
Abstract
Background
In this study, we aimed to analyze time-resolved plasma proteome changes in preterm neonates stratified by their gestational age to detect malfunctioning pathways that derive from the systemic immaturity of the neonate and to highlight those that are differentially regulated during the early development.
Methods
Preterm newborns were enrolled in three subgroups with different gestational ages: before 26 weeks of gestation (group 1), between 27 and 28 weeks of gestation (group 2), and between 29 and 30 (group 3) weeks of gestation. Plasma protein abundances were assessed at two time points (at preterm delivery and at the 36th week of post-menstrual age) by quantitative proteomics.
Result
The quantitative analysis of plasma proteome in preterm infants revealed a multitude of time-related differences in protein abundances between the studied groups. We report protein changes in several functional domains, including inflammatory domains, immunomodulatory factors, and coagulation regulators as key features, with important gestational age-dependent hemopexin induction.
Conclusion
The global trend emerging from our data, which can collectively be interpreted as a progression toward recovery from the perinatal perturbations, highlights the profound impact of gestation duration on the ability to bridge the gap in systemic homeostasis after preterm labor.
Similar content being viewed by others


Introduction
As shown by numerous observations, preterm delivery is associated with an increased incidence of complications, recognized as a set of accompanying complex diseases jointly defining the clinical outcome of a preterm-born neonate. These malfunctions derive from the systemic immaturity of the neonate and manifest the immanent adverse effects of implemented life-supporting intensive care procedures (a specific multitude of environmental risk factors). Emerging from such observations, the “fetal origin of disease hypothesis” (Barker’s hypothesis) links pregnancy complications or fetal development deficits with an infant’s increased risk of developing several cardiovascular and metabolic diseases in adult life.1,2,3 For instance, lower birth weight has been associated with higher risks of type 2 diabetes mellitus, syndrome X, hypertension, cerebrovascular and coronary heart disease in late adult life.1,2,3,4,5 However, the exact mechanisms of such “fetal disease programming” remain unconvincing or speculative. Importantly, the molecular processes that govern these effects characterize a perturbed proteome composition, which can be traced and deciphered by modern proteomic technologies. Of these technologies, plasma proteomics is of particular interest because it prospects within a tissue compartment that is at the crossroads of mutually percolating systemic signaling pathways. Moreover, time-resolved changes in the plasma proteome composition hold the promise of detecting malfunctioning pathways and highlighting those that are not restored during the early stages of development, possibly contributing to neonatal systemic complications. Indeed, to date, mass spectrometry-based proteomics have been applied in several studies (involving both maternal and fetal plasma specimens) related to pre-eclampsia,6 necrotizing enterocolitis and late-onset sepsis,7 markers of the premature rupture of fetal membranes,8 and the monitoring of the course of parturition.9
Thus, in the present study, we aimed to systematically compare (by iTRAQ quantitation) plasma proteome compositions in preterm infants at birth and at the 36th week of post-menstrual age in three gestational age-stratified patient subgroups to identify the affected proteins and related signaling pathways that are differentially regulated according to pregnancy duration.
Materials and methods
Patients
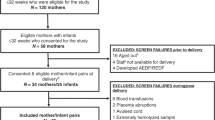
This prospective study was approved by the Jagiellonian University Bioethical Committee and was conducted between 1 September 2013 and 30 November 2015. Two cohorts of children were collected: a preterm group and a control group. The enrollment of three subgroups of preterm children was planned: subgroup 1—newborns born before or at 26 weeks of gestation; subgroup 2—newborns born between 27 and 28 weeks of gestation; and subgroup 3—newborns born between 29 and 30 weeks of gestation. The control group included healthy, full-term newborns. All preterm children were born in the level III perinatal center at Warsaw Medical University Hospital, and all control children were born in the level I city hospital at Cracow.
Detailed perinatal history (birth weight and gestational age) and maternal history were recorded after birth. The study was approved by the Ethics Committee of the Jagiellonian University. Written informed consent was obtained before birth from the parents of all study participants. Blood samples (0.5–1 mL) were collected at two time points: during delivery (umbilical cord blood) and at the 36th week of post-menstrual age. Blood samples were immediately centrifuged (2000 × g, 10 min, 4 °C) to separate the plasma and were then deep-frozen (−70 °C) and transferred to the laboratory at the Jagiellonian University in Cracow.
Sample preparation
A schematic graph representing the study design, sample management, and proteomics methodology is depicted in Fig. 1. The enrollment of preterm patients allowed for the formation of three experimental groups stratified by the time of sample collection and gestational age. ProteoMiner (Combinatorial Peptide Ligand Library beads, CPLL) were purchased from Bio-Rad Laboratories (Hercules, CA). For each patient, 0.2 mL of the plasma sample was diluted with deionized water (1:3) to reduce the physiological salt concentration below 50 mM and subsequently acidified to pH = 3 to increase the amount of protein harvested after enrichment. The samples were centrifuged (10,000 × g, 10 min) to eliminate any particles in suspension. Each plasma sample was intimately mixed with 100 µL of pre-equilibrated ProteoMiner in wash buffer (25 mM sodium chloride buffered with 25 mM acetate pH 4.0). The suspensions were gently shaken overnight at 4 °C. After supernatant separation by centrifugation (1000 × g, 1 min), the beads were washed twice with the wash buffer to remove any unbound protein excess. The captured proteins were eluted from the peptide libraries by three consecutive injections of 100 µL boiling 3% SDS, 3% DTT solution (95 °C, 5 min, vigorously mixed on thermomixer). After elution, the samples were precipitated overnight with ice-cold acetone (Sigma, St. Louis, MO) (1:6 v:v). Next, the protein pellets were dissolved in dissolution buffer (8 M urea, 4% CHAPS, 50 mM DTT in 0.1 M Tris-HCl pH 7.6) and centrifuged (10,000 × g, 10 min), after which the protein concentration of the harvested supernatant was determined using a Coomassie (Bradford) Protein Assay Kit (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions. Next, for data normalization to enable the comparison of samples across different iTRAQ 8plex labeling assemblies, an internal standard pool was generated by combining equal amounts of protein from all of the samples included in the analysis for use as a common reference. Forty micrograms of the measured protein content of each sample (and an aliquot containing 40 μg of the internal reference pool) was reduced and alkylated as recommended by the iTRAQ protocol (ABSciex, Framingham, MA). The proteins were then digested using modified trypsin (Thermo Scientific, Waltham, MA; 1:50 (w:w) ratio) at 37 °C overnight. The samples were randomly assigned to iTRAQ reagents and were labeled as recommended by the supplier. The labeled samples were then combined with the corresponding 8plex assemblies and dried in a vacuum concentrator (Eppendorf, Hamburg, Germany). Next, the trypsin-digested and labeled peptides were dissolved in 5% acetonitrile (ACN) containing 0.1% trifluoroacetic acid (TFA) and were purified with C18 MacroSpin Columns (Harvard Apparatus, Holliston, MA). The eluates were then dried in a vacuum concentrator, reconstituted in 5% ACN and 0.1% formic acid (FA), and subjected to strong cation exchange (SCX) fractionation. The samples were loaded onto previously conditioned SCX Macrospin columns (Harvard Apparatus), after which the flow-through fraction and 11 consecutive injections of the eluent buffer, comprising 5, 10, 20, 40, 60, 80, 100, 150, 200, 300, and 500 mM ammonium acetate in 5% ACN and 0.1% FA, were collected by centrifugation (2000 × g, 1 min). In this way, the labeled peptides from each 8plex assembly were distributed across 12 SCX fractions.

General overview of the study design and a summary of the results of the proteomic analysis. Altogether, 114 plasma samples were included in the analysis and were randomly assigned to 19 iTRAQ 8plex assemblies (a). On average, each iTRAQ set resulted in the identification of 20,000 peptides, allowing for the quantitation of 137 proteins (b)
2D-LC-nESI-MS/MS
Labeled peptides were injected onto a PepMap100 RP C18 75 µm i.d. × 15 cm column (Thermo Scientific, Waltham, MA) via a trap column PepMap100 RP C18 75 µm i.d. × 2 cm column (Thermo Scientific, Waltham, MA). Each peptide fraction was separated using a 65 min 7–45% B phase linear gradient (A phase—2% ACN and 0.1% FA; B phase—80% ACN, and 0.1% FA) operating at a flow rate of 300 nL/min on an UltiMate 3000 HPLC system (Thermo Scientific, Waltham, MA) and was applied to an online Velos Pro (Thermo Scientific, Waltham, MA) dual-pressure ion-trap mass spectrometer. The main working nano-electrospray ion source (Nanospray Flex, Thermo Scientific, Waltham, MA) parameters were as follows: ion spray voltage 1.9 kV and capillary temperature 250°C. Spectra were collected in full-scan mode (400–1500 Da), followed by five pairs of collisional-induced dissociation (CID) and higher-energy collisional dissociation (HCD) MS/MS scans of the five most intense precursor ions from the preceding survey full scan under dynamic exclusion criteria. The collected raw data were processed by EasierMGF software10 with modifications made by the authors that allow the recording of a series of reversed CID and HCD scans to generate hybrid HCD–CID spectra. These were analyzed by the X!Tandem (The GPM Organization) and Comet search engines and were statistically validated with PeptideProphet and integrated with the iProphet component of the Trans-Proteomic Pipeline (TPP) suite of software (Institute for Systems Biology, Seattle, WA). The search parameters were set as follows: taxonomy: human (UniProtKB/Swiss-Prot); enzyme: trypsin; missed cleavage sites allowed: 2; fixed modifications: methylthio (C), iTRAQ8plex (K), and iTRAQ8plex (N-term); variable modifications: methionine oxidation (M) and iTRAQ8plex (Y); parent mass error −1.5 to +3.0 Da; and peptide fragment mass tolerance: 0.7 Da. Quantitative peptide abundances (iTRAQ reported ion intensities) were extracted from the hybrid HCD–CID MS2 scans using Libra software (TPP). The peptide false discovery rate (FDR) was estimated using Mayu (TPP), and peptide identifications with FDR values below 1% were considered correct matches. DanteR software was used for the statistical analysis of iTRAQ-labeled peptides. In brief, for each 8plex iTRAQ assembly, the replicate peptides were aggregated across iTRAQ channels to unique peptides, while the corresponding reporter ion intensities were summed. Next, the aggregated reporter ion intensities were normalized to a corresponding internal reference. Then, the data set comprising all analyzed samples was incorporated to DanteR and quantile normalized. ANOVA was performed at the peptide level using a linear model with a minimum of 2 and a maximum of 100 peptides. Protein fold-change is reported as the mean value of the corresponding unique peptides. Finally, the Benjamini and Hochberg FDR correction was used to adjust p-values. Adjusted p values < 0.05 were considered significant for differentially regulated proteins.
Results
Fifty-seven preterm newborns (19 in each subgroup with two time points of blood collection) were included in the study, which resulted in the collection of a total of 114 plasma samples for proteomic analysis. The mean birth weight of the preterm cohort was 1002 g (SD: 250), and the mean gestational age was 27 weeks (SD: 1.0). The clinical characteristics of the preterm subgroups are presented in Table 1. The power analysis (http://www.dssresearch.com/toolkit/default.asp) indicated that with n = 19 patients in each preterm group, the estimated power of the study to validate the measured fold-change at the level of 1.15 was 0.98 (p = 0.05, 15.6% overall protein abundance variation).
In total, 1756 unique iTRAQ-labeled peptides were successfully sequenced and annotated with an FDR of less than 1% in the plasma samples during LC-MS/MS analysis, allowing for the identification of 245 proteins in CPLL-enriched plasma; of these, 98 were detected only as a single peptide (single hits). Thus, by excluding the decoy hits, we were able to quantify a total of 137 proteins. The complete list of all validated unique peptides (FDR < 1%) and the corresponding quantitative signals used for quantitation are presented in Supplemental Table S1 (online), while Supplemental Table S2 lists all peptide sequence matched (PSM) performed in the database search process (online). Unique peptides were additionally rolled-up to the protein level and are listed in Supplemental Table S3 (online). The abundance levels of 62, 56, and 55 for preterm groups 1, 2, and 3, respectively, were significantly different at the second time point compared to the cord blood samples collected at the time of preterm delivery. Differentially regulated plasma proteins are listed in Table 2 and Fig. 2, where statistically significant changes (adjusted p values < 0.05) are presented as the mean fold-changes.

Selected plasma proteins that were differentially secreted between the preterm groups. All presented protein plasma concentration differences are statistically significant (adjusted p < 0.05), highlighting the major discoveries among the main functional domains. Red (induction) or blue (repression) bars represent time-resolved protein abundance changes in the gestation groups (36 PMA to cord blood protein ratio). The data are means ± SDs
The main finding of our study is that preterm birth is associated with several protein abundance changes in plasma and can be successfully monitored by iTRAQ quantitation. The main processes related to the differentially regulated proteins encompassed inflammation, immunomodulation, coagulation, and complement activation, and the hemoglobin turnover network.
Most importantly, our proteomic analysis provides strong evidence for a time-dependent recovery from disturbances in the hemoglobin/heme turnover network in preterm neonates. All of the identified hemoglobin subunits, together with proteins that are secreted to handle aberrant hemoglobin release from erythrocytes, decrease (normalize) their abundance in samples obtained at the 36 PMA time-point (Figs. 2 and 3, Table 2). A key feature among these quantitative changes in protein levels is a gestational age-dependent induction of heme-scavenging hemopexin (Fig. 3, Table 2).

Changes in plasma hemoglobin turnover network proteins. The degree of the decrease in plasma hemoglobin subunit concentration depended on gestation age, indicating a correlation between heme/hemoglobin detoxification capacity and gestation duration. Hemoglobin changes were accompanied by a gestation-age-dependent, time-resolved induction of hemopexin—a free hemoglobin circulatory balance marker. All presented protein plasma concentration differences are statistically significant (adjusted p < 0.05). The data are means ± SDs
Next, our findings in relation to immune and inflammation-related proteins highlight gestation age- and time-related changes that can be collectively recognized as a partial recovery from the initial preterm birth-associated perturbations. We observed the induction of orosomucoid (ORM) 1 and 2 isoforms, amyloid P-component (SAP), peptidase inhibitor 16 (PI16), and L-selectin abundance, among others, in all of the preterm groups in the plasma samples collected at 36 PMA; together, these can be recognized as a manifold anti-inflammatory protein signature. On the other hand, we simultaneously detected the induction of leucine-rich alpha-2-glycoprotein (LRG1), which was accompanied by the repression of gelsolin, at 36 PMA; these observations suggest unresolved inflammation (Fig. 2, Table 2).
Additionally, we noted time-resolved changes in coagulation and complement activation cascades in preterm infants. All isoforms of fibrinogen chains concomitantly increased their plasma abundance in the 36-PMA samples (Fig. 2, Table 2). Contrarily, complement C3, C4-A, and coagulation factor V decreased markedly over time (Fig. 2, Table 2). These alterations were accompanied by a gestational age-related increase in the abundance of plasma protease C1 inhibitor (C1Inh). Moreover, our analysis revealed an interesting trend regarding the induction of alpha-2-macroglobulin (a2-M) in preterm neonates. A2-M was upregulated most notably in the first gestation group, while in the third group, the abundance of a2-M remained unchanged until sample collection at 36 PMA (Table 2).
Discussion
The plasma proteome represents a unique biological compartment containing proteins that are secreted form almost all tissues; thus, it possibly provides insight into personal health status. Unsurprisingly, a plethora of analytical efforts have been made in the proteomic field over the last decade to explore blood plasma in detail, resulting in the identification of a progressively increasing number of plasma proteins and the establishment of several dedicated data repositories.11,12 The Up-to-date Plasma Proteome Database contains information on 10546 proteins detected in human plasma,12 and the number of high-confidence proteins in the PeptideAtlas repository has been estimated to be 1929.11 However, obtaining this information usually requires multiple laborious and time-consuming orthogonal fractionation steps, which are unconceivable to incorporate in clinical studies encompassing a large cohort of patients. Here, we propose a quantification strategy based on two-dimensional chromatographic fractionation that is supported by the increased sample throughput obtained by iTRAQ multiplexing.
Preterm birth is associated with complex, mutually overlapping abnormalities resulting from systemic immaturity. Their comprehensive assessment requires a methodology allowing the simultaneous observation of the abundance changes in hundreds of proteins, thus providing insight into the biological processes in which these proteins are involved. Our study shows that despite the inherent difficulties arising from the dynamic range of abnormal plasma proteins (22 proteins constitute ~99% of the total plasma protein mass), modern proteomic quantitative methods (including iTRAQ isobaric labeling) can provide a thorough and detailed overview of aberrant premature plasma protein levels and their adaptive changes over time.
These benefits are particularly evident in our results involving proteins from the hemoglobin/heme turnover network in preterm neonates (Fig. 3). We were able to assess a variety of hemoglobin isoforms and proteins regulating aberrant hemoglobin release from erythrocytes and to follow their quantitative changes over time, including a pivotal gestational age-dependent induction of hemopexin. The latter is a heme-scavenging protein that is secreted to prevent the pro-oxidant and pro-inflammatory effects of heme and is considered a protein marker of free hemoglobin circulatory balance.13 The free plasma hemoglobin assessed in our study likely originated from erythrocyte hemolysis, which is a major cause of hyperbilirubinemia in premature and term neonates.14 Decreased erythrocyte survival and increased erythrocyte volume augment hemolysis, thus interfering with physiological bilirubin production and elimination.15 The ability of immature liver enzymes to handle unconjugated bilirubin is impaired in preterm infants compared with term neonates, and this predisposes infants to a high risk of neonatal jaundice.15 Our results suggest that the duration of the gestation period is a crucial determinant of the ability to effectively restore balance in circulating hemoglobin levels.
Chronic inflammatory and immunologic abnormalities occurring in the absence of well-defined triggers have been linked with preterm birth and preterm neonate complications.16 Moreover, fetal inflammation is strongly associated with impending preterm labor and with premature prelabor rupture of membranes (PPROM) and is an independent risk factor for subsequent neonatal morbidity.17 In line with these findings, our plasma proteomic data provide insights into specific markers of a disrupted balance between inflammation and immunomodulation status and the modulation of such markers over time, including orosomucoid (ORM) 1 and 2 isoforms, amyloid P-component (SAP), peptidase inhibitor 16 (PI16), and L-selectin. Orosomucoid (ORM) has been considered a part of many signaling pathways, in which it is considered an anti-inflammatory and immunomodulatory agent.18 ORM inhibits superoxide production by neutrophils, modulates cytokine production by monocytes, and promotes interleukin-1 receptor antagonist (IL1RA) secretion.18 Of note, ORM is an inducer of pro-resolution M2b macrophage polarization phenotype.19 Next, we report the induction of the inflammatory marker serum amyloid P-component (SAP), which correlated with the gestation age of the preterm infants (Fig. 2, Table 2). SAP can interact with DNA and histones and may scavenge nuclear material released from damaged circulating cells, while the amelioration of the acute inflammatory response and the monocyte differentiation to fibrocytes are believed to be crucial for SAP-mediated anti-fibrotic and wound healing activities.20 Our proteomic analysis also revealed the time-dependent induction of peptidase inhibitor 16 (PI16) and L-selectin (Table 2); these two proteins modulate the activity of immune cells. The former is a member of the CAP superfamily of proteins (cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins), which were found to be highly upregulated in cardiac disease.21,22 PI16 regulates the post-translational processing of chemerin, which has been shown to promote calcium mobilization and the chemotaxis of immature DCs and macrophages.23 L-selectin is a cell surface adhesion molecule but can simultaneously be released as a soluble isoform, which reduces leukocyte adhesion to the endothelium, thereby ameliorating immune cell infiltration during inflammation.24 Of note, low L-selectin plasma level have been associated with a poor responsiveness to surfactant therapy in preterm infants with respiratory distress and can serve as a predictive marker of a prolonged requirement for supplemental oxygen.25 According to our results, L-selectin induction was correlated with preterm gestation age (Table 2). Contrarily to the beneficial induction of ORM, SAP, PI16, and L-selectin in 36-PMA collected samples, we found that the concentration of leucine-rich alpha-2-glycoprotein (LRG1) increased over time (Table 2). Importantly, LRG1 induction has been considered a biomarker of several neoplastic and inflammatory disorders.26,27 The production of LRG1 increases in response to mediators of the acute-phase response, while serum LRG1 levels are elevatehrough by its secretion from activated neutrophils.28 Additionally, we identified gelsolin as being differentially regulated in the 36-PMA samples, and this molecule was decreased in abundance in all three groups of preterm neonates, with a fold-change of approximately −1.4 (Table 2). Gelsolin secretion has been implicated in the extracellular actin scavenging system and the presentation of lysophosphatidic acid and other inflammatory mediators to their receptors.29 Blood gelsolin levels decrease in a variety of clinical conditions and complications observed in preterm infants, such as acute respiratory distress syndrome, sepsis, trauma, prolonged hyperoxia, and liver injury.29,30 Moreover, the observed correlation between blood gelsolin levels and severe clinical conditions suggests that it has potential for use as a prognostic biomarker and may be a novel therapeutic target.30
The maturation process of the pediatric hemostatic system from the fetal to adult form—termed developmental hemostasis—has been described in several studies.31,32 In general, it has been demonstrated that most coagulation factors are decreased ~2-fold in neonates compared with adults, and these imbalances are even more pronounced in preterm newborns.31 Our results are consistent with these observations—we identified time-related alterations in coagulation and complement activation cascade proteins in preterm infants, as illustrated by the observed increase in fibrinogen isoform chains and marked decreases in complement C3, C4-A, and coagulation factor V abundances in the 36-PMA samples. These changes were accompanied with a gestational age-related increase in the abundance of plasma protease C1 inhibitor (C1Inh), which is an inhibitor for C1r and C1s and is thereby a key negative modifier of complement activation and kinin release.33 Intriguingly, our study highlights the induction of alpha-2-macroglobulin (a2-M) in preterm neonates. A2-M is known to regulate coagulation in response to antithrombin plasma levels. At physiological levels, a2-M is a pro-coagulant factor due to its ability to bind activated protein C (APC).34 Conversely, in antithrombin-deficient plasma, the anti-coagulant a2-M properties prevail due to the binding of free α-thrombin,35 a mechanism explaining the a2-M-mediated protection from thromboembolic events in antithrombin-deficient children.36 We are tempted to speculate that a2-M induction may represent a possible protective mechanism that originally developed to counteract disturbances in the regulation coagulation network, which it normalizes in a time-dependent manner as well as restoring hemostatic homeostasis.
Finally, we noted interesting changes in the plasma apolipoprotein composition; we found the induction of apolipoproteins A-I, A-IV, and D in the 36-PMA samples, while apolipoprotein E levels decreased in all patient groups (Fig. 2, Table 2). Apo-D, ApoA-I, and ApoA-IV are high-density lipoprotein (HDL) components with well-established anti-oxidative functions.37,38,39 Of note, ApoA-IV is a potent activator of phosphatidylcholine-sterol acyltransferase (LCAT),40 an observation that is directly confirmed by our results describing the induction of LCAT in plasma collected at 36 PMA in groups 2 and 3 (Table 2). Intriguingly, the apoE concentration in term neonates is comparable to that measured in adults and does not change until 30 days of life. However, during that period, apoE undergoes a dramatic redistribution from HDL (over 80% of the plasma apoE at birth) to very low-density lipoproteins.38 Thus, whether the apoE changes identified in our preterm population possess physiological significance and functionally influence the lipoprotein characteristics is unknown and remains an attractive hypothesis to be tested in a targeted prospective study.
Conclusions
In summary, the quantitative analysis of plasma proteome changes in preterm infants stratified by their gestation age revealed a multitude of temporal differences in protein abundances between the groups. We report changes in protein levels for several functional domains, and these can be collectively interpreted as a progression toward recovery from perinatal perturbations. In particular, our results point to inflammatory and immunomodulatory factors (ORM, SAP, PI16, ApoA-IV, Apo-D) and coagulation regulators (fibrinogen, C1Inh, and a2-M) as key features, with important gestational age-dependent hemopexin induction representing a gestational age-related marker of the improved heme/hemoglobin turnover network performance in preterm neonates. The latter protein exemplifies the global trend emerging from identified plasma protein composition differences that highlights, from different functional angles, the profound impact of gestation duration on the ability to bridge the gap in systemic homeostasis after preterm labor.
References
Barker, D. J., Osmond, C., Golding, J., Kuh, D. & Wadsworth, M. E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 298, 564–567 (1989).
Barker, D. J. et al. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36, 62–67 (1993).
Barker, D. J., Winter, P. D., Osmond, C., Margetts, B. & Simmonds, S. J. Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580 (1989).
Barker, D. J., Osmond, C. & Law, C. M. The intrauterine and early postnatal origins of cardiovascular disease and chronic bronchitis. J. Epidemiol. Community Health 43, 237–240 (1989).
Hales, C. N. & Barker, D. J. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35, 595–601 (1992).
Law, K. P., Han, T.-L., Tong, C. & Baker, P. N. Mass spectrometry-based proteomics for pre-eclampsia and preterm birth. Int. J. Mol. Sci. 16, 10952–10985 (2015).
Stewart, C. J. et al. Metabolomic and proteomic analysis of serum from preterm infants with necrotising entercolitis and late-onset sepsis. Pediatr. Res. 79, 425–431 (2016).
Vuadens, F. et al. Identification of biologic markers of the premature rupture of fetal membranes: proteomic approach. Proteomics 3, 1521–1525 (2003).
Phillips, R. J., Heesom, K. J., Trinder, J. & Bernal, A. L. Human maternal plasma proteomic changes with parturition. EuPA Open Proteom. 5, 10–20 (2014).
Gallardo, Ó., Ovelleiro, D., Gay, M., Carrascal, M. & Abian, J. A collection of open source applications for mass spectrometry data mining. Proteomics 14, 2275–2279 (2014).
Farrah, T. et al. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell Proteom. 10, M110.006353 (2011).
Nanjappa, V. et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 42, D959–D965 (2014).
Tolosano, E., Fagoonee, S., Morello, N., Vinchi, F. & Fiorito, V. Heme scavenging and the other facets of hemopexin. Antioxid. Redox Signal. 12, 305–320 (2010).
Christensen, R. D. & Yaish, H. M. Hemolysis in preterm neonates. Clin. Perinatol. 43, 233–240 (2016).
Kaplan, M. et al. Imbalance between production and conjugation of bilirubin: a fundamental concept in the mechanism of neonatal jaundice. Pediatrics 110, e47 (2002).
Cappelletti, M., Della Bella, S., Ferrazzi, E., Mavilio, D. & Divanovic, S. Inflammation and preterm birth. J. Leukoc. Biol. 99, 67–78 (2016).
Kemp, M. W. Preterm birth, intrauterine infection, and fetal inflammation. Front. Immunol. 5, 574 (2014).
Hochepied, T., Berger, F. G., Baumann, H. & Libert, C. Alpha(1)-acid glycoprotein: an acute phase protein with inflammatory and immunomodulating properties. Cytokine Growth Factor. Rev. 14, 25–34 (2003).
Nakamura, K., Ito, I., Kobayashi, M., Herndon, D. N. & Suzuki, F. Orosomucoid 1 drives opportunistic infections through the polarization of monocytes to the M2b phenotype. Cytokine 73, 8–15 (2015).
Pilling, D., Buckley, C. D., Salmon, M. & Gomer, R. H. Inhibition of fibrocyte differentiation by serum amyloid P. J. Immunol. 171, 5537–5546 (2003). 1950.
Gibbs, G. M., Roelants, K. & O’Bryan, M. K. The CAP superfamily: cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins--roles in reproduction, cancer, and immune defense. Endocr. Rev. 29, 865–897 (2008).
Regn, M. et al. Peptidase inhibitor 16 is a membrane-tethered regulator of chemerin processing in the myocardium. J. Mol. Cell. Cardiol. 99, 57–64 (2016).
Wittamer, V. et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 198, 977–985 (2003).
Schleiffenbaum, B., Spertini, O. & Tedder, T. F. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J. Cell Biol. 119, 229–238 (1992).
Koehne, P. S. et al. Soluble intercellular cell adhesion molecule-1 and L-selectin plasma concentrations and response to surfactant in preterm infants. Pediatr. Crit. Care Med. 3, 23–28 (2002).
Majek, P. et al. Proteome changes in the plasma of myelodysplastic syndrome patients with refractory anemia with excess blasts subtype 2. Dis. Markers 2014, 178709 (2014).
Kharbanda, A. B., Rai, A. J., Cosme, Y., Liu, K. & Dayan, P. S. Novel serum and urine markers for pediatric appendicitis. Acad. Emerg. Med. 19, 56–62 (2012).
Druhan, L. J. et al. Leucine rich α-2 glycoprotein: a novel neutrophil granule protein and modulator of myelopoiesis. PLoS ONE 12, e0170261 (2017).
Li, G. H., Arora, P. D., Chen, Y., McCulloch, C. A. & Liu, P. Multifunctional roles of gelsolin in health and diseases. Med. Res. Rev. 32, 999–1025 (2012).
Bucki, R., Levental, I., Kulakowska, A. & Janmey, P. A. Plasma gelsolin: function, prognostic value, and potential therapeutic use. Curr. Protein Pept. Sci. 9, 541–551 (2008).
Lippi, G., Franchini, M., Montagnana, M. & Guidi, G. C. Coagulation testing in pediatric patients: the young are not just miniature adults. Semin. Thromb. Hemost. 33, 816–820 (2007).
Andrew, M. Developmental hemostasis: relevance to hemostatic problems during childhood. Semin. Thromb. Hemost. 21, 341–356 (1995).
Matsushita, M., Thiel, S., Jensenius, J. C., Terai, I. & Fujita, T. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J. Immunol. 165, 2637–2642 (2000). 1950.
Hoogendoorn, H., Toh, C. H., Nesheim, M. E. & Giles, A. R. Alpha 2-macroglobulin binds and inhibits activated protein C. Blood 78, 2283–2290 (1991).
Sottrup-Jensen, L. Alpha-macroglobulins: structure, shape, and mechanism of proteinase complex formation. J. Biol. Chem. 264, 11539–11542 (1989).
Mitchell, L., Piovella, F., Ofosu, F. & Andrew, M. Alpha-2-macroglobulin may provide protection from thromboembolic events in antithrombin III-deficient children. Blood 78, 2299–2304 (1991).
Wong, W.-M. R. et al. Common variants of apolipoprotein A-IV differ in their ability to inhibit low density lipoprotein oxidation. Atherosclerosis 192, 266–274 (2007).
van Biervliet, J. P. et al. Apolipoprotein and lipid composition of plasma lipoproteins in neonates during the first month of life. Pediatr. Res. 20, 324–328 (1986).
Ganfornina, M. D. et al. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell 7, 506–515 (2008).
Duverger, N. et al. Functional characterization of human recombinant apolipoprotein AIV produced in Escherichia coli. Eur. J. Biochem. 201, 373–383 (1991).
Acknowledgements
The research leading to these results received funding from the Polish-Norwegian Research Programme, operated by the National Centre for Research and Development under the Norwegian Financial Mechanism 2009–2014 in the frame of Project Contract No Pol-Nor/196065/54/2013. The mass spectrometry measurements were performed at the Center for Medical Genomics OMICRON, Jagiellonian University Medical College.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Suski, M., Bokiniec, R., Szwarc-Duma, M. et al. Prospective plasma proteome changes in preterm infants with different gestational ages. Pediatr Res 84, 104–111 (2018). https://doi.org/10.1038/s41390-018-0003-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-018-0003-2
This article is cited by
-
Blood protein profiles related to preterm birth and retinopathy of prematurity
Pediatric Research (2022)
-
The use of proteomics for blood biomarker research in premature infants: a scoping review
Clinical Proteomics (2021)
-
Dramatic changes in blood protein levels during the first week of life in extremely preterm infants
Pediatric Research (2021)
-
Comparative two time-point proteome analysis of the plasma from preterm infants with and without bronchopulmonary dysplasia
Italian Journal of Pediatrics (2019)
