
Abstract
Background
Alterations in gastrointestinal microbial communities have been linked to human disease. Most studies use fecal samples as a proxy for the intestinal microbiota; however, the fecal microbiome is not fully representative of the mucosa-associated microbiota at the site of disease. While mucosal biopsies can be used instead, they often contain a high proportion of host DNA that can confound 16S ribosomal RNA (rRNA) gene sequencing studies.
Methods
To overcome these limitations, we sampled the mucosal–luminal interface (MLI) to study the mucosa-associated microbiota. We also employed a simple bioinformatics workflow to remove contaminants from 16S rRNA gene profiling results.
Results
Our results indicate that the microbial differences between individuals are greater than those between different microenvironments within the same individual. Moreover, biopsy samples frequently contained contaminants that could significantly impact biopsy profiling results.
Conclusions
Our findings highlight the utility of collecting MLI aspirates to complement biopsies and stools for characterizing human microbial communities.
Similar content being viewed by others
Introduction
In recent years, a large body of research has characterized the microbial composition of the gastrointestinal tract in relation to numerous human diseases including inflammatory bowel disease (IBD), irritable bowel disease, diabetes mellitus, obesity, colorectal cancer, malnutrition, and cardiovascular diseases.1 The majority of these studies used fecal samples as a proxy for the intestinal microbiota. Although close associations between the intestinal microbiota and human health and disease are now well established, a general mechanistic understanding of how the microbiome contributes to disease or health is lacking. A major challenge for understanding the intestinal microbiota’s role in disease is that the fecal microbiome is not fully representative of the mucosa-associated microbiota at the site of disease.2,3,4,5,6 Moreover, the gut microbiota is highly diverse and is substantially variable between individuals. Both these factors impair our ability to identify specific disease-associated microbes. For example, Gevers et al.7 demonstrated that the microbial dysbiosis seen in Crohn’s disease (CD) is more easily detected in mucosal biopsies than in stools. Another study found that early signs of intestinal dysbiosis in a mouse model of colitis could be detected in mucosal communities but not stools.2 Hence, it is tempting to speculate that the mucosa-associated microbiota will affect the host to a greater extent than the luminal bacteria.
Comparative studies of microbial compositions from mucosa-associated communities to fecal-based luminal communities have shown heterogeneous patterns characteristic of two distinct microbial niches.5,6,8,9,10,11,12 While the composition of the luminal microbial community is easily characterized by using stool samples, currently the main means of characterizing the mucosa-associated community are colonoscopic biopsies.
Gastrointestinal biopsies are collected by inserting biopsy forceps through the endoscope and pinching off a small amount of tissue. The gastrointestinal wall is often washed with sterile water prior to sample collection to visualize the mucosa and determine the optimal regions for biopsy collection. However, unless the mucosal surface is heavily washed, these biopsy samples would represent the microbial communities of both the loose and strongly adherent mucosal layers. In addition, biopsies represent a small area of the mucosa and might not be representative of the overall mucosa-associated microbiota, particularly in patchy diseases such as CD. Biopsy collection is also relatively invasive as compared to stool sampling, and biopsies contain high proportions of host DNA as compared to microbial DNA. High levels of human DNA concurrently with low levels of microbial DNA have been shown to affect high-throughput 16S ribosomal RNA (rRNA) gene-based sequencing datasets and confound their interpretation.13 Molecular biology reagents are also often contaminated with microbial DNA,14,15 and thus low biomass microbiotas (e.g., biopsies) are more sensitive to exogenous microbial DNA contamination. The impact of this exogenous DNA contamination on samples with low bacterial biomass remains underappreciated and often ignored during analysis.14,15,16 While the presence of contamination has been clearly demonstrated by running negative controls through experimental workflows and data analysis pipelines, such controls are often not performed, evaluated, or reported.15 A number of studies using low microbial biomass samples have identified statistically noteworthy taxa which are also known to be common exogenous contaminants from molecular biology reagents.14,15
Recent work has proposed using intestinal “lavage” (i.e., fluid remaining in the bowel after bowel prep) as a proxy for the mucosal communities.17 Lavage samples would thus be a mix of the microbial communities remaining in the lumen after bowel preparation and the loose mucus layer. Ideally, samples collected to profile the gastrointestinal microbiotas would be able to differentiate between the luminal, loosely adherent and strongly adherent mucosal communities.
An alternative procedure for collecting gastrointestinal tract microbiotas involve flushing the mucosal surface with sterile water and aspirating the resulting mixture of mucus.18,19,20,21,22 This technique allows for the mucosal–luminal interface (MLI) to be sampled by washing off and collecting the loose mucus layer on the surface of the intestinal wall. The adherent mucosal layer can then be sampled by taking biopsies at the site of washing. This approach allows for the microbes that are in close proximity to the host to be profiled and yields sufficient material for multi-omic experiments such as shotgun metagenomics and metaproteomics.20,21,22,23,24 Moreover, the MLI sampling approach has identified novel taxa involved in CD pathogenesis.19 However, MLI sampling has not been compared to biopsies or stools, and thus MLI samples are not routinely collected for microbiota studies.
This study builds on previous work from others on the impact of exogenous DNA contamination in microbiome studies13,14,15 by characterizing the cross-sectional biogeography of the human intestinal microbiome. Our aims were to demonstrate the usefulness of sampling the MLI to study the mucosa-associated microbiota while minimizing the effects of contaminants and to characterize the biogeography of the human colonic microbiome from the strongly adherent mucosal community (biopsies) to the more loosely mucosa-associated (MLI) and luminal (stools) microbiota. We used quantitative PCR (qPCR) to determine the biomass within our samples and describe a simple bioinformatics workflow to identify and remove contaminants from 16S rRNA amplicon-based datasets. We demonstrate that contaminants may significantly impact the microbiome results from intestinal biopsies and that MLI sampling constitutes a valuable source of biomass for characterizing mucosa-associated microbiota.
Methods
Ethics approval and consent to participate
Ethics approval for this study was obtained from the Research Ethics Board of the Children’s Hospital of Eastern Ontario (CHEO). Informed consent/assent was obtained from subjects and parents.
Human subjects and sample collection
Subjects were selected from children scheduled to undergo diagnostic colonoscopy at the CHEO (Supplemental Table S1). The visual appearance on endoscopy was normal on all subjects. Biopsies from these subjects were also histologically normal, and their final diagnosis was not for any known inflammatory intestinal disorder or any condition that is known to be associated with gut microbiota dysbiosis such as IBD. Patients who had used antibiotics, probiotics, or immunomodulatory medications within the last 4 weeks were excluded.
Children received a colonoscopy preparation the day before the endoscopy, following a previously reported standard protocol for Pico-Salix® that was shortened to one day rather than the 2-day preparation.25 At the time of endoscopy, loose fluid and debris were aspirated from the targeted location. Thereafter, sterile water was flushed onto the mucosa to remove the loosely adherent mucosal layer through the colonoscope working channel. There is never any damage to the intestinal mucosa using this technique. The resulting mixture was aspirated into a sterile container through the colonoscope working channel without difficulty and represents our MLI samples. We routinely obtain between 40 and 80 mL of MLI aspirates from colon endoscopies along the length of the region of colon of interest. After MLI collection, biopsy samples were collected at the same location. MLI aspirates were immediately placed on ice in the endoscopy suite, promptly transferred to the lab, mixed to ensure they were homogenous, aliquoted, and stored at −80 °C. Biopsies were flash frozen on dry ice in the endoscopy suite and stored at −80 °C until further processing. In total, 18 MLI and 10 matched biopsy samples were collected. To assess the potential impact of freezing on MLI aspirates, we collected samples from a separate cohort and processed the MLI aspirates immediately and after freezing at −80 °C.
We also collected stool samples from 11 patients as this is a readily available source of microbes that is commonly used in microbiota studies. Stools were collected by each subject, stored at −20 °C prior to transport to the CHEO and immediately stored at −80 °C until further processing.
Quantifying 16S rRNA gene copy numbers by qPCR
The copy number of 16 rRNA genes present in samples was determined by conducting absolute qPCR on metagenomic DNA using an Applied Biosystems 7500 and universal 16S rRNA primers 331F (5′-TCCTACGGGAGGCAGCAGT-3′) and 797R (5′-GGACTACCAGGGTATCTAATCCTGTT-3′).26 The standard curve for total 16S rDNA gene copy numbers was constructed from Fusobacterium nucleatum ATCC 10953 amplicons. DNA from a pure culture of F. nucleatum was extracted as done for the metagenomic samples, and the 16S sequence was amplified using the 331F/797R primers. The resulting amplicons were purified, quantified, and aliquots containing 107, 106, 105, 104, 103, and 102 copies prepared. Ct threshold values were determined for each concentration by qPCR. qPCR reaction conditions were as follows: 30 ng template DNA with 1 μM of each primer in 25 µL reaction mixture and QuantiFast SYBR Green PCR master mix (Qiagen). The amplification conditions were 5 min at 95 °C followed by 40 cycles of 95 °C for 10 s and data collection at 66 °C for 1 min. The number of 16S rRNA gene copies/µg DNA was determined for nine MLI aspirates and mucosal biopsies in duplicate. A Wilcoxon's matched-pair signed-rank test in GraphPad Prism (GraphPad, La Jolla, CA, USA) was used to statistically compare the results.
V6–16S rRNA gene library construction
Metagenomic DNA extraction, V6–16S rRNA gene library construction and sequencing was done as previously described.19 Briefly, metagenomic DNA was extracted using beads beating and V6–16S rRNA gene libraries constructed using two sequential PCR reactions. For MLI samples, 2 mL of collected aspirate was pelleted and used for metagenomic DNA extraction; for biopsies, the entire biopsy was used. All V6–16S rRNA gene libraries were constructed from 50 ng input DNA, except for the technical replicates used to identify the contaminate operational taxonomic units (OTUs). In these experiments, 10-fold serial dilutions of the input DNA were used instead (resulting in 50, 5, 0.5, and 0.05 ng inputs). Negative controls were constructed using sterile water as the template. Final amplicons were subsequently gel extracted (serial dilutions and negative controls were extracted even if no amplicon band was visible) and equal masses pooled for 100 bp paired-end sequencing on a HiSeq 2500 at the Center for Applied Genomics (Toronto, ON, Canada).
To assess the potential impact of freezing and sample volumes on MLI aspirate profiling results, we collected samples from a separate cohort and processed the MLI aspirates immediately and after freezing at −80 °C. We also profiled MLI microbial communities from aspirates volumes of 2, 4, 6, and 10 mL. These samples were processed separately from the matched sample types and subjected to V6 amplicon profiling using an Ion Torrent PGM as previously described.19
Read processing and OTU picking
Raw reads were processed as previously described.19 Briefly, raw sequencing reads were quality filtered and demultiplexed prior to OTUs picking. The demultiplexed reads are available at the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accesssion number PRJNA395534. QIIME 1.8.027 was used to identify OTU using a closed reference strategy against the Greengenes 13.5 database, OTUs with <2 counts were removed and the data were analyzed using phyloseq.28
Identifying potential contaminant OTUs
To identify potential technical contamination, we characterized the microbiota in four 10-fold serial dilutions of the DNA extracted from MLI aspirates (libraries were constructed from 50, 5, 0.5, and 0.05 ng input DNA). The OTU table with these samples was extracted and rarefied to 20,000 reads. Correlations between OTU relative abundances and the amount of DNA used for library construction was evaluated using Spearman, Kendall, and Pearson correlations. The DNA amount used for library construction was log10 transformed for the Pearson correlation. Correlations with a Benjamini and Hochberg false discovery rate corrected p < 0.05 were considered significant. The OTUs with significant negative correlations were iteratively removed from the negative controls based on their correlation values to assess their contribution to the reads seen in the negative controls (Supplemental Figure S1). Removing OTUs with a Spearman ρ < −0.7 reduced the number of reads in the negative control by >99% and these 26 OTUs were removed from all samples for subsequent analyses (Supplemental Figure S2, Supplemental Table S2). The raw data and annotated R code used to identify the potential contaminant OTUs are included as supplemental files (Supplemental Dataset 1, Supplemental Dataset 2).
Microbial community analyses
Differences between groups were assessed using either a Kruskal–Wallis test with Dunn’s post hoc or Mann–Whitney test as appropriate. Principal coordinate analysis (PCoA) was conducted to compare the β-diversity between samples rarefied to 20,000 reads and the variation explained by different metadata was assessed using adonis.29 When required, p values were corrected for multiple tests using the Benjamini and Hochberg approach and an FDR corrected p < 0.05 was considered significant.
LEfSe30 was used to identify differences in taxa abundance sample types. The raw OTU table was first filtered to remove OTUs that were not present in ≥10% of the samples within at least one group to reduce noise and the samples were then rarefied to 20,000 reads. A log10 linear discriminant analysis significant threshold ≥2 and p <0.05 was considered significant. The samples processed from both fresh and frozen MLI aspirates were rarefied to 8,000 reads and subjected to PCoA to assess the impact of freezing on MLI aspirate communities.
Results
Study cohort
A total of 18 children (5–18 years old) were included in the current study. The colonoscopy was medically indicated due to one or more symptoms such as weight loss, colonic polyp screening, prolonged unexplained non-infectious diarrhea, and/or abdominal pain. All recruited subjects exhibited a macroscopically normal mucosa with endoscopy and microscopically normal colon on histological examination of the biopsies (Supplemental Table S1).
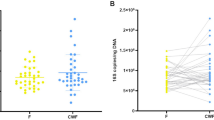
MLI aspirates generate higher microbial biomass as compared to mucosal microbiota
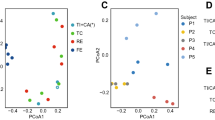
DNA extractions from individual biopsies yielded 10.7 ± 1.8 µg total DNA (mean ± SEM). In contrast, DNA extractions from 2 mL of our 40–80 mL MLI aspirate sample volumes typically resulted in 9.07 ± 0.32 µg total DNA. To test the efficiency of aspirates in collecting intestinal microbiota, we compared the 16S rRNA gene copy number/µg total DNA for nine paired MLI aspirates and mucosal biopsies using qPCR with universal primers. The average number of 16S rRNA gene copies/µg DNA was 104-fold greater in MLI aspirates than mucosal biopsies (Fig. 1). Samples with low microbial biomass are more vulnerable to contamination from downstream methodologies that may lead to inconsistencies.14,15 To test the impact of the microbial biomass content on the intestinal microbiota composition, we prepared serial dilutions of the DNA extracted from MLI aspirates and compared the microbiota composition retrieved from these dilutions to that of mucosal biopsies collected from the same children. We also constructed four negative control sequencing libraries where sterile water was used as the template. PCoA demonstrated that clustering of the microbiota was heavily biased on the amount of input DNA used for library construction (Fig. 2a, b). This visual clustering was confirmed by analyzing the variation explained by different covariates, with patient source and input DNA amount yielded fairly similar results (Table 1). Moreover, it was apparent that lowering the amount of input DNA in MLI aspirates resulted in the microbiota becoming more similar to the negative controls (Fig. 2b). In contrast to the undiluted MLI aspirates, many of the biopsy samples clustered with the negative controls and other highly diluted samples, suggesting that their composition was primarily derived from technical contaminants.

Mucosal–luminal interface (MLI) aspirates are efficient in collecting intestinal microbiota. The number of matched 16S ribosomal RNA (rRNA) gene copies from MLI aspirates or mucosal biopsies. The middle horizontal black line represents the median and pluses represent the mean. (**p < 0.01)

Removing potential contaminants reveals personal microbiotas. Principal coordinate analysis (PCoA) using the Bray–Curtis dissimilarities of patient-matched mucosal–luminal interface (MLI) aspirates, mucosal biopsies, diluted MLI aspirates, and negative sequencing controls. The raw results show a distinct trend for MLI samples to cluster with the negative controls as the amount of input DNA is diluted (a, b). Moreover, many biopsy samples also cluster with the negative controls. After contaminant removal, biopsies and MLI aspirates (including dilutions) cluster as function of donor patient (c, d)
We thus assessed whether the MLI/biopsy samples derived from low inputs of microbial biomass could be improved by removing technical contaminants. We tested the correlation between the OTU relative abundance and the amount of input DNA used for library construction using the Spearman correlation as previously proposed,13 along with the Kendall/Pearson correlations. OTUs with significant negative correlations (p < 0.05) were iteratively removed from the negative controls based on their coefficient values (Supplemental Figure S1). We found that the Spearman correlation was more sensitive in detecting potential outliers with significant negative correlations as compared to the Kendall/Pearson correlations. Removing the 26 OTUs with a Spearman ρ ≤ −0.7 was sufficient to remove >99% of the reads present in the negative controls, suggesting that these OTUs were true contaminants (Supplemental Table S2). Interestingly, repeating the PCoA analysis after filtering out these contaminant OTUs resulted in the MLI samples and mucosal biopsies clustering primarily by patient source and not by input DNA amount or sample type (Fig. 2c, d). In addition, the amount of variation explained by patient source became the dominant factor explaining the clustering pattern (Table 1). These results demonstrate that while low biomass samples such as biopsies can be heavily influenced by technical contamination, they can be highly representative of the original microbial community once this contamination is identified and removed. To note, contaminant OTUs represented 61.9 ± 9.67%, 0.06 ± 0.0005%, and 0.06 ± 0.0006% (mean ± SEM) of the total microbial population detected from mucosal biopsies, MLI aspirates, and stools, respectively, revealing that biopsies were the only sample type significantly affected by methodological contaminants.
We also sought to assess the impact of freezing on the MLI microbiome composition by comparing MLI aspirates processed for DNA extraction immediately vs. those that were frozen at −80 °C. PCoA demonstrated that freezing the samples had minimal impact on the microbiota composition when compared to either patient source or sampling location (Supplemental Figure S3, Supplemental Table S3). In addition, we tested whether the MLI aspirate volume used for the 16S rRNA gene amplicon library construction impacted the overall composition of the microbial community profiled (Supplemental Figure S4) and found that 2 mL aliquots were sufficient to accurately reflect the MLI aspirate microbial profile.
Patient microbiotas are highly personal across MLI aspirates, mucosal biopsies, and stool samples
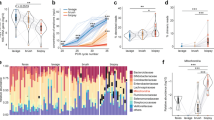
We then proceeded to assess microbial diversity in MLI aspirates, mucosal biopsies, and stool samples after removing contaminant OTUs. Stools showed a significantly higher richness (Chao1 index) and diversity (Shannon index) as compared to the biopsies or aspirates (p < 0.05; Fig. 3a, b). To gain a general overview of the gut microbiota similarity among the three sample types, we conducted PCoA and did not observe any separation between the samples based on the sample type (Fig. 3c). Instead, the samples appeared to cluster based on patient source (Fig. 3d). This was also apparent in the variation explained by each metadata factor, with patient source explaining the majority of the clustering (Table 1).

Diversity of the gut microbiota from mucosal–luminal interface (MLI) aspirates, mucosal biopsies, and stool samples. Richness (a) and diversity (b) of intestinal microbiota from mucosal biopsies, MLI aspirates, and stools. The middle horizontal black line represents the median and boxes the interquartile range. Principal coordinate analysis revealed that samples cluster by patient and by sample type (c, d). (*p < 0.05; **p < 0.01)
Given that interpatient variation was dominant in our samples, we sought to assess how similar the MLI aspirates were to both biopsy and stool samples by comparing matched patient samples after contaminant removal. We found that the MLI aspirates are significantly more similar to biopsies as compared to stool samples when using the Bray–Curtis dissimilarity (Fig. 4a) or weighted-Unifrac distance metric (Fig. 4b), but not when employing the unweighted-Unifrac distance metric (Fig. 4c).

Mucosal–luminal interface (MLI) aspirates and biopsies from the same patient are highly similar. The Bray–Curtis dissimilarity (a), weighted-Unifrac (b), and unweighted-Unifrac (c) distances from matched patient samples. Aspirates are significantly more similar to biopsies as compared to stool samples using the Bray–Curtis dissimilarity or weighted-Unifrac, but not when employing the unweighted-Unifrac distance metric (*p < 0.05; **p < 0.01). NS, not significant
We also compared the abundance of various taxa present in each sample type. The phyla Firmicutes, Bacteroidetes, and Proteobacteria accounted for 98% of the total relative abundance in each sample type (Fig. 5a). Firmicutes and Bacteroidetes showed an inverse relationship across the three sample types. Bacteroidetes showed a gradient decrease from biopsy to aspirate to stool samples, while Firmicutes showed an increasing gradient (p < 0.05). The high interpatient variation and our limited sample number precluded an OTU level analysis of the differentially abundant microbes. Instead, we employed LEfSe30 to identify taxa that were characteristic of each sample type at each phylogenic level. This analysis identified numerous taxa that were associated with each sample type (Fig. 6, Supplemental Table S4). The top feature for biopsy samples was an enrichment of Bacteroides spp., followed by enrichment of the order Bacillales and various Staphylococcus-related taxa (at the order, genus, and species levels). In contrast, the top features in aspirates were related to Lachnospiraceae or Enterococcus taxa. Stools showed general enrichment of Firmicutes taxa, especially unclassified taxa belonging to the Clostridiales order.

Taxonomic composition of biopsies, mucosal–luminal interface (MLI) aspirates, and stool microbial communities. Mean ± SEM relative abundance of dominant phyla from biopsies, MLI aspirates, and stools (a). The median relative and interquartile range of phyla with a median relative abundance >0.1% in at least one sample type (b). (*p < 0.05)

Microbial taxa that vary significantly in abundance between different sample types. Histogram of linear discriminant analysis effect size score of the top seven differentially abundant bacterial taxa between different sample types
Discussion
Currently, collecting mucosal biopsies is the most common methodology to sample mucosal microbiota at specific sites within the gastrointestinal tract. Stools are also frequently employed to survey microbiota communities as they are easily collected, non-invasive, and supply ample biological material for experimentation. However, it has become increasingly obvious that stools are poor proxies for regional changes that may occur in the gut.2,3,4,6,7 Stool is an amalgamation of microbiotas from the entire gastrointestinal tract and is most similar to the luminal microbiota from the descending colon.11 Indeed, stool is increasingly less representative of the gastrointestinal tract microbiota as one moves away from the rectum.11,31 While biopsies will profile a specific site within the gut, they are typically quite small and may not yield sufficient biological material for multiple experiments. In addition, there are several reports suggesting that the tightly adherent and loosely adherent mucosal communities of the gut contain distinct and functionally non-redundant bacterial communities.3,4,5
It should be noted that regardless of source, samples collected for microbial community profiling must have sufficient microbial biomass. We found that mucosal biopsies contain much less microbial biomass as compared to MLI aspirates (Fig. 1) and were often indistinguishable from negative controls and MLI samples that had been diluted 1000× (Fig. 2a, b). In fact, identifiable contaminants represented on average ~62% of the total microbiota identified from mucosal biopsies compared to ~0.06% in MLI aspirates or stool samples. Removing these contaminants from mucosal biopsies resulted in samples clustering by source individual as would be expected (Fig. 2c, d). Thus, it is likely that the low microbial biomass in mucosal biopsies is often swamped by reagent/laboratory contaminants, which may lead to inconsistent and misleading findings. Indeed, a recent report comparing colonic lavages to unwashed biopsy samples also noted the enrichment of likely contaminants in their biopsy microbiota results.17
There have been numerous reports describing the microbiota communities present in low biomass sample types such as breastmilk,32 blood,15,33 and the placenta.34,35 These attempts to characterize microbiota communities in low biomass samples haves highlighted some of the technical challenges inherent in using PCR-based profiling of microbial communities.14,15,36 Indeed, as others have previously noted,16 many of the “core” microbiota present in these samples are common contaminants within DNA extraction kits,14 PCR reagents,15 and ultra-pure water facilities.37 Many groups have begun to include positive controls to assess their microbiota profiling pipelines using in-house or commercially available mixtures. However, these controls are typically included under ideal conditions (i.e., at high concentration) that may not be representative of low biomass samples. Serially diluting representative samples known to contain high levels of bacterial biomass to assess the presence of contaminants can thus overcome these limitations. Moreover, we propose assessing the efficiency of contaminant removal to negative controls sequenced alongside true samples. We found that the Spearman correlation outperformed Kendall/Pearson correlations and that setting a threshold of ρ < −0.7 removed >99% of the reads present in our negative sequencing control. Each metric can be easily tested using the framework proposed above to find the optimal approach and threshold for each unique experimental setup. This approach would also be able to discriminate between true taxa that are part of the microbial community under study and phylogenetically related contaminants. Similarly, we note that existing positive control workflows could also be easily adapted to this approach.
The MLI aspirates and mucosal biopsies collected in this study exhibit lower microbial diversity as compared to stool samples (Fig. 3a, b), as previously reported for biopsies vs. stool samples.7,38,39 This is not surprising as stool microbiotas are composed of free luminal microbiota in addition to the shed mucosal microbiota from varying intestinal regions, whereas MLI aspirate/biopsies are microenvironments with specific microbial niches and communities. Our results indicate that the microbial differences between different individuals are greater than the differences between these microenvironments within the same individual (Fig. 3c, d). This finding is in agreement with other reports profiling mucosal biopsies and luminal microbial communities.6 By employing matched patient samples, we found that MLI aspirates were significantly more similar to biopsies as compared to stool samples when employing classical (Bray–Curtis) or relative abundance based (weighted-Unifrac) distance metrics (Fig. 4a, b). However, biopsy samples contain numerous rare microbial taxa that distinguish them from MLI aspirate samples (unweighted-Unifrac, Fig. 4c), highlighting the unique microbial communities that exist in close proximity to the gastrointestinal wall. It should also be noted that the biopsy samples may still harbor technical contaminants that were not able to be removed using our bioinformatic pipeline. As such, these rare microbes may actually be technical contaminants, and these could contribute to the differences seen between the biopsy samples and their matched stool/MLI aspirates. Additional carefully controlled studies (i.e., with appropriate negative and kit blanks etc.) with larger patient cohorts would be required to fully distinguish between “true” rare mucosal microbes and technical contamination.
The distinct microbial communities that exist within each sample type were further highlighted when comparing differentially abundant taxa. There was an inverse relationship between Bacteroidetes and Firmicutes across all three sample types with the presence of a gradient running from biopsies to aspirates to stools (Fig. 5b). As befitting their status as an intermediate between biopsies and stools, we identified fewer taxa as biomarkers for aspirates10 as compared to either biopsies or stools (>60 each) (Supplemental Table S4). In addition, other niche-specific factors might also contribute to the observed biogeographic microbial distribution including the host secretion of antimicrobial compounds and nutrient availability.8 These results suggest that aspirates are well poised to serve as an intermediary sample source that has features that straddle both biopsies and stools while still retaining unique information on the microbial composition in the human gastrointestinal tract.
Our collective findings reveal the utility of collecting MLI aspirates to complement biopsies and stools as for characterizing human microbial communities. Although the collection of MLI aspirates is still an invasive approach, it is far less traumatic to the intestinal epithelium, covers a larger surface area and generates a higher microbial biomass as compared to mucosal biopsies. The latter characteristic is important for avoiding technical contaminants and for performing multiple meta-omic studies on the same sample. Indeed, MLI sampling typically results in 40–80 mL of aspirate (as compared to 5 mL for colonic lavages17) and allows for the same sample to be profiled using multiple experimental setups including metaproteomics,20,21,22,23,24 16S rRNA gene profiling,19 shotgun metagenomics,22 and potentially other applications such as metatranscriptomics or metabonomics. Additional advantages of the MLI approach are that freezing MLI aspirates appears to have minimal impact on the microbial composition as assessed via 16S rRNA amplicon profiling (Supplemental Figure S3), and the rich biomass in MLI aspirates allows for a consistent microbial community profile to be maintained when storing aliquots (Supplemental Figure S4). This is an attractive feature for the clinical implementation of MLI samples in multi-center cohort studies to allow for batch sample processing.
In conclusion, sampling the MLI using aspirate washings during colonoscopy has the potential to combine the strengths of both stools and biopsies. In particular, MLI aspirates can mimic the regional specificity of biopsies, but still provide comparable biomass to stools. Moreover, MLI samples represent an important sample type in their own right to characterize human microbiota communities. Only by combining all three sample types will we achieve a complete picture of the human colonic microbiome, from strongly adherent mucosal communities (biopsies) to the more loosely mucosa-associated (MLI) and luminal (stool) microbiota.
References
Cani, P. D. Gut microbiota—at the intersection of everything?. Nat. Rev. Gastroenterol. Hepatol. 14, 321–322 (2017).
Glymenaki, M. et al. Compositional changes in the gut mucus microbiota precede the onset of colitis-induced inflammation. Inflamm. Bowel Dis. 23, 912–922 (2017).
Heinsen, F. A. et al. Dynamic changes of the luminal and mucosa-associated gut microbiota during and after antibiotic therapy with paromomycin. Gut Microbes 6, 243–254 (2015).
Huse, S. M. et al. Comparison of brush and biopsy sampling methods of the ileal pouch for assessment of mucosa-associated microbiota of human subjects. Microbiome 2, 5 (2014).
Galley, J. D. et al. The structures of the colonic mucosa-associated and luminal microbial communities are distinct and differentially affected by a prolonged murine stressor. Gut Microbes 5, 748–760 (2014).
Lavelle, A., et al. Spatial variation of the colonic microbiota in patients with ulcerative colitis and control volunteers. Gut 64, 1553–1561 (2015).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014).
Donaldson, G. P., Lee, S. M. & Mazmanian, S. K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14, 20–32 (2016).
Tang, M. S. et al. Inferred metagenomic comparison of mucosal and fecal microbiota from individuals undergoing routine screening colonoscopy reveals similar differences observed during active inflammation. Gut Microbes 6, 48–56 (2015).
Ringel, Y. et al. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes 6, 173–181 (2015).
Yasuda, K. et al. Biogeography of the intestinal mucosal and lumenal microbiome in the Rhesus macaque. Cell Host Microbe 17, 385–391 (2015).
Parthasarathy, G. et al. Relationship between microbiota of the colonic mucosa vs feces and symptoms, colonic transit, and methane production in female patients with chronic constipation. Gastroenterology 150, 367–379.e361 (2016).
Jervis-Bardy, J. et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome 3, 19 (2015).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B. & Chiodini, R. J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8, 24 (2016).
Perez-Munoz, M. E., Arrieta, M. C., Ramer-Tait, A. E. & Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome 5, 48 (2017).
Watt, E. et al. Extending colonic mucosal microbiome analysis-assessment of colonic lavage as a proxy for endoscopic colonic biopsies. Microbiome 4, 61 (2016).
McHardy, I. H. et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome 1, 17 (2013).
Mottawea, W. et al. Altered intestinal microbiota–host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 7, 13419 (2016).
Presley, L. L. et al. Host–microbe relationships in inflammatory bowel disease detected by bacterial and metaproteomic analysis of the mucosal–luminal interface. Inflamm. Bowel Dis. 18, 409–417 (2012).
Li, X. et al. A metaproteomic approach to study human–microbial ecosystems at the mucosal luminal interface. PLoS ONE 6, e26542 (2011).
Zhang, X. et al. MetaPro-IQ: a universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome 4, 31 (2016).
Deeke, S. A. et al. Mucosal–luminal interface proteomics reveals biomarkers of pediatric inflammatory bowel disease-associated colitis. Am. J. Gastroenterol. 113, 713–724 (2018).
Zhang, X. et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 9, 2873 (2018).
Jimenez-Rivera, C., Haas, D., Boland, M., Barkey, J. L. & Mack, D. R. Comparison of two common outpatient preparations for colonoscopy in children and youth. Gastroenterol. Res. Pract. 2009, 518932 (2009).
Bourne, D. G., Muirhead, A. & Sato, Y. Changes in sulfate-reducing bacterial populations during the onset of black band disease. ISME J. 5, 559–564 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Oksanen, J. et al. vegan: Community Ecology Package. R package (2016). https://CRAN.R-project.org/package=vegan, Accessed 31 May 2018.
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Momozawa, Y., Deffontaine, V., Louis, E. & Medrano, J. F. Characterization of bacteria in biopsies of colon and stools by high throughput sequencing of the V2 region of bacterial 16S rRNA gene in human. PLoS ONE 6, e16952 (2011).
Hunt, K. M. et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE 6, e21313 (2011).
Paisse, S. et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56, 1138–1147 (2016).
Aagaard, K. et al. The placenta harbors a unique microbiome. Sci. Transl. Med 6, 237ra265 (2014).
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E. & Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6, 23129 (2016).
Lauder, A. P. et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4, 29 (2016).
Kulakov, L. A., McAlister, M. B., Ogden, K. L., Larkin, M. J. & O’Hanlon, J. F. Analysis of bacteria contaminating ultrapure water in industrial systems. Appl. Environ. Microbiol. 68, 1548–1555 (2002).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
van den Bogert, B., de Vos, W. M., Zoetendal, E. G. & Kleerebezem, M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl. Environ. Microbiol. 77, 2071–2080 (2011).
Acknowledgements
We thank Ruth Singleton and Amanda Mack for their assistance in collecting patient clinical data and samples. This work was funded by Genome Canada and the Ontario Genomics Institute (OGI-067), CIHR (GPH-129340, MOP-114872, ECD-144627), and the Ontario Ministry of Economic Development and Innovation (REG1-4450). D.M. and A.S. acknowledge funding from the IBD Foundation of Canada, Crohn’s and Colitis Canada, the CHEO foundation, the CHEO Research Institute. and the University of Ottawa Faculty of Medicine. W.M. acknowledges funding from Mansoura University, Egypt. T.A. was supported by a scholarship from King Abdulaziz University, through the Saudi Arabian Cultural Bureau in Canada. J.B. is the recipient of a CIHR/CAG Postdoctoral Award. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
W.M., D.M., and A.S. designed the study. D.M. collected the samples. W.M., J.L., T.A., J.M., and J.B. performed the experiments. W.M., J.B., and A.S. analyzed the data. W.M., J.B., and A.S. wrote the manuscript. D.M. and A.S. contributed equally to the work. All the authors contributed to editing and revising the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A.S. and D.M. co-founded Biotagenics, a clinical microbiomics company. The other authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mottawea, W., Butcher, J., Li, J. et al. The mucosal–luminal interface: an ideal sample to study the mucosa-associated microbiota and the intestinal microbial biogeography. Pediatr Res 85, 895–903 (2019). https://doi.org/10.1038/s41390-019-0326-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0326-7
