
Abstract
Genetic anomalies have a role in autism spectrum disorders (ASD). Each genetic factor is responsible for a small fraction of cases. Environment factors, like preterm delivery, have an important role in ASD. Preterm infants have a 10-fold higher risk of developing ASD. Preterm birth is often associated with maternal/fetal inflammation, leading to a fetal/neonatal inflammatory syndrome. There are demonstrated experimental links between fetal inflammation and the later development of behavioral symptoms consistent with ASD. Preterm infants have deficits in connectivity. Most ASD genes encode synaptic proteins, suggesting that ASD are connectivity pathologies. Microglia are essential for normal synaptogenesis. Microglia are diverted from homeostatic functions towards inflammatory phenotypes during perinatal inflammation, impairing synaptogenesis. Preterm infants with ASD have a different phenotype from term born peers. Our original hypothesis is that exposure to inflammation in preterm infants, combined with at risk genetic background, deregulates brain development leading to ASD.
Similar content being viewed by others


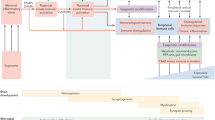
Introduction
The focus of this review is brain injury due to preterm birth and a set of neurodevelopmental disorders strongly associated with preterm birth, the autism spectrum disorders (ASD). Indeed, infants born preterm have a 10-fold higher risk to develop signs of ASD when compared to term infants.1,2,3 Of important note, preterm and term infants with ASD have not completely similar clinical phenotypes as preterm infants generally present a subcategory of ASD with social and communication inabilities but no repetitive/stereotypic behaviors.4 In term infants, it is largely accepted that ASD is linked to a combination of genetic and environmental factors, the relative weight of each being different for almost every infant.5 In preterm infants, brain injury (the so-called “encephalopathy of prematurity” or “EP”) is often secondary to neuroinflammation, which is mediated by microglia and astrocytes. EP is characterized in part by diffuse white-matter injury and synaptic abnormalities.6 As a great number of preterm born infants, relative to term born infants, will develop ASD, our overarching argument is that exposure to neuroinflammatory insult during the last trimester is an environmental factor of great importance to the development of ASD (Fig. 1). To provide support to our argument, we will provide a brief introduction to preterm birth and the brain injuries associated with early birth and the ASDs. We will then point to the similarities and differences in the etiology of these three disorders (preterms with cognitive impairment but no ASD, preterms with ASD, and terms with ASD), in the neurophenotypic outcomes, and also compare and contrast their neurobehavioural profiles. These differences will be key to understand for the development of viable strategies to improve the outcomes for all infants with ASDs.

Schematic representation of the potential interplay between environmental, epigenetic, and genetic factors in the appearance of ASD signs in preterm infants
Preterm birth
The World Health Organization (WHO) estimates that every year there are at least 15 million infants that are born prematurely, birth before 37 of 40 weeks completed gestation or gestational week (GW). This is ~1 in 10 infants globally that are born too early. Of these 15 million preterm born infants, close to 1 million will not survive and the majority of survivors will be left with permanent neurobehavioral deficits.7,8 The incidence of preterm birth is increasing in developed countries and the cause of this is unknown,9 although environmental factors such as air pollution are strong candidates.10 People who were born preterm tend to have lower IQ and to perform worse than aged-matched, full-term born controls in several cognitive areas such as executive functioning, language processing and working memory.11,12,13,14,15,16 These deficits become apparent in early childhood and are permanent, with observations having been made in large cohorts of infants born preterm through adolescence and into late adulthood.17,18 The severity of outcomes for individuals born preterm is directly related to the severity of the preterm birth although this is modulated by a host of factors to be discussed below (i.e., inflammation, stress, genetics). Specifically, preterm birth can be subdivided further based on gestational week at delivery as: extremely preterm (before GW 28), very preterm, (between GW 28 to 32) and moderate to late preterm (from GW 32 to 37). The majority (84%) of preterm births are classified as moderate to late preterm.9
The etiology of preterm birth
Approximately 45% of preterm deliveries occur with no obvious precipitating factors, i.e., spontaneously, and 25% follow a premature rupture of membrane (PPROM).19 The remaining 30% of preterm birth are due to induced labor or caesarean due to maternal or fetal conditions. More specifically, several clinical and demographic factors have been associated with preterm birth and are outlined in Table 1.
We would like to highlight a genetic paradigm is an important contributor as women are at higher risk of having a preterm delivery if they have previously experienced preterm delivery, or if they have close family members who have had a preterm delivery.20 Although no candidate genes for preterm birth have yet been reported from genome-wide association studies (GWAS), another approached has been to determine associations between single-nucleotide polymorphism (SNP) and preterm birth. More than 30 SNPs in genes known to have functions in inflammation and tissue remodeling have been associated with preterm birth, PPROM or the severity of brain injury after preterm birth across a number of studies.21,22,23,24 More recently, Strauss et al. reviewed the literature and presented a suite of studies that support that ’preterm birth has a polygenic basis that involves rare mutations or damaging variants in multiple genes involved in innate immunity and host defense mechanisms against microbes and their noxious products.’ These studies include whole-exome sequencing (WES) highlighting candidate genes linked to PPROM, and predominately innate immunity genes.25 This year another study has linked HSPA1L, Heat Shock Protein 70, with spontaneous preterm birth.26 Mutations in HSPA1L have also been recently linked with bowel inflammation.27
Psychosocial risk factors such as stress, anxiety and depression, individually or as comorbid conditions increase the risk of preterm delivery.28 Also linked to the incidence of preterm birth are environmental factors such as work environment, ambient and household air pollution10,29 and natural disasters,28 although any link between preterm birth and exposure to disaster (expected to increase stress levels) such as terrorist attack and hurricane was not supported in a meta-analysis.30
However, the single main risk factor for preterm birth is maternal–fetal infection/inflammation, with 25–40% of preterm delivery linked to infections either subsequent to PPROM or often with no prior diagnosis. The site of infection can include the choriodecidual space, chorion, placenta, amniotic fluid, umbilical cord, or the fetus. Infection triggers a cascade of events leading to preterm labor,31,32 that we understand involves the release of pro-inflammatory chemokines and cytokines which promote the production of prostaglandins that stimulate uterine contractions and the degradation of fetal membranes.28,31,32 It is also obvious that there is an interaction between genetic variance, i.e., single-nucleotide polymorphisms (SNPs) and preterm labor. Cohort studies have identified in various settings associations between variants in SNPs in cyclooxygenase-2 (PTGS2) and interleukin-6 receptor 1 (IL-6R) and preterm birth22,24 and specifically [reviewed in refs.33,34]. Moreover, in a US-based case-control study, researchers demonstrated that mothers carrying a polymorphism in the promoter of the tumor necrosis factor alpha gene (TNFα) have increased odds of preterm delivery following infection compared to women carrying the same polymorphism and not subjected to infection.28 In addition, preterm birth may be associated with intrauterine growth restriction (IUGR), also known as fetal growth restriction (FGR). IUGR/FGR is characterized by a fetus that does not grow to its genetically destined potential and the clinical diagnostic criteria for IUGR/FGR is when the birth weight is below the 10th percentile for the sex and GW of the infant.35
Encephalopathy of prematurity (EP)
Between GW 18 and 40, fetal white-matter (WM) volume shows a 22-fold increase in volume, the cortical grey matter (GM) undergoes a 21-fold increase and the deep subcortical structure volume increases by 10-fold. As such, preterm birth itself is associated with disruption of the normal developmental programs via neural circuit organization36 and also with events harmful to the brain directly such as inflammatory mediators (discussed further below). Specifically, MRI and post-mortem studies show in preterm born infants a constellation of injuries with diffuse white and grey matter abnormalities, including poor oligodendrocyte (OL) maturation and delayed myelination, reduced neurite formation and glial activation. In addition, generalized microstructural changes in the cortex are found in preterm infants although the structural underpinnings of these signals are poorly understood.37,38 Collectively these constellation of changes is known as encephalopathy of prematurity. We expand on the descriptions of these white and grey matter injuries in following sections.
In addition, it is worth mentioning that the last half of gestation is also fundamental for the growth and maturation of peripheral organs. As such, many preterm born infants suffer from a wide range of complications including respiratory, cardiovascular, and gastrointestinal deficits.39 In addition, necessary daily treatments and surgical procedures for the health of the infant can be painful. Pain has been suggested to itself impact on later cerebral and psychosocial development, but has not been linked specifically to ASD.40,41 It requires complex confounding variable analyses to link pain exposure to neurodevelopmental outcomes in preterm born infants as it is the sickest infants that need the greatest number of painful interventions. However, in the rat early life inflammatory pain exposure causes social behavior defects that mimics facets of human ASD behaviors.42
Introduction to causes of EP
There are several factors that are thought to contribute to the evolution of EP that include the disturbance of normal development and damaging extrinsic factors. The first postulated cause of EP relates to the facts that preterm born infants have immature respiratory and cardiovascular systems. The in utero environment in which normal brain development take place has a far lower oxygen tension than the oxygen tension found ex utero. This high (relatively) oxygen environment in which premature infants then undergo the remainder of their brain development. This ex utero hyperoxia is itself considered to be detrimental for brain development, and the negative effects of hyperoxia on the developing brain has been successfully modelled.43,44 However, a meta-analysis has found that target oxygen saturation ranges of 85–89% versus 91–95% has no effect on mortality and neurodevelopmental impairment at 18–24 months.45 It is also described in some cases that the ability of preterm infants to autoregulate their cerebral oxygen saturation is impaired.46,47 This is suggested to lead to periods of hypoxia–ischemia and reperfusion associated with the production of reactive oxygen species (ROS). However, poor cerebral autoregulation is not always observed in cohorts of preterm born infants and it is unclear what the true proportions of infants suffering from poor cerebral autoregulation is, see references and discussion in ref.48 The primary risk factor for preterm birth and predictor for outcome is exposure to infection or inflammation1,2,49,50, and for a lively discussion on the causal factors of injury in preterm infants please see ref.51 In response to infection and inflammation, the systemic immune cells produce a plethora of cytokines,52,53 which are a broad and loose category of small proteins (~5–20 kDa) that are important in cell signaling, including the chemotactic cytokines, chemokines. Chemokines and cytokines play not only a role in the bodies response to injury and infection, but also in the carefully curates steps of normal development, such as OL maturation.54,55 Chief among the cytokines known to have a role in injury to the developing brain is TNFa, which has direct effects in high concentrations to injure OL and neurons.56 As such, it is to be expected that inflammatory cytokines and chemokines have a chief role in the white and grey matter damage of EP. Cytokines are produced within the brain predominantly by microglia and astrocytes and the specific role of inflammation and these glial cells will be elaborated on below.
White matter defects associated with preterm birth
At the beginning of the twentieth century and through to the 1990’s in high income health care systems, white-matter injury (WMI) was the most obvious and predominantly studied facet of brain damage in premature infants.6 In 1962, Banker and Larroche reported, in 51 infants, the presence of periventricular leukomalacia (PVL), a severe form of cystic WMI, but relative sparing of the GM. Thanks to the advances in neonatal care in high income health care systems there are fewer preterm born infants demonstrating large severe cystic WMI, and also steep reduction in mortality among very and extremely preterm born infants.57 Limited access to health care, both antenatal and postnatal, is still associated with worse brain injury, higher mortality and poorer outcomes for preterm born infants, especially in developing countries.58
The panorama of injury in preterm born infants in developed countries is now described to principally involve either: (1) moderate WMI with small necrotic lesions and/or glial scars, referred to as “punctate white-matter lesion” (PWML) and diffuse signal abnormalities; or (2) mild to indeterminate WMI, showing microscopic or no necrotic lesions.59,60,61 Because there is heterogeneity in the types of WMI, the outcome for infants can vary from fine to severe motor deficits, sensory deficits, cognitive and learning impairments and behavioral disturbances.60,62
The predominate cause of WMI in preterm born infants is considered to be due to the maturation arrest of OL in the absence of overt oligodendrocyte progenitor cell (OPC) death. This has been shown in human preterm cohort studies,63,64,65 in studies of moderate inflammatory injury,66 and in studies of more severe experimental injury.67,68,69 In a cohort of human cases with severe WMI,70,71 OL maturation arrest has also been shown secondary to initial OPC death and progenitor proliferation. Numerous studies have demonstrated that the period between GW 28 and 32, which coincides with preterm delivery, is critical for OL maturation because pre-OL are highly vulnerable to environmental factors including inflammatory products such as cytokines and chemokines, for comprehensive reviews on this topic please refer to refs.72,73
Grey matter injury associated with preterm birth
As mention above, grey matter injury (GMI) includes a reduction in the growth of cortical and subcortical regions in EP, including the basal ganglia, thalamus, hippocampus, orbitofrontal lobe, posterior cingulate cortex, centrum semiovale and cerebellum.74,75,76 Previously, these abnormalities in preterm infants were attributed to injury related to severe WMI. However, despite a reduction in the severity of WMI (i.e., less cystic lesions), many preterm born infants still suffer from significant GM abnormalities. The presence of GMI in the absence of cystic lesions demonstrates that these GM changes are primarily development disturbances rather than secondary to destructive processes. Although it is known that during the 3rd trimester the cerebral cortex undergoes a dramatic increase in volume and gyration, results concerning GM from MRI after birth are equivocal. Some studies observed a delay in microstructural development of the GM in multiple cortical regions of brains from infants born preterm.37,38,77,78,79 Of note, it is not yet totally clear what are the neuropathological correlates of these abnormal cortical microstructure.80 Advances in MRI-based tractography have made it possible to define a “connectome”, which is a map of neuronal connections in the brain, and compare connectivity of preterm born brains to term born brains. This has been demonstrated a connectivity reduction between thalamus and cortex in preterm infants compared to term infants.81 Moreover, infants born at term demonstrated a strong resting states network in sensorimotor cortex and cerebellum, which in preterm infants at term-equivalent age were weaker.76,81,82,83 One hypothesis concerning the correlation between white matter and grey matter damage is that WMI in part due to pre-OL maturation blockade may activate astrogliosis and axonal myelination defects in preterm brains associated with neuroinflammation. These axonal defect lead to GMI due to abnormality of the axonal physical tension.77,82
Moreover, data suggest that there is also a reduction in the number of interneurons (inhibitory neurons) in preterm post-mortem brain (H. Stolp, unpublished data) and in rabbit studies.84,85 One hypothesis concerning the GM development delay is that preterm birth affects production, migration, survival and/or differentiation of some subsets of interneurons as well as a blockade of neuronal maturation linked to altered dendritic spine morphology and density.82 It has been demonstrated in several studies that this blockade in neuronal maturity is linked to less complexity of dendrites and less synaptic activity due to disrupted maturation of dendrites.6,74,75,82
Preterm birth and neuropsychiatric disorders
Infants who were born preterm have a 3-fold increase risk of developing a number of neuropsychiatric diseases, including those characterized by deficits in communication, attention and hyperactivity, social skills, and the severity of these are correlated to the degree of prematurity.86,87,88 Preterm born children and adults also have significantly higher scores on measures of high-intensity pleasure, perceptual sensitivity, and attention problems, and lower scores on discomfort, cuddliness, and attentional focusing.89,90,91 Overall these observations in infants born preterm are thought to be correlated to an impairment of the sensory-neuronal circuits.92
Autism spectrum disorders
According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-5), infants with ASD among other symptoms exhibit difficulty with communication and interaction with others and also show restricted interests and repetitive behaviors. ASD include infants diagnosed with PDD-NOS (Pervasive Developmental Disorder, Not Otherwise Specified), autistic disorder, and asperger syndrome. Indeed, ASD present with deficits that range in severity and are generally considered to be composed of deficits across two domains: social-communication deficits and restricted and repetitive behaviors (RRB), that can be further subdivided into repetitive sensory motor (RSM) and insistence on sameness (IS) behaviors.93 Kim et al.94 proposed recently a structural hierarchy of ASD with subcategories that distinguished between the two domains of the spectrum.
In ~35% of cases of ASD, inheritance follow a Mendelian pattern or are directly related to a genetic syndrome. The majority however of ASD is attributed to environmental factors, such as preterm delivery.5 Genes strongly associated with an increase of ASD risk include, fragile X mental retardation 1 (FMR1), neuroligin (NLGN), neurexin (NRXN), and SHANK.95 These and numerous additional genes whose mutations have been associated with ASD are involved in the control of neuronal activity, and epigenetic and transcriptional regulations (see below).
Early diagnosis of ASD, during early childhood development, are now possible through several behavioral observations and clinic evaluations such as the Autism Diagnostic Observation Schedule (ADOS)96,97 and the Modified Checklist for Autism In Toddlers (M-CHAT).98,99 To highlight the prevalence of ASD in preterm born infants, positive screening using the M-CHAT is close to 6% in children who were born at term, in in contrast, to 21% of children who were at an extremely low gestational age.100,101
ASD in infants born preterm
As outlined above, a major neuropsychiatric deficit associated with preterm birth are the ASDs. Several studies have demonstrated an increased risk for ASD of 7- to 10-fold in preterm born infants when compared to term born infants.1,2,3,102 Specifically, preterm infant in which preterm birth was associated with chorioamnionitis have a 16-fold increased risk to develop an ASD.3 This risk remains increased even when adjusting for IQ, given that intellectual disability (ID) is increased among infants born preterm.1 However, as ID is a common co-morbidity of ASD even among infants not born preterm the utility of excluding these patients probably gives an underestimate of the true rates of ASD in those born preterm. It is worth noting however that an association between preterm birth and ASD was not found in a cohort of Danish children born between 1990 and 1999,103 or in an Australian cohort born between 1980 and 1995.104 These differences may relate to changes in perinatal care, as the associations between ASD and prematurity are found more reliably in cohorts born after the year 2000 [see references and discussion in ref.105], where the spectrum of injury has shifted away from gross injury to diffuse and microstructural injury. A pertinent discussion on the problems of comparing early developmental milestones in infants and children born preterm with term born peers discussed whether any increase in rates of ASD might relate to delayed development rather than ASD.106 However, not only are more preterm children diagnosed with ASD in infancy and as toddlers, but these diagnoses are maintained into childhood4 and at least into young adulthood.107
The ASD phenotype for children born preterm is reported to differ from those born at term. Specifically, children born preterm have been reported to have significantly more impairment in social interactions, communication and total score, but not impairments associated with stereotypic behaviors,4 greater sensory problems,108 and in another cohort have more comorbid presentation with attention-deficient hyperactivity disorder (ADHD).109 A paper from this year attempted to identify whether repetitive behaviors could be used as a screening tool in toddlers born across a spectrum of gestational ages and weights.110 In comparison to the studies mentioned above, this paper sought to ascertain risk of suboptimal behavioral/psychological outcomes in otherwise undiagnosed children, rather than to clarify the proportion of behaviors in people with ASD based on gestational age at birth. Sifre and colleagues observed that gestational length alone did not predict the occurrence of repetitive or restricted behaviors, but that together with birth weight there was an association. However, the authors point out that their study was on a small cohort of relatively healthy born preterm infants and greater work in children at higher rick may reveal stronger links between birth timing and behavioral risk factors for ASD. A recent review has brought together these phenotypic differences together with observations on brain development,87 to outline how preterm birth causes an unbalanced maturation rate across the brain leading to neuropsychiatric disorders such as ASD. Phenotypic subtypes related to characteristics including sensory processing are well established in ASD.111,112 Nuanced mathematical approaches to using rich behavioral information such as these from patients is working towards better classifications based on environmental factors such as preterm birth and genetic influences.94
Similarities in the brain regions affected by EP and associated with ASD
Brain regions predominantly affected by preterm birth are those involved in sensory perception and social interactions,87 see Table 2. Of note, preterm born infants show a reduction in the volume of the orbitofrontal region, an important regulator of self-monitoring and emotional self-regulation.76 In term born infant diagnosed with ASD, the same regions are also affected.113,114,115
The corpus callosum, also known as the callosal commissure is the largest WM structure in the human brain, consisting of 200–250 million axonal projections. The corpus callosum increases in length by approximately 25% during the last trimester, reflecting axonal growth and myelination. Thus, this region is strongly and permanently affected in the brains of infants born preterm primarily due to effects on OL maturation. Furthermore, an underdeveloped corpus callosum is commonly reported in infants with ASD116, and was associated with impairment in social competence and additional deficits linked to autism, specifically in measures of social responsiveness.117
The amygdala processes social information including emotional responses (see review118). Infants born preterm demonstrate a larger amygdala volume but a reduced magnitude of activation, as assessed via MRI compare to infants born at term.92,119,120,121,122 In term infant diagnosed with ASD, clinicians also observed an increased amygdala volume and a reduction of WM connectivity in the right amygdala.123,124
The cerebellum is involed in the motor control but congential cebellar abnormalities are often associated with cognitive impairment. Similarly, in preterm infants, cerebellar damage was associated with poor neurological outcome including very increased risk for ASD.125 In addition several studies have showed abnormal cerebellar structures in term infants with ASD.126
We would also like to specifically highlight that there are shared physical characteristics between patient with ASD and those who were born preterm and suffered EP listed in Table 2. Specifically, these shared physical characteristics include fine motor deficits, poor coordination and toe walking.127 Toe walking is considered a co-morbidity with ASD rather than a diagnostic criterion, and although what causes this is not completely understood it is possibly due to a dysfunctional vestibular system or sensory processing issues. Sensory processing issues are also common in preterm born infants,128 possibly linking the increased presentation of toe walking in these two patient population.
Neuroinflammation
Neuroinflammation is defined as the mechanism of central nervous system (CNS) inflammation that occurs in response to trauma, infections, and/or neurodegenerative diseases.129 Two key cells involved in neuroinflammation are microglia and astrocyte, described below. In addition, immune cells from the peripheral system also produce cytokines and can themselves invade the brain following blood–brain barrier disruptions and participate in neuroinflammation.130
Microglia
Microglia are the resident immune cells of brain and spinal cord derived from erythromyeloid precursors from the yolk sac that migrate to the neuroepithelium during early embryonic development to become effectors of brain development and brain homeostasis.131,132,133 Microglial express a plethora of receptors such as recepetors of cytokines/chemokines, damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) membrane receptor to detect environmental modifications (see below). Microglia have been found to exist in all species with a nervous system.134
In the human and the mouse, microglia initially reside in the areas of the developing WM tracts, and proliferation in these zones begins early in development.135,136 In humans, there is proliferation of these immature amoeboid microglia during GW 10–16 and diffusion of these cells throughout the developing brain from the middle of the second trimester (approximately week 25).137 In the mouse proliferation occurs during the first two postnatal weeks with a peak at postnatal day (P)14 and adult numbers around P21. In the mouse and the human the turnover of microglia is very slow, and these cells live for at least 3 years in the uninjured mouse,138 and up to 20 years in the human.139 The peak window of vulnerability for EP is a critical period for microglia because they are still located in the developing WM.135,140
During development, immature amoeboid microglia adopt a ramified homeostatic state and form in adulthood a network specialized for local surveillance. The long cellular processes of microglia continuously scan their environment to detect disruptions in homeostasis using their battery of receptors for DAMPS, PAMPS, cytokines and factors in the extracellular matrix. Microglia are capable of switching rapidly to an activated profile if any modification is detected. The concept of activation includes a loss of homeostatic functions, to be discussed below. In response to altered homeostasis, such as brain injury or disease, activated microglia can exhibit graduated morphological transformation toward an amoeboid appearance that resembles the phagocytic macrophage. Via striking shifts in the transcriptional program of the cell, microglia also change their expression of receptors, enzymes and trophic factors and can migrate towards an area of injury and proliferate. It is increasingly recognized that there is considerable heterogeneity of microglial activation states in the brain. As such, the specific nomenclature of activation states and their specific roles in the brain are matters of debate,141 and this undoubtedly arises from developmental differences in microglial responses142 and also differences dependent on the specific nature of the brain injury (compare and contrast143,144). However, to aid study and description of these plastic cells we can distinguish three main phenotypes: (i) The pro-inflammatory phenotypes with increased expression of several pro-inflammatory enzymes or cytokines used as markers: NO synthase inducible 2 (Nos2), Cyclooxygenase 2 (Ptgs2) and Tnfa; (ii) The anti-inflammatory and pro-regenerative phenotype, which overexpress marker such Cd206, lectin galactosidase-binding soluble 3 (Lgals3) and insulin-like growth factor 1 (Igf1); and (iii) The immuno-regulatory phenotype often associated with pro-inflammatory stimulation probably to resolve inflammation expressing different markers such as Il1rn, Suppressor of cytokines signaling 3 (Socs3) and Sphk1.145 Importantly, these phenotypes can overlap and transition between them is possible, if not always complete, at least in vitro.145
In addition to their immune functions, microglia have specific and critical roles during normal brain development and thought out life. During neurogenesis, microglia promote selective neural precursor cell (NPC) death in several regions by producing ROS and by phagocytosis of the extraneous cells;146 but in contrast, microglia also support the survival of layer V neurons136 and support OL maturation by expressing IGF1.147 In the WM, microglia cells are also known to promote axon fasciculation, whereby neighboring axons travelling together adhere to each other and to be important in normal myelination.147 Microglia also contribute to circuit assembly by controlling the laminar position of somatosensory interneurons.136,148,149 Another major function of microglia during development are their crosstalk with mechanisms of synapse formation and pruning. Recent evidences demonstrated that microglia at P8-10 promote filopodia formation (future synapse) by contact-induced Ca2+.150 Moreover, from P8 to P21, microglia promote synaptic pruning by tagging weak synapses with complement system that are then phagocytosed.151 This increases the strength and the plasticity of synapses. It is worth noting that a disturbance in each of these normal neurodevelopmental functions have been linked to ASD.133,150,152,153,154,155,156,157 In preterm post-mortem brains, our team previously demonstrated that around cystic lesions microglia cell number is increased compare to term brains. This suggest that microglial activation maybe a key component of WMI and we propose also GMI.64,158,159,160 This association between microgliosis and injury has been observed in models of perinatal brain injury66,161,162,163,164,165,166,167 and reviewd in ref.168 Transcriptomic analysis of microglia in our mouse model of EP showed that not only they were polarized towards a pro-inflammatory phenotype but also that many transcripts corresponding to normal function of microglia during brain development were significantly downregulated.169 This suggests that these immature microglia abandon their developmental and homeostatic functions due to an inflammatory stimulus
Astrocytes
Astrocyte are the most numerous glial cell type. Neocortical astrocytes arise from two separate pools.170 Astrocytes found in the deep cortical layers and the WM arise from radial glial cells towards the end of neural proliferation.171 These radial glia are neural stem cells and have a role in guiding migrating neurons. Astrocytes of the superficial cortical layers are primarily derived from glial precursors that multiply in the subventricular zone and then migrate into the cortex.
In the human neocortex, astrocytic proliferation begins at around GW 24, with a peak at around GW 26–28. The exact date at which the production of astrocytes ends is not known, but it could be supposed that the major part of astrocytic production is over by the end of a normal pregnancy. This peak in the production of astrocytes at around GW 26–28 could be of particular importance for preterm newborns. Indeed, astrocytes have important roles in development and homeostasis, including in axonal guidance, the stimulation of neuronal growth, synapse formation, the transfer of metabolites between blood vessels and neurons, the establishment of the pattern of certain brain structures, the production of extracellular matrix components, the production of trophic factors, neuronal survival, myelination, and the maintenance of the blood–brain barrier. For example, experimentally blocking astrocyte production temporarily in the neocortex of rodents induces an increase in programmed cell death in neurons and long-term changes in neocortical synaptic equipment.172 As expected, based on the human data, in the mouse astrocytes are present before birth and proliferate during the first week and then elaborate their characteristic fine processes, the time equivalent to the human last trimester. Astrocytic processes are then in contact with the newly forming synapse.173
Astrocytes express receptors for DAMPs and PAMPs, such as bacterial and viral pathogens.174,175 In a healthy brain, astrocytes release modulatory factor including TGF-β that are thought to promote an anti-inflammatory environment.176 However, in response to injury or infection astrocytes will participate in neuroinflammation by releasing pro-inflammatory cytokines in response to microglial activation. Reactive astrocytes are also involved in the protecting neurons during neuroinflammation by reducing elevated extracellular glutamate177 and by modulating microglial activation. However, by contributing to the release of toxic molecules, they can also induce brain damage.173,178,179,180,181 Furthermore, there is a well-recognized interplay between microglia and astrocytes in both driving180 and resolving176 brain injury dependent on the circumstance. In developing brain, systemic inflammation activates maturing astrocytes that directly impairs OL maturation180 supporting that inflammation is a deleterious environmental factor leading to brain maturation impairments and neurodevelopmental disorders.
Neuroinflammation in EP and ASD
As previously described above, preterm delivery is often induced by maternal–fetal infection/inflammation.19,32,182 Parturition itself is a process mediated by inflammatory products.183,184 As such, it is overt stimulation of early parturition by aberrant inflammation that has a role in linking maternal–fetal infection/inflammation and preterm birth. In addition to driving preterm birth, exposure to maternal–fetal infection/inflammation is also a leading factor in the presence and severity of brain injury/neurodevelopmental disturbance in the infant2,185,186 and reviewd in refs.72,187 The maternal immune system reacts to the threat by releasing pro-inflammatory chemokines and cytokines in the blood vessel. These molecules then reach the developing fetus, which responds by expressing the same pro-inflammatory molecules even if not directly exposed to the infection. Thus the fetus will develop a systemic inflammation, that leads to a neuroinflammation.72 In preterm infants, neuroinflammation is the main cause of WMI: the pro-inflammatory molecules released by microglia is extremely toxic to pre-OL as describe above, and pro-inflammatory microglia profile disrupt its normal developmental function described below.156 Recently, some clinical data suggest that there is a correlation between neonatal increase of blood pro-inflammatory cytokines in term born and developing ASD later on.156,188,189 Moreover, data from post-mortem brains on term born diagnose with ASD demonstrate an increase in microglial and astroglial activation in brains tissues in cortex, cerebellum and WM.190 These data are additional arguments that underlie perinatal systemic inflammation as a major environmental factor in the development of ASD.
Similar causal factors in preterm birth and ASD
Sex dimorphism in ASD and preterm infant brain injury
Sexual dimorphism in outcome is common in infants with ASD and those born preterm.109,191,192 The “male disadvantage hypothesis” of Naeye et al.193 refers to the observation that females have a better survival rate compared to males facing same environmental factors. During development, there are two key levels of sexual dimorphism: (1) primary sex determination at the fertilization; (2) the secondary sex determination, after birth, occur in response to hormone secreted by sexual organs.194 During pregnancy, genetic and epigenetic differences between the sexes modulate placentogenesis. This sex-dependent variations in placental structure and function can lead to a differential response to stressors.195
Two recent studies in Australia and the UK have shown no difference in the rates of preterm birth between the sexes.196,197 However, clinicians routinely report developmental difference with males have a higher incidence of brain lesions and respiratory and cardiovascular deficits.191,198,199 Moreover, there is also a sex disparity to infection and inflammatory response in preterm birth.200 It is worth noting that there are striking sex differences in the microglia201,202,203 and astrocytes204,205,206 of mice and humans, that is independent of sex hormones. Given the important role of neuroinflammation in brain injury of the preterm infant these differences might explain in part the propensity of males to do worse than females.
ASD, like many neurodevelopmental disorders including consequences of prematurity, shows a clear sex bias with male-to-female ratio around 4:1.207 The real basis for this sex-ratio is still unknown but studies have mostly focused on genetic and epigenetic factors controlling primary sex determination period reviewed in Schaafsma and Pfaff.208 On the other hand, factors affecting the secondary sex determination period such as hormones and environmental stress during pregnancy could also be at play.208,209 As a consequence of these mechanisms, girls could be either better protected against the initial insult and/or they could have a better resilience than boys (reviewed in Szatmari209).
Epigenetic factors
We would like to highlight that epigenetic factors such as microRNA (miRNA) and chromatin remodeling proteins are emerging to increase the robustness of developmental programming. miRNA are a class of non-coding RNA that regulate gene expression and there is increasing evidence that they have a critical role in conferring robustness to cell fate programs in other cell types.210,211 Evolutionary, miRNA have been demonstrated to respond strongly to environmental stress; thus, a modification in miRNA expression due to environmental factors during development could impact their function.210,211,212
In EP, and more specifically in OPCs, this could lead to a sustained inhibition of myelination transcription factors, resulting in a blockade of the differentiation process, and thereby WMI. More generally, recent explorations have strongly supported a role for epigenetic mechanisms in OL differentiation and integrity in sustaining major changes in transcriptional networks, nuclear chromatin components, histone acetylation and methylation events, actors of DNA methylation and miRNA, all of which are sensitive to environmental insults.213,214,215,216,217,218,219,220 For a specific and detailed description of the molecular mechanisms regulating OL maturation in the context of WMD in preterm infants, please see ref. 73
In ASD, an epigenetic dimension have also been add to the risk factors including MeCP2 or other methyl binding proteins (MBDs), histone deacetylases (HDACs), chromodomain-helicase-DNA-binding Protein 8 (CHD8) or imprinted loci (IGF-1/H19) (reviewed in ref.221).
Animal modeling of neuroinflammation and signs of ASD
As described, numerous genes predominantly involved in synapse function have been associated with the development of ASD. Genetically modified mouse models for each of these genes has shown that these capture some aspects of behavioral defects that are reminiscent of clinical signs observed in ASD, even if transposing behavioral abnormalities observed in mice to infants with ASD is challenging. These studies have also verified synaptopathies and connectivity deficits and many describe specific regional alteration such as the hippocampus and cortex.222 However, the cerebellum is strongly associated in poor outcomes in preterm born infants223 and with ASD224, but only few animal studies have looked for cerebellum alteration in mouse models for ASD. Future studies should have stronger interest in cerebellum dysfunction in ASD mouse model.
Over the past 20 years as the spectrum of injuries in preterm born infants has reduced in severity and our epidemiological studies have improved in their specificity, new models of injury to the preterm born infant have been developed. These have revolved around inflammation,66 hyperoxia225 and hypoxia226 and there have been indications that the outcomes in these models has relevance for the neuropathology and outcomes in infants with ASD. Specifically, low level exposure to inflammation during development is associated with changes in genes associated with synapse structure and function,227 and synaptic density is reduced by hyperoxia and chronic hypoxia. Furthermore, behaviors including reduced social play behavior and increased repetitive grooming are impaired in a recently described model combining the injuries of inflammation and hypoxia.228 A limitation so far has been that a great majority of testing for outcomes in models of perinatal brain injury has focused on learning and memory with a lack of social interaction testing that could potentially make stronger relevance for ASD.
Conclusion
In conclusion, we formulated an argument that the development of ASD is linked to genetic and environmental factors. One of environmental factor is preterm birth and the associated processes of brain developmental disturbance induced by neuroinflammation that is observed in many preterm born infants. We specifically highlight that behavioral testing indicates that infants born preterm with ASD, when compared to term infants with ASD, have a subcategory of ASD with social and communication inabilities but no repetitive/stereotypic behaviors. Understanding these differences will be key to understand for the development of viable strategies to improve the outcomes for all infants with ASDs. The first step in moving towards this understanding is building enhanced animal models that encompass genetic and environmental factors. These studies need to include comprehensive neuropathological, imaging and behavioral outcome measures looking for similarities and differences relevant to term and preterm born infants with ASD.
Disclaimer
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. The first author (C.B.) is a PhD student affiliated to the PhD School Bio Sorbonne Paris Cité.
References
Joseph, R. M. et al. Prevalence and associated features of autism spectrum disorder in extremely low gestational age newborns at age 10 years. Autism Res. 10, 224–232 (2017).
Leviton, A. et al. The risk of neurodevelopmental disorders at age 10 years associated with blood concentrations of interleukins 4 and 10 during the first postnatal month of children born extremely preterm. Cytokine 110, 181–188 (2018).
Limperopoulos, C. et al. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics 121, 758–765 (2008).
Johnson, S. et al. Autism spectrum disorders in extremely preterm children. J. Pediatr. 156, 525–531 e2 (2010).
Kim, Y. S. & Leventhal, B. L. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol. Psychiatry 77, 66–74 (2015).
Kinney, H. C. The encephalopathy of prematurity: one pediatric neuropathologist’s perspective. Semin. Pediatr. Neurol. 16, 179–190 (2009).
Harrison, M. S. & Goldenberg, R. L. Global burden of prematurity. Semin. Fetal Neonatal Med. 21, 74–79 (2016).
Liu, L. et al. Global, regional, and national causes of under-5 mortality in 2000-15: an updated systematic analysis with implications for the sustainable development goals. Lancet 388, 3027–3035 (2016).
Blencowe, H. et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet 379, 2162–2172 (2012).
Qian, Z. et al. Ambient air pollution and preterm birth: a prospective birth cohort study in Wuhan, China. Int J. Hyg. Environ. Health 219, 195–203 (2016).
Cheong, J. L. et al. Association between moderate and late preterm birth and neurodevelopment and social-emotional development at age 2 years. JAMA Pediatr. 171, e164805 (2017).
Spittle, A. J. et al. Neurobehaviour at term-equivalent age and neurodevelopmental outcomes at 2 years in infants born moderate-to-late preterm. Dev. Med. Child Neurol. 59, 207–215 (2017).
Raju, T. N. K. et al. Adults born preterm: a review of general health and system-specific outcomes. Acta Paediatr. (2017).
Doyle, L. W. et al. Biological and social influences on outcomes of extreme-preterm/low-birth weight adolescents. Pediatrics 136, e1513–e1520 (2015).
Pascoe, L. et al. Preventing academic difficulties in preterm children: a randomised controlled trial of an adaptive working memory training intervention—IMPRINT study. BMC Pediatr. 13, 144 (2013).
Moore, T. et al. Neurological and developmental outcome in extremely preterm children born in England in 1995 and 2006: the EPICure studies. BMJ 345, e7961 (2012).
Healy, E. et al. Preterm birth and adolescent social functioning-alterations in emotion-processing brain areas. J. Pediatr. 163, 1596–1604 (2013).
Heinonen, K. et al. Late preterm birth and neurocognitive performance in late adulthood: a birth cohort study. Pediatrics 135, e818–e825 (2015).
Goldenberg, R. L. et al. Epidemiology and causes of preterm birth. Lancet 371, 75–84 (2008).
D’Onofrio, B. M. et al. Preterm birth and mortality and morbidity: a population-based quasi-experimental study. JAMA Psychiatry 70, 1231–1240 (2013).
Gravett, M. G. et al. Global report on preterm birth and stillbirth (2 of 7): discovery science. BMC Pregnancy Childbirth 10(Suppl 1), S2 (2010).
Romero, R. et al. Identification of fetal and maternal single nucleotide polymorphisms in candidate genes that predispose to spontaneous preterm labor with intact membranes. Am. J. Obstet. Gynecol. 202, 431.e1–431.e34 (2010).
Boardman, J. P. et al. Common genetic variants and risk of brain injury after preterm birth. Pediatrics 133, e1655–e1663 (2014).
Harding, D. R. et al. Cognitive outcome and cyclo-oxygenase-2 gene (-765 G/C) variation in the preterm infant. Arch. Dis. Child Fetal Neonatal Ed. 92, F108–F112 (2007).
Modi, B. P. et al. Mutations in fetal genes involved in innate immunity and host defense against microbes increase risk of preterm premature rupture of membranes (PPROM). Mol. Genet. Genom. Med. 5, 720–729 (2017).
Huusko, J. M. et al. Whole exome sequencing reveals HSPA1L as a genetic risk factor for spontaneous preterm birth. PLoS Genet. 14, e1007394 (2018).
Takahashi, S. et al. De novo and rare mutations in the HSPA1L heat shock gene associated with inflammatory bowel disease. Genome Med. 9, 8 (2017).
Yamamoto, S. & Premji, S. The role of body, mind, and environment in preterm birth: mind the gap. J. Midwifery Women’s. Health 62, 696–705 (2017).
Padula, A. M. et al. Environmental pollution and social factors as contributors to preterm birth in Fresno County. Environ. Health 17, 70 (2018).
Harville, E., Xiong, X. & Buekens, P. Disasters and perinatal health:a systematic review. Obstet. Gynecol. Surv. 65, 713–728 (2010).
Barros, F. C. et al. The distribution of clinical phenotypes of preterm birth syndrome: implications for prevention. JAMA Pediatr. 169, 220–229 (2015).
Nadeau, H. C., Subramaniam, A. & Andrews, W. W. Infection and preterm birth. Semin. Fetal Neonatal Med. 21, 100–105 (2016).
Holst, D. & Garnier, Y. Preterm birth and inflammation—the role of genetic polymorphisms. Eur. J. Obstet. Gynecol. Reprod. Biol. 141, 3–9 (2008).
Moura, E. et al. Inflammatory cytokine gene polymorphisms and spontaneous preterm birth. J. Reprod. Immunol. 80, 115–121 (2009).
Miller, S. L., Huppi, P. S. & Mallard, C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J. Physiol. 594, 807–823 (2016).
Andescavage, N. N. et al. Complex trajectories of brain development in the healthy human fetus. Cereb. Cortex 27, 5274–5283 (2017).
Eaton-Rosen, Z. et al. Investigating the maturation of microstructure and radial orientation in the preterm human cortex with diffusion MRI. Neuroimage 162, 65–72 (2017).
Ball, G. et al. Development of cortical microstructure in the preterm human brain. Proc. Natl Acad. Sci. USA 110, 9541–9546 (2013).
Chehade, H. et al. Preterm birth: long term cardiovascular and renal consequences. Curr. Pediatr. Rev. (2018).
Vinall, J. et al. Invasive procedures in preterm children: brain and cognitive development at school age. Pediatrics 133, 412–421 (2014).
Brummelte, S. et al. Procedural pain and brain development in premature newborns. Ann. Neurol. 71, 385–396 (2012).
Lee, J. H. et al. Neonatal inflammatory pain and systemic inflammatory responses as possible environmental factors in the development of autism spectrum disorder of juvenile rats. J. Neuroinflamm. 13, 109 (2016).
Scheuer, T. et al. Oligodendroglial maldevelopment in the cerebellum after postnatal hyperoxia and its prevention by minocycline. Glia 63, 1825–1839 (2015).
Endesfelder, S. et al. Caffeine protects neuronal cells against injury caused by hyperoxia in the immature brain. Free Radic. Biol. Med 67, 221–234 (2014).
Manja, V., Saugstad, O. D., & Lakshminrusimha, S. Oxygen saturation targets in preterm infants and outcomes at 18–24 months: a systematic review. Pediatrics 139 (2017).
Nuntnarumit, P., Rojnueangnit, K. & Tangnoo, A. Oxygen saturation trends in preterm infants during the first 15 min after birth. J. Perinatol. 30, 399–402 (2010).
Schwaberger, B. et al. Do sustained lung inflations during neonatal resuscitation affect cerebral blood volume in preterm infants? A randomized controlled pilot study. PLoS ONE 10, e0138964 (2015).
Kooi, E. M. W. et al. Measuring cerebrovascular autoregulation in preterm infants using near-infrared spectroscopy: an overview of the literature. Expert Rev. Neurother. 17, 801–818 (2017).
Hagberg, H. et al. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208 (2015).
Kuban, K. C. et al. The breadth and type of systemic inflammation and the risk of adverse neurological outcomes in extremely low gestation newborns. Pediatr. Neurol. 52, 42–48 (2015).
Gilles, F. et al. Hypoxia–ischemia is not an antecedent of most preterm brain damage: the illusion of validity. Dev. Med. Child Neurol. (2017).
Menon, R. et al. Diversity in cytokine response to bacteria associated with preterm birth by fetal membranes. Am. J. Obstet. Gynecol. 201, 306.e1–306.e6 (2009).
McElrath, T. F. et al. Blood protein profiles of infants born before 28 weeks differ by pregnancy complication. Am. J. Obstet. Gynecol. 204, 418.e1–418.e12 (2011).
Gottle, P. et al. Activation of CXCR7 receptor promotes oligodendroglial cell maturation. Ann. Neurol. 68, 915–924 (2010).
Vela, J. M. et al. Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Mol. Cell. Neurosci. 20, 489–502 (2002).
Andrews, T., Zhang, P. & Bhat, N. R. TNFalpha potentiates IFNgamma-induced cell death in oligodendrocyte progenitors. J. Neurosci. Res. 54, 574–583 (1998).
Carducci, B. & Bhutta, Z. A. Care of the growth-restricted newborn. Best Pract. Res. Clin. Obstet. Gynaecol. (2018).
Rajaratnam, J. K. et al. Neonatal, postneonatal, childhood, and under-5 mortality for 187 countries, 1970-2010: a systematic analysis of progress towards Millennium Development Goal 4. Lancet 375, 1988–2008 (2010).
Tusor, N. et al. Punctate white matter lesions associated with altered brain development and adverse motor outcome in preterm infants. Sci. Rep. 7, 13250 (2017).
van Tilborg, E. et al. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white-matter injury. Glia 66, 221–238 (2018).
Volpe, J. J. Confusions in nomenclature: “periventricular leukomalacia” and “white matter injury”-identical, distinct, or overlapping? Pediatr. Neurol. 73, 3–6 (2017).
Counsell, S. J., Ball, G. & Edwards, A. D. New imaging approaches to evaluate newborn brain injury and their role in predicting developmental disorders. Curr. Opin. Neurol. 27, 168–175 (2014).
Billiards, S. S. et al. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 18, 153–163 (2008).
Verney, C. et al. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white-matter injury in very preterm infants. J. Neuropathol. Exp. Neurol. 71, 251–264 (2012).
Buser, J. R. et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 71, 93–109 (2012).
Favrais, G. et al. Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 70, 550–565 (2011).
Segovia, K. N. et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 63, 520–530 (2008).
Schmitz, T. et al. Cellular changes underlying hyperoxia-induced delay of white matter development. J. Neurosci. 31, 4327–4344 (2011).
Dean, J. M. et al. Delayed cortical impairment following lipopolysaccharide exposure in preterm fetal sheep. Ann. Neurol. 70, 846–856 (2011).
Back, S. A. et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann. Neurol. 58, 108–120 (2005).
Haynes, R. L. et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J. Neuropathol. Exp. Neurol. 62, 441–450 (2003).
Bennet, L. et al. Chronic inflammation and impaired development of the preterm brain. J. Reprod. Immunol. 125, 45–55 (2018).
van Tilborg, E. et al. Impaired oligodendrocyte maturation in preterm infants: potential therapeutic targets. Prog. Neurobiol. 136, 28–49 (2016).
Back, S. A. Brain injury in the preterm infant: new horizons for pathogenesis and prevention. Pediatr. Neurol. 53, 185–192 (2015).
Penn, A. A. et al. Controversies in preterm brain injury. Neurobiol. Dis. 92(Pt A), 90–101 (2016).
Ball, G. et al. The effect of preterm birth on thalamic and cortical development. Cereb. Cortex 22, 1016–1024 (2012).
Garcia, K. E. et al. Dynamic patterns of cortical expansion during folding of the preterm human brain. Proc. Natl Acad. Sci. USA 115, 3156–3161 (2018).
Kelly, C. E. et al. Brain structural and microstructural alterations associated with cerebral palsy and motor impairments in adolescents born extremely preterm and/or extremely low birthweight. Dev. Med. Child Neurol. 57, 1168–1175 (2015).
Vinall, J. et al. Slower postnatal growth is associated with delayed cerebral cortical maturation in preterm newborns. Sci. Transl. Med. 5, 168ra8 (2013).
Stolp, H. B. et al. Voxel-wise comparisons of cellular microstructure and diffusion-MRI in mouse hippocampus using 3D bridging of optically-clear histology with neuroimaging data (3D-BOND). Sci. Rep. 8, 4011 (2018).
Ball, G. et al. The influence of preterm birth on the developing thalamocortical connectome. Cortex 49, 1711–1721 (2013).
Dean, J. M. et al. What brakes the preterm brain? An arresting story. Pediatr. Res. 75, 227–233 (2014).
Smyser, C. D. et al. Longitudinal analysis of neural network development in preterm infants. Cereb. Cortex 20, 2852–2862 (2010).
Malik, S. et al. Neurogenesis continues in the third trimester of pregnancy and is suppressed by premature birth. J. Neurosci. 33, 411–423 (2013).
Tibrewal, M. et al. Disruption of interneuron neurogenesis in premature newborns and reversal with estrogen treatment. J. Neurosci. 38, 1100–1113 (2018).
Cassiano, R. G., Gaspardo, C. M. & Linhares, M. B. Prematurity, neonatal health status, and later child behavioral/emotional problems: a systematic review. Infant Ment. Health J. 37, 274–288 (2016).
Fenoglio, A., Georgieff, M. K. & Elison, J. T. Social brain circuitry and social cognition in infants born preterm. J. Neurodev. Disord. 9, 27 (2017).
Sciberras, E. et al. Prenatal risk factors and the etiology of ADHD-review of existing evidence. Curr. Psychiatry Rep. 19, 1 (2017).
Kroll, J. et al. A dimensional approach to assessing psychiatric risk in adults born very preterm. Psychol. Med. 48, 1738–1744 (2018).
Heinonen, K. et al. Common mental disorders in young adults born late-preterm. Psychol. Med. 46, 1–12 (2016).
Saigal, S. & Doyle, L. W. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet 371, 261–269 (2008).
Rogers, E. E. & Hintz, S. R. Early neurodevelopmental outcomes of extremely preterm infants. Semin Perinatol. 40, 497–509 (2016).
Bishop, S. L. et al. Subcategories of restricted and repetitive behaviors in children with autism spectrum disorders. J. Autism Dev. Disord. 43, 1287–1297 (2013).
Kim, H. et al. Structural hierarchy of autism spectrum disorder symptoms: an integrative framework. J. Child Psychol. Psychiatry 59, 30–38 (2018).
Fakhoury, M. Imaging genetics in autism spectrum disorders: linking genetics and brain imaging in the pursuit of the underlying neurobiological mechanisms. Prog. Neuropsychopharmacol. Biol. Psychiatry 80(Pt B), 101–114 (2018).
Falkmer, T. et al. Diagnostic procedures in autism spectrum disorders: a systematic literature review. Eur. Child Adolesc. Psychiatry 22, 329–340 (2013).
Kamp-Becker, I. et al. Diagnostic accuracy of the ADOS and ADOS-2 in clinical practice. Eur. Child Adolesc. Psychiatry (2018).
Ben-Sasson, A., Robins, D. L. & Yom-Tov, E. Risk assessment for parents who suspect their child has autism spectrum disorder: machine learning approach. J. Med. Internet Res. 20, e134 (2018).
Mukherjee, S. B. Autism spectrum disorders—diagnosis and management. Indian J. Pediatr. 84, 307–314 (2017).
Kuban, K. C. et al. Positive screening on the modified checklist for autism in toddlers (M-CHAT) in extremely low gestational age newborns. J. Pediatr. 154, 535–540.e1 (2009).
Robins, D. L. et al. The modified checklist for autism in toddlers: an initial study investigating the early detection of autism and pervasive developmental disorders. J. Autism Dev. Disord. 31, 131–144 (2001).
Agrawal, S. et al. Prevalence of autism spectrum disorder in preterm infants: a meta-analysis. Pediatrics 142 (2018).
Maimburg, R. D. & Vaeth, M. Perinatal risk factors and infantile autism. Acta Psychiatr. Scand. 114, 257–264 (2006).
Glasson, E. J. et al. Perinatal factors and the development of autism: a population study. Arch. Gen. Psychiatry 61, 618–627 (2004).
Meldrum, S. J. et al. Autism spectrum disorder in children born preterm-role of exposure to perinatal inflammation. Front Neurosci. 7, 123 (2013).
Hofheimer, J. A., Sheinkopf, S. J. & Eyler, L. T. Autism risk in very preterm infants—new answers, more questions. J. Pediatr. 164, 6–8 (2014).
Wolford, E. et al. Autism spectrum traits and visual processing in young adults with very low birth weight: the Helsinki Study of Very Low Birth Weight adults. J. Dev. Orig. Health Dis. 8, 161–167 (2017).
Salk, A. Phenotype of term vs. preterm children in the autism treatment network database. J. Am. Acad. Child Adolesc. Psychiatry 55, S100 (2016).
Bowers, K. et al. Phenotypic differences in individuals with autism spectrum disorder born preterm and at term gestation. Autism 19, 758–763 (2015).
Sifre, R. et al. Restricted, repetitive, and reciprocal social behavior in toddlers born small for gestation duration. J. Pediatr. 200, 118–124 e9 (2018).
Tomchek, S. D. et al. Sensory subtypes in preschool aged children with autism spectrum disorder. J. Autism Dev. Disord. 48, 2139–2147 (2018).
Ausderau, K. K. et al. Sensory subtypes in children with autism spectrum disorder: latent profile transition analysis using a national survey of sensory features. J. Child Psychol. Psychiatry 55, 935–944 (2014).
Wee, C. Y. et al. Diagnosis of autism spectrum disorders using regional and interregional morphological features. Hum. Brain Mapp. 35, 3414–3430 (2014).
Dean, D. C. 3rd et al. Investigating the microstructural correlation of white matter in autism spectrum disorder. Brain Connect. 6, 415–433 (2016).
McAlonan, G. M. et al. Mapping the brain in autism. A voxel-based MRI study of volumetric differences and intercorrelations in autism. Brain 128(Pt 2), 268–276 (2005).
Prigge, M. B. et al. Corpus callosum area in children and adults with autism. Res. Autism Spectr. Disord. 7, 221–234 (2013).
Alexander, A. L. et al. Diffusion tensor imaging of the corpus callosum in autism. Neuroimage 34, 61–73 (2007).
Janak, P. H. & Tye, K. M. From circuits to behaviour in the amygdala. Nature 517, 284–292 (2015).
Adolphs, R. What does the amygdala contribute to social cognition?. Ann. N. Y. Acad. Sci. 1191, 42–61 (2010).
Gousias, I. S. et al. Magnetic resonance imaging of the newborn brain: manual segmentation of labelled atlases in term-born and preterm infants. Neuroimage 62, 1499–1509 (2012).
Scheinost, D. et al. Prenatal stress alters amygdala functional connectivity in preterm neonates. Neuroimage Clin. 12, 381–388 (2016).
Cismaru, A. L. et al. Altered amygdala development and fear processing in prematurely born infants. Front. Neuroanat. 10, 55 (2016).
Gibbard, C. R. et al. Structural connectivity of the amygdala in young adults with autism spectrum disorder. Hum. Brain Mapp. 39, 1270–1282 (2018).
Herrington, J. D. et al. Negative valence in autism spectrum disorder: the relationship between amygdala activity, selective attention, and co-occurring anxiety. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2, 510–517 (2017).
Limperopoulos, C. et al. Cerebral hemodynamic changes during intensive care of preterm infants. Pediatrics 122, e1006–e1013 (2008).
Khan, A. J. et al. Cerebro-cerebellar resting-state functional connectivity in children and adolescents with autism spectrum disorder. Biol. Psychiatry 78, 625–634 (2015).
Stephens, B. E. et al. Screening for autism spectrum disorders in extremely preterm infants. J. Dev. Behav. Pediatr. 33, 535–541 (2012).
Machado, A. et al. Sensory processing during childhood in preterm infants: a systematic review. Rev. Paul. Pediatr. 35, 92–101 (2017).
Shastri, A., Bonifati, D. M. & Kishore, U. Innate immunity and neuroinflammation. Mediat. Inflamm. 2013, 342931 (2013).
Hagberg, H., Gressens, P. & Mallard, C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 71, 444–457 (2012).
Kierdorf, K. et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 16, 273–280 (2013).
Ginhoux, F. et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010).
Tay, T. L. et al. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J. Physiol. 595, 1929–1945 (2017).
Biber, K., Owens, T. & Boddeke, E. What is microglia neurotoxicity (Not)? Glia 62, 841–854 (2014).
Verney, C. et al. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J. Anat. 217, 436–448 (2010).
Ueno, M. et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16, 543–551 (2013).
Monier, A. et al. Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J. Comp. Neurol. 499, 565–582 (2006).
Mouton, P. R. et al. Age and gender effects on microglia and astrocyte numbers in brains of mice. Brain Res. 956, 30–35 (2002).
Reu, P. et al. The lifespan and turnover of microglia in the human brain. Cell Rep. 20, 779–784 (2017).
Verney, C. et al. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J. Neuropathol. Exp. Neurol. 71, 251–264 (2012).
Ransohoff, R. M. A polarizing question: do M1 and M2 microglia exist?. Nat. Neurosci. 19, 987–991 (2016).
Butovsky, O. et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143 (2014).
Chhor, V. et al. Role of microglia in a mouse model of paediatric traumatic brain injury. Brain Behav. Immun. 63, 197–209 (2017).
Hellstrom Erkenstam, N. et al. Temporal characterization of microglia/macrophage phenotypes in a mouse model of neonatal hypoxic-ischemic brain injury. Front. Cell Neurosci. 10, 286 (2016).
Chhor, V. et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 32, 70–85 (2013).
Sierra, A. et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495 (2010).
Wlodarczyk, A. et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. (2017).
Pont-Lezica, L. et al. Microglia shape corpus callosum axon tract fasciculation: functional impact of prenatal inflammation. Eur. J. Neurosci. 39, 1551–1557 (2014).
Squarzoni, P. et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 8, 1271–1279 (2014).
Miyamoto, A. et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 7, 12540 (2016).
Paolicelli, R. C. et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011).
Fernandez de Cossio, L. et al. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav. Immun. 63, 88–98 (2017).
Jawaid, S. et al. Alterations in CA1 hippocampal synapses in a mouse model of fragile X syndrome. Glia 66, 789–800 (2018).
Li, Q. & Barres, B. A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242 (2018).
Pierre, W. C. et al. Neonatal microglia: the cornerstone of brain fate. Brain Behav. Immun. 59, 333–345 (2017).
Tay, T. L. et al. Microglia gone rogue: impacts on psychiatric disorders across the lifespan. Front Mol. Neurosci. 10, 421 (2017).
Thion, M. S. & Garel, S. On place and time: microglia in embryonic and perinatal brain development. Curr. Opin. Neurobiol. 47, 121–130 (2017).
Hagberg, H., David Edwards, A. & Groenendaal, F. Perinatal brain damage: the term infant. Neurobiol. Dis. 92(Pt A), 102–112 (2016).
Verney, C. et al. Neuronal damage in the preterm baboon: impact of the mode of ventilatory support. J. Neuropathol. Exp. Neurol. 69, 473–482 (2010).
Hagberg, H., Peebles, D. & Mallard, C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment. Retard. Dev. Disabil. Res. Rev. 8, 30–38 (2002).
Baud, O. et al. Gestational hypoxia induces white matter damage in neonatal rats: a new model of periventricular leukomalacia. Brain Pathol. 14, 1–10 (2004).
Tahraoui, S. L. et al. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol. 11, 56–71 (2001).
Ophelders, D. R. et al. Neuroinflammation and structural injury of the fetal ovine brain following intra-amniotic Candida albicans exposure. J. Neuroinflamm. 13, 29 (2016).
Schmitz, T. et al. Minocycline protects the immature white matter against hyperoxia. Exp. Neurol. 254, 153–165 (2014).
Kaur, C., Rathnasamy, G. & Ling, E. A. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J. Neuroimmune Pharmacol. 8, 66–78 (2013).
Fleiss, B. et al. Neuroprotection by the histone deacetylase inhibitor trichostatin A in a model of lipopolysaccharide-sensitised neonatal hypoxic-ischaemic brain injury. J. Neuroinflamm. 9, 70 (2012).
Dean, J. M. et al. Cerebellar white matter injury following systemic endotoxemia in preterm fetal sheep. Neuroscience 160, 606–615 (2009).
Baburamani, A. A. et al. Microglia toxicity in preterm brain injury. Reprod. Toxicol. 48, 106–112 (2014).
Krishnan, M. L. et al. Integrative genomics of microglia implicates DLG4 (PSD95) in the white matter development of preterm infants. Nat. Commun. 8, 428 (2017).
Gressens, P. et al. The germinative zone produces the most cortical astrocytes after neuronal migration in the developing mammalian brain. Biol. Neonate 61, 4–24 (1992).
Kanski, R. et al. A star is born: new insights into the mechanism of astrogenesis. Cell Mol. Life Sci. 71, 433–447 (2014).
Zupan, V. et al. Prenatal blockade of vasoactive intestinal peptide alters cell death and synaptic equipment in the murine neocortex. Pediatr. Res. 47, 53–63 (2000).
Farhy-Tselnicker, I. & Allen, N. J. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev. 13, 7 (2018).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Orre, M. et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 35, 1–14 (2014).
Norden, D. M. et al. TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia 62, 881–895 (2014).
Nijboer, C. H. et al. Astrocyte GRK2 as a novel regulator of glutamate transport and brain damage. Neurobiol. Dis. 54, 206–215 (2013).
Joe, E. H. et al. Astrocytes, microglia, and Parkinson’s disease. Exp. Neurobiol. 27, 77–87 (2018).
Ponath, G., Park, C. & Pitt, D. The role of astrocytes in multiple sclerosis. Front. Immunol. 9, 217 (2018).
Shiow, L. R. et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia 65, 2024–2037 (2017).
Stephenson, J. et al. Inflammation in CNS neurodegenerative diseases. Immunology (2018).
Wu, H. C. et al. Subclinical histologic chorioamnionitis and related clinical and laboratory parameters in preterm deliveries. Pediatr. Neonatol. 50, 217–221 (2009).
Christiaens, I. et al. Inflammatory processes in preterm and term parturition. J. Reprod. Immunol. 79, 50–57 (2008).
Norman, J. E. et al. Inflammatory pathways in the mechanism of parturition. BMC Preg. Childbirth 7(Suppl 1), S7 (2007).
Anblagan, D. et al. Association between preterm brain injury and exposure to chorioamnionitis during fetal life. Sci. Rep. 6 (2016).
Leviton, A. et al. The development of extremely preterm infants born to women who had genitourinary infections during pregnancy. Am. J. Epidemiol. 183, 28–35 (2016).
Malaeb, S. & Dammann, O. Fetal inflammatory response and brain injury in the preterm newborn. J. Child Neurol. 24, 1119–1126 (2009).
Zerbo, O. et al. Neonatal cytokines and chemokines and risk of autism spectrum disorder: the Early Markers for Autism (EMA) study: a case-control study. J. Neuroinflamm. 11, 113 (2014).
Krakowiak, P. et al. Neonatal cytokine profiles associated with autism spectrum disorder. Biol. Psychiatry 81, 442–451 (2017).
Vargas, D. L. et al. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81 (2005).
O’Driscoll, D. N. et al. Gender disparities in preterm neonatal outcomes. Acta Paediatr. (2018).
Zagni, E., Simoni, L. & Colombo, D. Sex and gender differences in central nervous system-related disorders. Neurosci. J. 2016, 2827090 (2016).
Naeye, R. L. et al. Neonatal mortality, the male disadvantage. Pediatrics 48, 902–906 (1971).
Stevant, I., Papaioannou, M. D. & Nef, S. A brief history of sex determination. Mol. Cell Endocrinol. 468, 3–10 (2018).
Gabory, A. et al. Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biol. Sex. Differ. 4, 5 (2013).
Atwell, K. et al. Selection bias and outcomes for preterm neonates. Pediatrics (2018).
Teoh, P. J. et al. Gender and preterm birth: Is male fetal gender a clinically important risk factor for preterm birth in high-risk women?. Eur. J. Obstet. Gynecol. Reprod. Biol. 225, 155–159 (2018).
Peacock, J. L. et al. Neonatal and infant outcome in boys and girls born very prematurely. Pediatr. Res. 71, 305–310 (2012).
Johnston, M. V. & Hagberg, H. Sex and the pathogenesis of cerebral palsy. Dev. Med. Child Neurol. 49, 74–78 (2007).
O’Driscoll, D. N., Greene, C. M. & Molloy, E. J. Immune function? A missing link in the gender disparity in preterm neonatal outcomes. Expert Rev. Clin. Immunol. 13, 1061–1071 (2017).
Villa, A. et al. Sex-specific features of microglia from adult mice. Cell Rep. 23, 3501–3511 (2018).
Caplan, H. W., Cox, C. S. & Bedi, S. S. Do microglia play a role in sex differences in TBI?. J. Neurosci. Res. 95, 509–517 (2017).
VanRyzin, J. W. et al., Temporary depletion of microglia during the early postnatal period induces lasting sex-dependent and sex-independent effects on behavior in rats. eNeuro 3 (2016).
Jaber, S. M. et al. Sex differences in the mitochondrial bioenergetics of astrocytes but not microglia at a physiologically relevant brain oxygen tension. Neurochem Int 117, 82–90 (2018).
Morrison, H. W. & Filosa, J. A. Sex differences in astrocyte and microglia responses immediately following middle cerebral artery occlusion in adult mice. Neuroscience 339, 85–99 (2016).
Santos-Galindo, M. et al. Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol. Sex. Differ. 2, 7 (2011).
Fombonne, E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J. Autism Dev. Disord. 33, 365–382 (2003).
Schaafsma, S. M. & Pfaff, D. W. Etiologies underlying sex differences in autism spectrum disorders. Front Neuroendocrinol. 35, 255–271 (2014).
Szatmari, P. Risk and resilience in autism spectrum disorder: a missed translational opportunity?. Dev. Med. Child Neurol. 60, 225–229 (2018).
Mehta, A. & Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 16, 279–294 (2016).
Ebert, M. S. & Sharp, P. A. Roles for microRNAs in conferring robustness to biological processes. Cell 149, 515–524 (2012).
Mendell, J. T. & Olson, E. N. MicroRNAs in stress signaling and human disease. Cell 148, 1172–1187 (2012).
Dugas, J. C. et al. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron 65, 597–611 (2010).
Liu, J. & Casaccia, P. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 33, 193–201 (2010).
Liu, J. et al. Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J. Neurosci. 35, 352–365 (2015).
Moyon, S. et al. Functional characterization of DNA methylation in the oligodendrocyte lineage. Cell Rep. 15, 748–760 (2016).
Ntranos, A. & Casaccia, P. Bromodomains: translating the words of lysine acetylation into myelin injury and repair. Neurosci. Lett. 625, 4–10 (2016).
Schang, A. L., Saberan-Djoneidi, D. & Mezger, V. The impact of epigenomic next-generation sequencing approaches on our understanding of neuropsychiatric disorders. Clin. Genet. 93, 467–480 (2018).
Shen, S. et al. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 11, 1024–1034 (2008).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 16, 551–563 (2015).
Qiu, S., Aldinger, K. A. & Levitt, P. Modeling of autism genetic variations in mice: focusing on synaptic and microcircuit dysfunctions. Dev. Neurosci. 34, 88–100 (2012).
Limperopoulos, C. et al. Injury to the premature cerebellum: outcome is related to remote cortical development. Cereb. Cortex 24, 728–736 (2014).
Rogers, T. D. et al. Is autism a disease of the cerebellum? An integration of clinical and pre-clinical research. Front. Syst. Neurosci. 7, 15 (2013).
Felderhoff-Mueser, U. et al. Oxygen causes cell death in the developing brain. Neurobiol. Dis. 17, 273–282 (2004).
Ment, L. R. et al. Association of chronic sublethal hypoxia with ventriculomegaly in the developing rat brain. Brain Res. Dev. Brain Res. 111, 197–203 (1998).
Fleiss, B. et al. Inflammation-induced sensitization of the brain in term infants. Dev. Med Child Neurol. 57(Suppl 3), 17–28 (2015).
van Tilborg, E. et al. Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism-like behavior in a rat model of diffuse white matter injury. Glia 66, 78–93 (2018).
Acknowledgements
The studies undertaken by us and cited in this review were supported by grants from Inserm, Université Paris Diderot, Université Sorbonne-Paris-Cité, Investissement d’Avenir (ANR-11-INBS-0011, NeurATRIS), ERA-NET Neuron (Micromet), DHU PROTECT, Association Robert Debré, PremUP, Fondation de France, Fondation pour la Recherche sur le Cerveau, Fondation des Gueules Cassées, Roger de Spoelberch Foundation, Grace de Monaco Foundation, Leducq Foundation, Action Medical Research, and Cerebral Palsy Alliance Research Foundation Australia. We wish to acknowledge the support of the Department of Perinatal Imaging & Health, King’s College London. In addition, we acknowledge financial support from the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bokobza, C., Van Steenwinckel, J., Mani, S. et al. Neuroinflammation in preterm babies and autism spectrum disorders. Pediatr Res 85, 155–165 (2019). https://doi.org/10.1038/s41390-018-0208-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-018-0208-4
This article is cited by
-
Chorioamnionitis accelerates granule cell and oligodendrocyte maturation in the cerebellum of preterm nonhuman primates
Journal of Neuroinflammation (2024)
-
Targeting the brain 5-HT7 receptor to prevent hypomyelination in a rodent model of perinatal white matter injuries
Journal of Neural Transmission (2023)
-
Epigenetic priming of immune/inflammatory pathways activation and abnormal activity of cell cycle pathway in a perinatal model of white matter injury
Cell Death & Disease (2022)
-
Placental programming, perinatal inflammation, and neurodevelopment impairment among those born extremely preterm
Pediatric Research (2021)
-
The immune-inflammatory response of oligodendrocytes in a murine model of preterm white matter injury: the role of TLR3 activation
Cell Death & Disease (2021)
