648 (2018).
Gomez-Roig, M. D. et al. Placental 11B-hydroxysteroid dehydrogenase type 2 mRNA levels in intrauterine growth restriction versus small-for-gestational-age fetuses. Fetal Diagn. Ther. 39, 147–151 (2016).
Brosens, I. et al. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 204, 193–201 (2011).
Adelman, D. M. et al. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 14, 3191–3203 (2000).
Gheorghe, C. P. et al. Gene expression in the placenta: maternal stress and epigenetic responses. Int. J. Dev. Biol. 54, 507–523 (2010).
Januar, V. et al. Epigenetic regulation of human placental function and pregnancy outcome: considerations for causal inference. Am. J. Obstet. Gynecol. 213, S182–S196 (2015).
Mina, T. H. et al. Maternal distress associates with placental genes regulating fetal glucocorticoid exposure and IGF2: role of obesity and sex. Psychoneuroendocrinology 59, 112–122 (2015).
Davis, E. P., Waffarn, F. & Sandman, C. A. Prenatal treatment with glucocorticoids sensitizes the HPA axis response to stress among full-term infants. Dev. Psychobiol. 53, 175–183 (2011).
Straughen, J. K. et al. The association between placental histopathology and autism spectrum disorder. Placenta 57, 183–188 (2017).
Chang, J. M. et al. Autism risk classification using placental chorionic surface vascular network features. BMC Med. Inform. Decis. Mak. 17, 162 (2017).
Park B. Y. et al., National Children’s Study Council. Placental gross shape differences in a high autism risk cohort and the general population. PLoS ONE 13, e0191276 (2018).
Schmidt, R. J. et al. Self-reported pregnancy exposures and placental DNA methylation in the MARBLES prospective autism sibling study. Environ. Epigenet. 2, pii: dvw024 (2016).
Schroeder, D. I. et al. Placental methylome analysis from a prospective autism study. Mol. Autism 7, 51 (2016).
Bilbo, S. D. et al. Beyond infection—maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp. Neurol. 299, 241–251 (2018).
Wong, H. & Hoeffer, C. Maternal IL-17A in autism. Exp. Neurol. 299, 228–240 (2018).
Wu, W. L. et al. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav. Immun. 62, 11–23 (2017).
Ponzio, N. M. et al. Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann. NY Acad. Sci. 1107, 118–128 (2007).
Ali, A. et al. The placental immune response is dysregulated developmentally vitamin D deficient rats: relevance to autism. J. Steroid Biochem. Mol. Biol. 180, 73–80 (2018).
Vuillermot, S. et al. Vitamin D treatment during pregnancy prevents autism-related phenotypes in a mouse model of maternal immune activation. Mol. Autism 8, 9 (2017).
Anderson, P. & Doyle, L. W., Victorian Infant Collaborative Study Group. Neurobehavioral outcomes of school-age children born extremely low birth weight or very preterm in the 1990s. JAMA 289, 3264–3272 (2003).
Whitehouse, A. J. et al. Maternal vitamin D levels and the autism phenotype among offspring. J. Autism Dev. Disord. 43, 1495–1504 (2013).
Vinkhuyzen, A. A. E. et al. Gestational vitamin D deficiency and autism-related traits: the Generation R Study. Mol. Psychiatry 23, 240–246 (2018).
Vinkhuyzen, A. A. E. et al. Gestational vitamin D deficiency and autism spectrum disorder. BJPsych Open 3, 85–90 (2017).
Magnusson, C. et al. Maternal vitamin D deficiency and the risk of autism spectrum disorders: population-based study. BJPsych Open 2, 170–172 (2016).
Stubbs, G., Henley, K. & Green, J. Autism: Will vitamin D supplementation during pregnancy and early childhood reduce the recurrence rate of autism in newborn siblings? Med Hypotheses 88, 74–78 (2016).
Chawla, D. et al. Early prenatal vitamin D concentrations and social-emotional development in infants. J. Matern. Fetal Neonatal Med. 32, 1441–1448 (2019).
Garcia-Serna, A. M. & Morales, E. Neurodevelopmental effects of prenatal vitamin D in humans: systematic review and meta-analysis. Mol. Psychiatry (2019). https://doi.org/10.1038/s41380-019-0357-9.
Werling, D. M. & Geschwind, D. H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 26, 146–153 (2013).
Robinson, E. B. et al. Examining and interpreting the female protective effect against autistic behavior. Proc. Natl. Acad. Sci. USA 110, 5258–5262 (2013).
Mueller, B. R. & Bale, T. L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 28, 9055–9065 (2008).
Pankevich, D. E. et al. Prenatal stress programming of offspring feeding behavior and energy balance begins early in pregnancy. Physiol. Behav. 98, 94–102 (2009).
Bronson, S. L. & Bale, T. L. The placenta as a mediator of stress effects on neurodevelopmental reprogramming. Neuropsychopharmacology 41, 207–218 (2016).
Bale, T. L. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialog. Clin. Neurosci. 18, 459–464 (2016).
Bronson, S. L., Chan, J. C. & Bale, T. L. Sex-specific neurodevelopmental programming by placental insulin receptors on stress reactivity and sensorimotor gating. Biol. Psychiatry 82, 127–138 (2017).
Ratto, A. B. et al. What about the girls? Sex-based differences in autistic traits and adaptive skills. J. Autism Dev. Disord. 48, 1698–1711 (2018).
Tang, S. et al. Altered forebrain functional connectivity and neurotransmission in a kinase-inactive met mouse model of autism. Mol. Imaging 18, 1536012118821034 (2019).
Sandman, C. A., Glynn, L. M. & Davis, E. P. Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. J. Psychosom. Res. 75, 327–335 (2013).
Cumberland, A. L. et al. Increased anxiety-like phenotype in female guinea pigs following reduced neurosteroid exposure in utero. Int. J. Dev. Neurosci. 58, 50–58 (2017).
Fatemi, S. H. & Folsom, T. D. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 35, 528–548 (2009).
Fatemi, S. H. et al. The viral theory of schizophrenia revisited: abnormal placental gene expression and structural changes with lack of evidence for H1N1 viral presence in placentae of infected mice or brains of exposed offspring. Neuropharmacology 62, 1290–1298 (2012).
Fineberg, A. M. & Ellman, L. M. Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol. Psychiatry 73, 951–966 (2013).
Shen, Q. et al. The role of pro-inflammatory factors in mediating the effects on the fetus of prenatal undernutrition: implications for schizophrenia. Schizophr. Res. 99, 48–55 (2008).
Cannon, M., Jones, P. B. & Murray, R. M. Obstetric complications and schizophrenia: historical and meta-analytic review. Am. J. Psychiatry 159, 1080–1092 (2002).
Plitman, E. et al. Kynurenic acid in schizophrenia: a systematic review and meta-analysis. Schizophr. Bull. 43, 764–777 (2017).
Beggiato, S. et al. Maternal genotype determines kynurenic acid levels in the fetal brain: implications for the pathophysiology of schizophrenia. J. Psychopharmacol. 32, 1223–1232 (2018).
Stroud, L. R. et al. Prenatal major depressive disorder, placenta glucocorticoid and serotonergic signaling, and infant cortisol response. Psychosom. Med. 78, 979–990 (2016).
Capron, L. E., Ramchandani, P. G. & Glover, V. Maternal prenatal stress and placental gene expression of NR3C1 and HSD11B2: the effects of maternal ethnicity. Psychoneuroendocrinology 87, 166–172 (2018).
Monk, C. et al. Distress during pregnancy: epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am. J. Psychiatry 173, 705–713 (2016).
Raikkonen, K. et al. Maternal depressive symptoms during pregnancy, placental expression of genes regulating glucocorticoid and serotonin function and infant regulatory behaviors. Psychol. Med. 45, 3217–3226 (2015).
Cottrell, E. C. et al. Foetal and placental 11beta-HSD2: a hub for developmental programming. Acta Physiol. (Oxf.) 210, 288–295 (2014).
Wyrwoll, C. S. & Holmes, M. C. Prenatal excess glucocorticoid exposure and adult affective disorders: a role for serotonergic and catecholamine pathways. Neuroendocrinology 95, 47–55 (2012).
Bonnin, A. et al. A transient placental source of serotonin for the fetal forebrain. Nature 472, 347–350 (2011).
Janssen, A. B. et al. Maternal prenatal depression is associated with decreased placental expression of the imprinted gene PEG3. Psychol. Med. 46, 2999–3011 (2016).
Aarnoudse-Moens, C. S. et al. Executive function in very preterm children at early school age. J. Abnorm. Child Psychol. 37, 981–993 (2009).
Baron, I. S. et al. Cognitive deficit in preschoolers born late-preterm. Early Hum. Dev. 87, 115–119 (2011).
Talge, N. M. et al. Late-preterm birth by delivery circumstance and its association with parent-reported attention problems in childhood. J. Dev. Behav. Pediatr. 33, 405–415 (2012).
Penn, A. A. et al. Controversies in preterm brain injury. Neurobiol. Dis. 92, 90–101 (2016).
Guttmacher, A. E., Maddox, Y. T. & Spong, C. Y. The Human Placenta Project: placental structure, development, and function in real time. Placenta 35, 303–304 (2014).
Monk, D. Genomic imprinting in the human placenta. Am. J. Obstet. Gynecol. 213, S152–S162 (2015).
Simoes L. R., et al. Maternal immune activation induced by lipopolysaccharide triggers immune response in pregnant mother and fetus, and induces behavioral impairment in adult rats. J. Psychiatr Res. 100, 71–83 (2018).
Howerton, C. L., Morgan, C. P., Fischer, D. B., Bale, T. L. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl Acad. Sci. USA 110, 5169–5174 (2013).
Guidotti, A., Grayson, D. R., Caruncho, H. J. Epigenetic RELN Dysfunction in Schizophrenia and Related Neuropsychiatric Disorders. Front Cell Neurosci. 10, 89 (2016).
Acknowledgements
This work was supported by NIH R01 (R01HD092593) and Simons Foundation Autism Research Initiative (SFARI) grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Kratimenos, P., Penn, A.A. Placental programming of neuropsychiatric disease. Pediatr Res 86, 157–164 (2019). https://doi.org/10.1038/s41390-019-0405-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0405-9
This article is cited by
-
Elevated circulating adiponectin levels do not prevent anxiety-like behavior in a PCOS-like mouse model
Scientific Reports (2024)
-
Short-term neonatal and long-term neurodevelopmental outcome of children born term low birth weight
Scientific Reports (2024)
-
PIBF1 regulates trophoblast syncytialization and promotes cardiovascular development
Nature Communications (2024)
-
Prenatal delta-9-tetrahydrocannabinol exposure is associated with changes in rhesus macaque DNA methylation enriched for autism genes
Clinical Epigenetics (2023)
-
Placental DNA methylation profile as predicting marker for autism spectrum disorder (ASD)
Molecular Medicine (2023)
