
Abstract
Background
Environmental tobacco smoke (ETS) is a known risk factor for severe respiratory syncytial virus (RSV) infections, yet the mechanisms of ETS/RSV comorbidity are largely unknown. Cystathionine γ-lyase regulates important physiological functions of the respiratory tract.
Methods
We used mice genetically deficient in the cystathionine γ-lyase enzyme (CSE), the major H2S-generating enzyme in the lung to determine the contribution of H2S to airway disease in response to side-stream tobacco smoke (TS), and to TS/RSV co-exposure.
Results
Following a 2-week period of exposure to TS, CSE-deficient mice (KO) showed a dramatic increase in airway hyperresponsiveness (AHR) to methacholine challenge, and greater airway cellular inflammation, compared with wild-type (WT) mice. TS-exposed CSE KO mice that were subsequently infected with RSV exhibited a more severe clinical disease, airway obstruction and AHR, enhanced viral replication, and lung inflammation, compared with TS-exposed RSV-infected WT mice. TS-exposed RSV-infected CSE KO mice had also a significant increase in the number of neutrophils in bronchoalveolar lavage fluid and increased levels of inflammatory cytokines and chemokines.
Conclusion
This study demonstrates the critical contribution of the H2S-generating pathway to airway reactivity and disease following exposure to ETS alone or in combination with RSV infection.
Similar content being viewed by others


Introduction
Second-hand tobacco smoke (TS), also known as environmental TS (ETS), is a complex mixture of gases and particles that include smoke from the burning cigarette, cigar, or pipe tip (side-stream smoke, SS) and exhaled mainstream smoke (MS). It is involuntarily inhaled by nonsmokers, lingers in the air for hours after cigarettes have been extinguished, and can induce or exacerbate a wide range of diseases, from cancer to respiratory infections, chronic obstructive pulmonary disease (COPD), and asthma.1,2 It is estimated that close to 50% of children worldwide under the age of 5 years are exposed to ETS.3 Younger children represent the most vulnerable population with regard to severe lung infections caused by respiratory syncytial virus (RSV), an enveloped, negative-sense single-stranded RNA virus of the Pneumoviridae family that is recognized as a leading cause of lower respiratory tract illness in young children. It is estimated that RSV produces over 64 million (M) cases of acute infections globally, and is responsible for acute emergency room visits for an estimated 2.1 M children under 5 years of age annually in the United States.4 The clinical spectrum of RSV-caused disease in children ranges from a relatively mild “cold” to acute lower respiratory tract infections, which may be so severe to require mechanical ventilation (intensive care). Although infants with certain risk factors (prematurity, chronic lung disease, congenital heart disease, or immunodeficiencies) have an increased risk for more severe RSV disease, the large majority of infants with RSV infections that require hospitalization are previously healthy.5 Therefore, the spectrum of disease in infants points to host determinants, viral-specific factors, and environmental insults that combined may lead to enhanced viral replication, inflammation of the airway mucosa, and lung injury, ultimately determining the severity of RSV infections.6,7 With regard to environmental factors, different studies have reported that exposure to ETS may be associated with more severe manifestations of RSV infection.8 A study of more than 200 young children has revealed that maternal cigarette smoking, especially postnatal, is associated with the severity of RSV bronchiolitis infection in infants.9 Lanari et al.10 demonstrated that exposure to TS, in general, seems to worsen the severity of the bronchiolitis. Moreover, a study by Gurkan et al.,11 in which cotinine levels were measured at the index of hospitalization, showed that infants admitted to the hospital with severe RSV bronchiolitis were exposed more recently to TS than infants hospitalized for non-respiratory diseases. However, the mechanism(s) underlying such increased severity of RSV infection in subjects exposed to ETS are largely unknown.
Hydrogen sulfide (H2S) is a cellular gaseous transmitter with known biological activities, including anti-inflammatory properties.12 Synthesis of H2S involves two pyridoxal-5′-phosphate-dependent enzymes, cystathionine β-synthase (CBS), and cystathionine γ-lyase (CSE), that are involved in the metabolic pathway of l-cysteine. A third enzyme, 3-mercaptopyruvate sulfurtransferase, is also involved in a cysteine aminotransferase pathway.13 H2S modulates ion channel and cellular function by a multitude of signaling pathways, including protein modification by S-sulfhydration and scavenging reactive oxygen species, which affects protein function, cellular localization, stability, and resistance to oxidative damage (reviewed in refs. 14,15). In the respiratory tract, endogenous H2S has been shown to participate in the regulation of important physiological functions, such as airway tone, pulmonary circulation, and cell proliferation and apoptosis, and to modulate lung fibrosis, oxidative stress, and inflammation.16 Based on this premise, the goal of our study was to investigate in an experimental mouse model the contribution of endogenous H2S to airway responsiveness and inflammation in response to side-stream TS as a surrogate of ETS exposure and its function in TS-enhanced RSV pathogenesis.
Methods
RSV preparation
RSV (long strain) was grown in HEp-2 cells and purified by centrifugation on discontinuous sucrose gradients, and viral pools were titered in plaque-forming units (PFU)/ml using a methylcellulose plaque assay (see Supplementary Material).17
Ethics statement
Animal care and use were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch at Galveston (Protocols: 9001002 and 1304014). The TS exposure was performed in the Inhalation Toxicology Core, UTMB. Mice were euthanized with a high dose of anesthetic ketamine and xylazine by an intraperitoneal injection.
TS exposure and infection of mice
CSE-deficient mice (CSE KO) were used to examine the role of endogenous H2S in the pathogenesis of TS exposure. Ten- to twelve-week-old male and female C57BL/6J mice (wild type, WT) used in this work were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). CSE KO mice on C57BL/6J background were generously provided by Dr. Solomon Snyder (Johns Hopkins University, Baltimore, MD, USA). Both male and female CSE KO and WT age-matched mice were used in the experiments. Groups of CSE−/− (KO) and WT control mice were subjected to whole-body exposure of either cigarette-generated TS (as side stream, TS) or filtered air (5 h per day, 5 days per week, for 2 weeks). The Standard Reference Cigarettes 3R4F used in this study were obtained from the Tobacco and Health Institute at the University of Kentucky (http://www.ca.uky.edu/refcig). In some experiments, at the end of the 2-week exposure period, groups of mice were infected intranasally with 50 μl of RSV diluted in phosphate-buffered saline (PBS), at a dose of 106 PFU per mouse or were mock inoculated using the same volume of PBS (sham infection). Daily determination of body weight, illness score, cytokine and chemokine concentration and cell differential counts in bronchoalveolar lavage (BAL) fluid, and airway function tests (see below) were performed exactly as we previously described.18 Mouse strains, side-stream cigarette smoke or air exposure, and infection with RSV or sham are identified in the paper as WT or CSE KO, TS or air, and RSV or PBS. The time line of the experimental protocol for air/smoke exposure and air/smoke exposure, followed by either PBS or RSV infection is shown in Supplemental Fig. S1a, S1b, respectively.
Statistical analysis
All results are expressed as mean ± SEM for each experimental group. Data were analyzed using the GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA, USA).
Statistical significance was calculated between groups by one-way analysis of variance, followed by Tukey’s post hoc test for samples with unequal variances. P < 0.05 value was selected to indicate significance. All experiments were repeated at least three times, data in figures are shown from a representative experiment.
Results
CSE gene deficiency enhances TS-induced AHR and lung inflammation
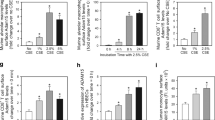
To establish the role of endogenous H2S in a TS surrogate of ETS exposure, we first measured airway function (AHR), cytokine and chemokine concentration, and inflammatory cell recruitment in BAL fluid of mice lacking CSE, a key enzyme in the biosynthesis pathway of H2S.19 For pulmonary function, groups of CSE KO and WT mice were assessed for AHR in response to methacholine (MCh) by whole-body barometric plethysmograph (BuxcoTM) at approximately 24 h after the last exposure to TS (overall 2-week-long exposure). As shown in Fig. 1a, AHR measured as enhanced pause (Penh) values was similar between air- and TS-exposed WT mice. On the other hand, significant dose-dependent increase in AHR to MCh challenge was observed in CSE KO mice. Specifically, CSE KO mice had increased AHR when compared with WT mice even in conditions of air exposure (p < 0.05 at 25 mg/ml and p < 0.01 at 50 mg/ml MCh doses). After exposure to TS, CSE KO mice showed a twofold increase in Penh values at doses of 6 and 12.5 mg/ml, respectively, and threefold increase at doses of 25 and 50 mg/ml of MCh, compared with air-exposed CSE KO mice. Compared with WT mice, CSE KO mice had an even more dramatic increase in AHR, specifically a sixfold increase in Penh values at 6 and 12.5 mg/ml, and a tenfold increase in MCh doses at 25 and 50 mg/ml.

Cystathionine γ-lyase enzyme (CSE) gene deficiency enhances tobacco smoke (TS)-induced airway hyperresponsiveness (AHR) and lung inflammation. Groups of CSE-deficient mice (KO) and wild-type (WT) mice were exposed to side-stream TS or air (5 h per day, 5 days per week, for 2 weeks). TS/CSE KO mice have enhanced AHR after methacholine (MCh) challenge (a), influx of total cells (b), macrophages (c), neutrophils (d), and lymphocyte (e) in bronchoalveolar lavage (BAL) fluid, increased interleukin-6 (IL-6), chemokine (C–X–C motif) ligand 1 (CXCL1), C–C motif chemokine ligand 2 (CCL2), and CCL4 (f) compared with TS/WT mice and air/CSE KO mice, and CSE messenger RNA (mRNA) levels in the lung (g). Data are expressed as mean ± SEM (n = 4 mice (2 male and 2 female)/group and are representative of three independent experiments). *P < 0.05,**p < 0.01, and ***p < 0.001, CSE KO TS vs. WT TS, CSE KO TS vs. CSE KO air, CSE KO air vs. WT air, and WT TS vs. WT air
The total cell number in BAL fluid was not different between air CSE KO and WT mice (Fig. 1b). On the other hand, the total number of BAL cells was significantly elevated both in WT and CSE KO mice after 2 weeks of TS exposure, compared with the respective air control groups (Fig. 1b), and in TS CSE KO compared with TS WT mice. In particular, macrophage cell counts in BAL increased with TS exposure compared with the respective air controls, but significantly more so in CSE KO mice than in WT mice (Fig. 1c, p < 0.05), and in TS CSE KO mice compared with TS WT mice. Exposure to TS also resulted in a significant increase in the number of neutrophils (p < 0.01) and lymphocytes (p < 0.05) in CSE KO mice compared with air CSE KO mice, and also relative to TS WT mice (Fig. 1d, e). In general, we did not observe significant differences in BAL neutrophil and lymphocyte numbers between air and TS WT animals.
To determine the profile of major inflammatory molecules that could explain the increased influx of cells into the lungs of CSE KO mice, we measured a panel of inflammatory cytokines in samples of BAL fluids by a Bio-Plex array, 24 h after the 2-week period of TS or control-filtered air exposure. Most of the cytokines/chemokines included in the 18-cytokine array were undetectable in BAL of WT or CSE KO mice, with the exception of those presented in Fig. 1f (interleukin-6 (IL-6), chemokine (C–X–C motif) ligand 1 (CXCL1), C–C motif chemokine ligand 2 (CCL2), and CCL4). Baseline levels (i.e., air mice) of IL-6 and CXCL1 were significantly increased in the BAL fluid of CSE KO mice compared with WT mice (p < 0.05 for IL-6, and p < 0.001 for CXCL1). No significant differences were observed for any of these four cytokines between smoke and air WT mice. On the other hand, increased levels of CCL2 and CCL4 were observed in TS compared with air CSE KO mice (p < 0.05). In addition, CSE KO mice exposed to TS showed significantly increased BAL concentration of IL-6 (p < 0.05), CXCL1, (p < 0.001), CCL2 (p < 0.05), and CCL4 (p < 0.05), compared with WT mice.
Next, we determined whether cigarette smoke exposure alters the expression of the H2S-producing enzyme CSE in WT mice. Lungs samples were harvested at day 1 post infection to assess messenger RNA (mRNA) levels of CSE. As shown in Fig. 1g, mRNA levels of the H2S-producing enzyme CSE are significantly increased in TS-exposed WT mice when compared with air WT mice (p < 0.05).
CSE deficiency and TS exposure enhance RSV pathogenicity
To investigate the role of endogenous H2S in an experimental model of smoke exposure and RSV infection, we exposed CSE KO and WT mice to TS or air as described in the previous section, followed 24 h later by infection with RSV. Using daily body weight as an indicator of clinical disease, we found that early body weight loss after RSV inoculation was similar between air- and TS-exposed WT mice (which peaked at day 1, Fig. 2a). In control condition of air exposure, RSV-infected CSE KO mice exhibited enhanced body weight loss at the peak of the clinical disease when compared with infected WT mice (p < 0.05). Following exposure to TS, RSV-infected CSE KO mice showed significant greater body weight loss beginning at day 1, peaking at day 2 and delayed recovery to baseline weight, compared with WT mice and also compared with air RSV-infected CSE KO mice (p < 0.05, Fig. 2a). No differences in body weight loss were observed among groups of WT and CSE KO mice either air- or TS-exposed, which were PBS inoculated (data not shown). Next, we investigated the role of CSE deficiency and smoke exposure on RSV-mediated airway obstruction and AHR. As shown in Fig. 2b, RSV-infected CSE KO mice that were exposed to TS had significantly increased baseline airway obstruction compared with both air RSV- and CSE KO-infected mice or air RSV-infected WT mice (at day 1 after infection, returning to baseline at day 5 post infection [p < 0.05]). TS RSV-infected CSE KO mice also exhibited significantly greater AHR in response to aerosolized MCh (day 5 post infection), compared with air RSV-infected CSE KO as well as with air or TS RSV-infected WT mice (Fig. 2c).

Tobacco smoke (TS) and cystathionine γ-lyase enzyme (CSE) gene deficiency exacerbate clinical disease, viral replication, and airway hyperresponsiveness (AHR) following respiratory syncytial virus (RSV) infection. Groups of CSE-deficient mice (KO) and wild-type (WT) mice were exposed to TS or air as in Fig. 1, followed by infection with RSV as described in the Methods section. a Effect of TS exposure on RSV-induced disease. Change in body weight was measured over a period of 5 days after infection. b Airway obstruction represented by baseline enhanced pause (Penh) at days 1 and 5 after RSV infection. c AHR in response to methacholine (MCh) challenge at day 5 after RSV infection. d Increased RSV replication in the lung of TS/CSE KO mice, at day 5 after RSV infection lungs measured by plaque assay. e Lungs were harvested at day 5 after infection, fixed for slide preparation, and hematoxylin and eosin (H&E) stained. Alveolitis and bronchiolitis inflammatory scores of prepared slides (scored as described in the Methods section). Data are expressed as mean ± SEM (n = 4 mice (2 male and 2 female)/group and are representative of three independent experiments). *P < 0.05, **p < 0.01, and ***p < 0.001 CSE KO TS/RSV vs. WT TS/RSV, CSE KO TS/RSV vs. CSE KO air/RSV, CSE KO air/RSV vs. WT air/RSV, and WT TS/RSV vs. WT air/RSV
To determine whether CSE deficiency and exposure to smoke would alter viral replication, groups of mice were killed at day 5 after RSV infection and lung tissue collected to determine virus titer by a plaque assay. As shown in Fig. 2d, no significant difference was observed in peak viral titer between air- and TS-exposed RSV-infected WT mice. On the other hand, air RSV-infected CSE KO mice had significantly higher peak titers in the lung compared with air-infected WT mice (p < 0.001). Overall, the highest peak titers were found in the lung of CSE KO mice exposed to TS and subsequently infected with RSV, which had significantly higher peak virus titers compared with both air RSV-infected CSE KO mice (p < 0.05) and with smoke-exposed RSV-infected WT mice (p < 0.01).
For histopathology studies, lungs from mock- and RSV-infected CSE KO and WT mice, TS or air, were collected at day 5 after infection, formalin-fixed, H&E-stained, and qualitatively and quantitatively analyzed using an established grading score (from 0, absent to 4, severe). Mock-infected (PBS) animals from TS or air had no inflammatory infiltrates in the lung. For RSV-infected mice, quantification of pulmonary inflammation showed no significant differences between TS and air WT mice (Fig. 2e). On the other hand, lungs from TS-exposed and RSV-infected CSE KO mice showed diffuse inflammation with perivasculitis, peribronchiolitis, alveolitis, and vasculitis when compared with all other groups. Overall, quantification of the pulmonary inflammation by alveolitis and bronchiolitis scores indicated significantly greater airway pathology at day 5 in TS-exposed RSV-infected CSE KO mice compared with WT mice (p < 0.05, Fig. 2e). Representative images for each group at day 5 after infection are shown in Fig. 3a, b.

Tobacco smoke (TS) and cystathionine γ-lyase enzyme (CSE) gene deficiency exacerbates pulmonary inflammation. TS- or air-exposed CSE-deficient mice (KO) and wild-type (WT) mice were infected with respiratory syncytial virus (RSV) or sham. Lungs were harvested at day 5 after infection, fixed for slide preparation, and hematoxylin and eosin (H&E) stained. a Representative photomicrographs of lung sections from WT and CSE KO mice exposed to either air or TS and then mock infected with phosphate-buffered saline (PBS). The lung sections are mostly unremarkable. b Representative photomicrographs of lung sections from WT and CSE KO mice exposed to either air or TS and then infected with RSV. The sections from CSE KO TS/RSV mice show moderate-to-severe alveolitis, compared with minimal focal alveolitis observed in the other groups. Original magnification ×20
CSE deficiency and TS increase airway inflammatory cells following infection
Overall, the number of total inflammatory cells, neutrophils, and lymphocyte subpopulations was greater in BAL fluids of TS/CSE KO mice. As shown in Fig. 4a, a significant greater number of total BAL cells was observed in TS/CSE KO/PBS mice, compared with all other PBS groups (day 1, p < 0.05). The difference in the total number of cells persisted at day 5 (p < 0.05). Differential H&E staining revealed a slight increase (nonsignificant) of the number of macrophages in BAL at day 1 post infection for smoke-exposed CSE KO/PBS mice (Fig. 4b), but significantly more neutrophils (Fig. 4c) and lymphocytes (Fig. 4d) at both days 1 and 5 in these animals compared with all other PBS groups. Exposure to air or TS followed by RSV infection was characterized by increase in the total number of cells, macrophages, neutrophils, and lymphocytes in BAL of CSE KO and WT mice compared with air/PBS mice (Fig. 4a–d). There were no statistically significant differences between smoke and air/WT RSV mice for total cells or number of macrophages and neutrophils at day 1, respectively, day 5 post infection. At day 5 post infection, we observed a significant increase in the number of lymphocytes in the BAL for smoke-exposed vs. air RSV-infected WT mice (p < 0.05, Fig. 4d).

Tobacco smoke (TS) and cystathionine γ-lyase enzyme (CSE) deficiency increases airway inflammatory cells following infection. TS- or air-exposed CSE-deficient mice (KO) and wild-type (WT) mice were infected with respiratory syncytial virus (RSV) or sham. Bronchoalveolar lavage (BAL) fluid was collected at different time points after infection and cell preparations were stained (Hema 3 stain, Fisher Scientific) and counted under the microscope (200 cells per slide). TS increased the influx of total cells (a), macrophages (b), neutrophils (c), and lymphocyte (d) into the BAL of CSE KO phosphate-buffered saline (PBS) mice, which was further exacerbated in the RSV-infected CSE KO mice, compared with WT animals. Data are expressed as mean ± SEM (n = 4 mice (2 male and 2 female)/group and are representative of three independent experiments). *P < 0.05, **p < 0.01, and ***p < 0.001 CSE KO TS/RSV vs. WT TS/RSV, CSE KO TS/RSV vs. CSE KO air/RSV, CSE KO air/RSV vs. WT air/RSV, WT TS/RSV vs. WTair/RSV, CSE KO TS/PBS vs. WT TS/PBS, and CSE KO TS/PBS vs. CSE KO air/PBS, WT air/RSV day 1 post infection (pi) vs. WT air/RSV day 5 pi, WT TS/RSV day 1 pi vs. WT TS/RSV day 5 pi, CSE KOair/RSV day 1 pi vs. CSE KO air/RSV day 5 pi, CSE KO TS/RSV day 1 pi vs. CSE KO TS/RSV day 5 pi
However, the total cells in BAL were significantly increased at day 1 compared with day 5 post infection for air- and TS-exposed WT and CSE KO mice after RSV infection (p < 0.001).
The most robust cell inflammation was observed as early as day 1 and persisting at day 5 following RSV infection in CSE KO mice that had been exposed to TS, in regard to total cells (p < 0.001, Fig. 4a), number of neutrophils (p < 0.001, Fig. 4c), and lymphocytes (p < 0.01, Fig. 4d), compared with other RSV-infected groups (Fig. 4c, d). The percentage of BAL macrophages, neutrophils, and lymphocytes are presented in Supplemental Fig. S2.
CSE deficiency and TS enhance BAL cytokines and chemokines following RSV infection
RSV infection is a potent inducer of cytokine and chemokine production, and increased chemokine release has been shown to play an important role in RSV-induced lung inflammation and to correlate with disease severity. We have recently shown that in the absence of CSE, RSV infection induced significantly higher levels of the cytokines IL-6, IL-12(p40), and tumor necrosis factor-α (TNF-α), and chemokines CCL2, CCL3, CCL4, and CCL5 compared with CSE-competent infected mice.20 To investigate whether a 2-week period of exposure to TS along with CSE deficiency affected RSV-induced cytokine response, BAL samples were collected at days 1 and 5 after infection from CSE KO and WT mice and were assessed for cytokines and chemokine levels by a Multi-Plex array. TS exposure and RSV infection in WT mice was associated with significant increase levels for IL-12(p40), granulocyte colony-stimulating factor (p < 0.05), CCL4 (p < 0.05), and CCL5 (p < 0.01) when compared with air/WT RSV-infected group at day 1 after infection. BAL concentrations of IL-6, IL-12(p40), and TNF-α in TS/CSE KO RSV-infected mice were significantly higher at day 1 compared with air/CSE KO RSV-infected mice (p < 0.05), and with TS/WT RSV-infected mice (p < 0.01) (Fig. 5a). Concentrations of IL-6 and IL-12(p40) remained higher at day 5 in TS/CSE KO RSV-infected mice compared with TS/WT RSV-infected mice. In addition, exposure to TS followed by RSV infection in CSE KO mice leads to an enhanced production of the chemokines CCL2 (p < 0.001), CCL3 (p < 0.001), CCL4 (p < 0.01), and CCL5 (p < 0.001), compared with RSV-infected WT mice at day 1 post infection (Fig. 5b). Similar findings were observed with the chemokines CCL3 (p < 0.01), CCL4 (p < 0.01), and CCL5 (p < 0.05) when compared with air/CSE KO RSV-infected mice.

Tobacco smoke (TS) and cystathionine γ-lyase enzyme (CSE) deficiency enhances bronchoalveolar lavage (BAL) cytokines and chemokines following respiratory syncytial virus (RSV) infection. TS- or air-exposed CSE-deficient mice (KO) and wild-type (WT) mice were infected with RSV or sham. BAL fluid was collected at different time points after infection to measure cytokines and chemokines by a Bio-Plex array. TS/CSE KO RSV mice had significantly increased levels of cytokines interleukin-6 (IL-6), IL-12(p40), tumor necrosis factor-α (TNF-α), and granulocyte colony-stimulating factor (G-CSF) (a) and chemokines C–C motif chemokine ligand 2 (CCL2), CCL3, CCL4, and CCL5 (b) compared with TS/WT RSV and air/CSE KO RSV mice. Data are expressed as mean ± SEM (n = 4 mice (2 male and 2 female)/group and are representative of three independent experiments). *P < 0.05, **p < 0.01, and ***p < 0.001 CSE KO TS/RSV vs. WT TS/RSV, CSE KO TS/RSV vs. CSE KO air/RSV, CSE KO air/RSV vs. WT air/RSV, and WT TS/RSV vs. WT air/RSV
Discussion
This study shows that in the absence of CSE mice that have been exposed to TS for a short period of 2 weeks had a dramatic increase in airway reactivity to MCh challenge (24 h after last smoke exposure) (Fig. 1). Even in mice exposed solely to filtered air, airway reactivity was significantly more pronounced than in WT mice with intact H2S-generating capacity. CSE is a key enzyme that controls production of H2S in the lung16,21 and CSE KO mice exhibit a profound depletion of H2S in several peripheral tissues.19 In other rodent studies, levels of endogenous H2S were found to be decreased in the lung of rats challenged with ovalbumin. Serum levels of H2S correlated positively with H2S levels in lung tissues and peak expiratory flow, and negatively with allergic inflammation and airway smooth muscle hyperplasia.22 In a mouse model of asthma, lack of CSE expression resulted in increased AHR after MCh challenge and some evidence of lung T-helper type 2 (Th2)-driven inflammation.23 In our study, levels of Th1 or Th2 cytokines [(i.e., interferon-γ (IFN-γ) and IL-4)] in BAL samples of TS or air mice, either WT or CSE KO, were at the lowest level of detection or were not significantly different among different groups following infection with RSV (data not shown). One report examining the effect of TS alone on airway function/reactivity in mice has shown that side-stream TS exposure for a short period (days) resulted in increased AHR to MCh challenge; however, mouse strain (ICR) as well as the source of TS (Marlboro) were different from those in our study.24
We also show for the first time that compared with control WT animals, a relatively short period of TS exposure enhances RSV pathogenicity in mice that are CSE deficient, including: (1) peak of clinical disease as measured by body weight loss, as well as delayed recovery; (2) increased airway obstruction, measured at day 1 after RSV inoculation; (3) enhanced AHR to MCh challenge at day 5 post infection; (4) greater viral replication in the lung; and (5) more severe lung inflammation, characterized by peribronchiolitis and alveolitis. These pathogenic responses in TS-exposed/RSV-infected CSE KO mice were associated with an increase in concentration of inflammatory cytokines IL-6, IL-12(p40), and TNF-α and chemokines CCL2, CCL3, CCL4, and CCL5. Other studies in C57BL mice have demonstrated the effect of co-exposure to TS (direct exposure) and RSV on parameters of lung fibrosis, chronic inflammation, protease, and cytokine expression, but using longer protocols of exposure to smoke25 and repeated viral infections, as models of COPD.26 Airway function was not examined in those studies. One report examining the effect of TS alone on airway function/reactivity in mice has shown that side-stream TS exposure for a short period (days) resulted in increased AHR to MCh challenge; however, mouse strain (ICR) as well as the source of TS (Marlboro) were different from those in our study.24 Previous work by others in a BALB/c mouse model of double infection with RSV (neonatal/adult challenge) has shown that exposure to SS from birth to 35 days was associated with an increase in eosinophils and reduced levels of Th1 cytokines in BAL following RSV challenge.3 In our study, levels of IFN-γ and IL-4 and Th1/Th2 ratio in BAL samples at day 5 post infection by a highly sensitive enzyme-linked immunosorbent assay were no significantly different in any of the treatment groups, including those exposed to TS and CSE KO (data not shown).
Endogenous H2S is involved in critical physiological functions of the respiratory tract and its dysregulation appears to be involved in diseases such as COPD and asthma.16 For example, smokers have lower levels of H2S, when compared with nonsmokers with acute exacerbation of COPD or healthy controls.27 Moreover, in patients with stable COPD, serum H2S levels were found to be significantly lower in those with more severe obstruction, correlated positively with the percentage of predicted forced expiratory volume in 1 s values, and negatively with neutrophils in sputum.27 In other studies, patients with COPD have been shown to have lower levels of expression of CSE protein and CBS mRNA.28 Patients with COPD who had lower levels of exhaled H2S had greater number of eosinophils in sputum, worse lung function, and more frequent exacerbations.29 In asthma, several studies have shown that both children and adult patients have lower levels of serum and exhaled H2S (reviewed in ref. 16), with lower H2S levels being correlated with more altered pulmonary function tests and severity of their disease.30,31 Elegant studies in mice have also shown that H2S contributes to lung development, expression of CSE and CBS enzymes in airway epithelium and lung vessels, and demonstrated their critical function in the process of alveolarization and pulmonary vascular development.32 The same group has shown that systemic administration of the H2S slow-releasing donor GYY4137 partially restored arrested alveolarization in an experimental model of bronchopulmonary dysplasia.33
Recent investigations in our laboratory have identified a novel antiviral role for H2S.34 In airway epithelial cells, we showed that RSV inhibits the expression of CSE and H2S, and on the other hand, increased its degradation. By studies using the CSE inhibitor propargylglycine, we demonstrated an increase in viral replication and cytokine production. On the other hand, when cells were treated with a slow-releasing H2S chemical, GYY4137, RSV replication was significantly reduced. We have also shown that CSE KO have enhanced clinical disease AHR, viral replication, and lung inflammation compared with WT mice.20 Although the exact mechanism(s) leading to increased AHR in CSE KO mice exposed to TS alone and/or after RSV infection are not fully understood, our studies suggest that the H2S pathway in the lung is critical in relaxing airway smooth muscle. Some studies have shown that H2S relaxes vascular smooth muscle by increasing ATP-sensitive K+ channel (KATP) currents and hyperpolarizing the cell membrane,35 or by KATP channel-independent mechanisms, such as inhibition of Ca2+ release through the inositol-1,4,5-triphosphate receptor.36
The association between parental or household TS exposure and incidence of lower respiratory tract infections in infancy and the contribution of these factors to RSV disease is not fully clear.37,38 However, when the severity of RSV bronchiolitis has been examined, male sex and tobacco smoking by a household member were associated with the need for both supplemental oxygen and mechanical ventilation.39 Studies in premature infants, newborns, and children have shown that CSE expression increases with age,40 as levels of this enzyme would be developmentally delayed at the time of the most severe manifestations of RSV infection in vulnerable patients. We have previously shown that H2S plays a critical role in experimental infection of mice by RSV, by reducing inflammation and viral replication in the lung, as demonstrated by treatment with an H2S donor that reduced lung neutrophilia and pathology.20 Based on the results of this study, we suggest that TS, the most common and important indoor environmental pollutant, may contribute to more severe manifestations of RSV infections in subjects who may be genetically predisposed due to a lower capacity to produce endogenous H2S, that is, young infants with immature H2S-genarating enzymes or those who are more susceptible to the effect of RSV on reduction/degradation of H2S.34
References
Moshammer, H. et al. Parental smoking and lung function in children: an international study. Am. J. Respir. Crit. Care Med 173, 1255–1263 (2006).
Pauwels, R. A. & Rabe, K. F. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 364, 613–620 (2004).
Phaybouth, V. et al. Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L222–L231 (2006).
Hall, C. B. et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 360, 588–598 (2009).
Wang, E. E., Law, B. J. & Stephens, D. Pediatric Investigators Collaborative Network on Infections in Canada (PICNIC) prospective study of risk factors and outcomes in patients hospitalized with respiratory syncytial viral lower respiratory tract infection. J. Pediatr. 126, 212–219 (1995).
Devincenzo, J. P., El Saleeby, C. M. & Bush, A. J. Respiratory syncytial virus load predicts disease severity in previously healthy infants. J. Infect. Dis. 191, 1861–1868 (2005).
El Saleeby, C. M., Bush, A. J., Harrison, L. M., Aitken, J. A. & Devincenzo, J. P. Respiratory syncytial virus load, viral dynamics, and disease severity in previously healthy naturally infected children. J. Infect. Dis. 204, 996–1002 (2011).
Difranza, J. R., Masaquel, A., Barrett, A. M., Colosia, A. D. & Mahadevia, P. J. Systematic literature review assessing tobacco smoke exposure as a risk factor for serious respiratory syncytial virus disease among infants and young children. BMC Pediatr. 12, 81 (2012).
Bradley, J. P. et al. Severity of respiratory syncytial virus bronchiolitis is affected by cigarette smoke exposure and atopy. Pediatrics 115, e7–e14 (2005).
Lanari, M. et al. Prevalence of respiratory syncytial virus infection in Italian infants hospitalized for acute lower respiratory tract infections, and association between respiratory syncytial virus infection risk factors and disease severity. Pediatr. Pulmonol. 33, 458–465 (2002).
Gurkan, F., Kiral, A., Dagli, E. & Karakoc, F. The effect of passive smoking on the development of respiratory syncytial virus bronchiolitis. Eur. J. Epidemiol. 16, 465–468 (2000).
Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 6, 917–935 (2007).
Paul, B. D. & Snyder, S. H. H2S: a novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 40, 687–700 (2015).
Kimura, H. Physiological roles of hydrogen sulfide and polysulfides. Handb. Exp. Pharmacol. 230, 61–81 (2015).
Wallace, J. L. & Wang, R. Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 14, 329–345 (2015).
Bazhanov, N., Ansar, M., Ivanciuc, T., Garofal, R. P. & Casola, A. Hydrogen sulfide: a novel player in airway development, pathophysiology of respiratory diseases, and antiviral defenses. Am. J. Respir. Cell. Mol. Biol. 57, 403–410 (2017).
Olszewska-Pazdrak, B. et al. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J. Virol. 72, 4756–4764 (1998).
Ivanciuc, T., Sbrana, E., Casola, A. & Garofalo, R. P. Protective role of nuclear factor erythroid 2-related factor 2 against respiratory syncytial virus and human metapneumovirus infections. Front. Immunol. 9, 854 (2018).
Yang, G. et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590 (2008).
Ivanciuc, T. et al. Hydrogen sulfide is an antiviral and antiinflammatory endogenous gasotransmitter in the airways. role in respiratory syncytial virus infection. Am. J. Respir. Cell. Mol. Biol. 55, 684–696 (2016).
Chen, Y. & Wang, R. The message in the air: hydrogen sulfide metabolism in chronic respiratory diseases. Respir. Physiol. Neurobiol. 184, 130–138 (2012).
Chen, Y. H. et al. Endogenous hydrogen sulfide reduces airway inflammation and remodeling in a rat model of asthma. Cytokine 45, 117–123 (2009).
Zhang, G., Wang, P., Yang, G., Cao, Q. & Wang, R. The inhibitory role of hydrogen sulfide in airway hyperresponsiveness and inflammation in a mouse model of asthma. Am. J. Pathol. 182, 1188–1195 (2013).
Glynos, C. et al. Comparison of the effects of e-cigarette vapor with cigarette smoke on lung function and inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 315, 662–672 (2018).
Mebratu, Y. A., Smith, K. R., Agga, G. E. & Tesfaigzi, Y. Inflammation and emphysema in cigarette smoke-exposed mice when instilled with poly (I:C) or infected with influenza A or respiratory syncytial viruses. Respir. Res. 17, 75 (2016).
Foronjy, R. F., Dabo, A. J., Taggart, C. C., Weldon, S. & Geraghty, P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PLoS ONE 9, e90567 (2014).
Chen, Y. H. et al. Endogenous hydrogen sulfide in patients with COPD. Chest 128, 3205–3211 (2005).
Sun, Y. et al. Metabolic changes of H2S in smokers and patients of COPD which might involve in inflammation, oxidative stress and steroid sensitivity. Sci. Rep. 5, 14971 (2015).
Zhang, J., Wang, X., Chen, Y. & Yao, W. Exhaled hydrogen sulfide predicts airway inflammation phenotype in COPD. Respir. Care 60, 251–258 (2015).
Tian, M. et al. Correlation between serum H2S and pulmonary function in children with bronchial asthma. Mol. Med. Rep. 6, 335–338 (2012).
Zhang, J., Wang, X., Chen, Y. & Yao, W. Correlation between levels of exhaled hydrogen sulfide and airway inflammatory phenotype in patients with chronic persistent asthma. Respirology 19, 1165–1169 (2014).
Madurga, A. et al. The H2S-generating enzymes cystathionine beta-synthase and cystathionine gamma-lyase play a role in vascular development during normal lung alveolarization. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L710–L724 (2015).
Madurga, A. et al. Systemic hydrogen sulfide administration partially restores normal alveolarization in an experimental animal model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L684–L697 (2014).
Li, H. et al. Role of hydrogen sulfide in paramyxovirus infections. J. Virol. 89, 5557–5568 (2015).
Fitzgerald, R. et al. H2S relaxes isolated human airway smooth muscle cells via the sarcolemmal K(ATP) channel. Biochem. Biophys. Res. Commun. 446, 393–398 (2014).
Castro-Piedras, I. & Perez-Zoghbi, J. F. Hydrogen sulphide inhibits Ca2+ release through InsP3 receptors and relaxes airway smooth muscle. J. Physiol. 591, 5999–6015 (2013).
Jones, L. L. et al. Parental and household smoking and the increased risk of bronchitis, bronchiolitis and other lower respiratory infections in infancy: systematic review and meta-analysis. Respir. Res. 12, 5 (2011).
Simoes, E. A. F. Environmental and demographic risk factors for respiratory syncytial virus lower respiratory tract disease. J. Pediatr. 143, 118–126 (2003).
Semple, M. G., Taylor-Robinson, D. C., Lane, S. & Smyth, R. L. Household tobacco smoke and admission weight predict severe bronchiolitis in infants independent of deprivation: prospective cohort study. PLoS ONE 6, e22425 (2011).
Vina, J. et al. l-Cysteine and glutathione metabolism are impaired in premature infants due to cystathionase deficiency. Am. J. Clin. Nutr. 61, 1067–1069 (1995).
Acknowledgements
We thank the Inhalation Toxicology Core (ITC) Facility, University of Texas Medical Branch, Galveston, TX, for their assistance with the cigarette TS exposure, Kimberly Palkowetz for technical assistance, and Cynthia Tribble for assistance in manuscript submission. This work was partially supported by NIH grants ES026782 (R.P.G.), AI125434 (A.C.), ES006676 (A.C., NIEHS), and AI062885 (R.P.G. and A.C.).
Author information
Authors and Affiliations
Contributions
Each author listed on the paper have met the Pediatric Research authorship requirements.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ivanciuc, T., Sbrana, E., Casola, A. et al. Cystathionine γ-lyase deficiency enhances airway reactivity and viral-induced disease in mice exposed to side-stream tobacco smoke. Pediatr Res 86, 39–46 (2019). https://doi.org/10.1038/s41390-019-0396-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0396-6
