
Abstract
Successful antitumor immunity largely relies on efficient T cell priming by antigen-presenting cells (APCs); however, the capacity of APCs is found to be defective in many cancers. Metabolically reprogrammed cancer cells support the energetic and biosynthetic demands of their high proliferation rates by exploiting nutrients available in the tumor microenvironment (TME), which in turn limits proper metabolic reprogramming of APCs during recruitment, differentiation, activation and antigen presentation. Furthermore, some metabolites generated by the TME are unfavorable to antitumor immunity. This review summarizes recent studies on the metabolic features of APCs and their functionality in the TME. Particularly, we will describe how APCs respond to altered TME and how metabolic byproducts from cancer and immunomodulatory cells affect APCs. Finally, we introduce the current status of APC-oriented research and clinical trials targeting metabolic features to boost efficient immunotherapy.
Similar content being viewed by others
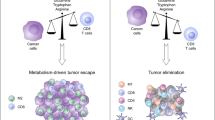
Introduction
Tumor is akin to a chronic infection where immune cells are constantly exposed to an antigen that cannot be cleared. Upon the encounter of antigens, the innate immune cells, such as dendritic cells (DCs) and macrophages, play a role as the first-line of defense against antigens by uptaking in an indiscreet manner. DCs, macrophages, and B cells are usually referred to as professional antigen-presenting cells (APCs) and are critical “matchmakers” which awaken antigen-specific adaptive immunity via presenting peptides loaded on major histocompatibility complex I or II (MHCI/MHCII) to T lymphocytes in the lymph nodes (LNs). Cells referred to as “non-professional APCs” by expressing MHCII in an inducible manner, for example, innate lymphoid cells, fibroblast, or epithelial cells, also participate in the activation of adaptive immunity [1].
The ability of T cells to kill tumor cells in an antigen-specific manner is conferred by APCs as so in infection, therefore, the role of APCs is critical for the successful immunosurveillance of cancer. Although clonal expansion and differentiation of T cells majorly occur within lymphoid organs, a positive correlation between tertiary lymphoid structures (TLS) for priming intratumoral T cells with better response to therapy or prognosis implies the importance of APCs within TME [2,3,4]. However, immunosurveillance is not always successful because tumor cells render TME felicitous to themselves by depleting nutrients and secreting metabolites.
Immune cells co-habitating with tumor cells compete for nutrients with limited availability, and are constantly communicating with tumor cells within the TME. Competition for nutrients and exposure to TME-derived metabolites lead to the metabolic adaptation of immune cells, and consequently result in dysfunctionality in antitumor immunity. DCs frequently show disrupted function, and tumor-associated macrophages (TAM) rather play pro-tumorigenic roles within the TME. Therefore, targeting metabolic crosstalk in the TME is rising as a novel therapeutic strategy. In this review, we will address the immunometabolic features of APCs, particularly focused on DCs and TAMs, and their crosstalk within the TME. Additionally, we will also discuss the studies of adaptation and functional alteration of APCs by tumor metabolites and the impact on antitumor immunity. We will further discuss the therapeutic approaches targeting the metabolic features of APCs to restore their antitumor immunity.
Cell biology of APCs in cancer
Macrophages
Although TAMs adopt more diverse phenotypes than dichotomized status as their heterogeneity within TME has been actively discussed in recent studies, the activation status of macrophages has long been oversimplified into two categories: classically activated, pro-inflammatory M1 macrophages and alternatively activated, anti-inflammatory M2 macrophages [3,4,5]. M1 macrophages are believed to be anti-tumorigenic by secreting pro-inflammatory cytokines such as tumor-necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6, while M2 macrophages are known to be pro-tumorigenic due to their expression of anti-inflammatory molecules such as IL-4, IL-10, and transforming growth factor beta (TGFβ). M1 macrophages and M2 macrophages adopt different metabolic features. Dependency on glycolysis is a crucial feature for M1 macrophages, since perturbation of glycolysis alters their polarization status into M2 macrophages [6, 7]. The pentose phosphate pathway (PPP) enhanced in M1 macrophages provides nicotinamide adenine dinucleotide phosphate (NADPH) for the production of nitric oxide (NO) and reactive oxygen species (ROS) [8]. In contrast, M2 macrophages adopt oxidative phosphorylation fueled by fatty acid oxidation (FAO) and glutamine metabolism. Due to unfavorable conditions to support the M1 polarization of macrophages, TAMs are often featured as M2 macrophages. How TME alter the polarization status of macrophages will be discussed further in the following sections.
The abundance of TAMs has been associated with poor prognosis in different types of solid tumors, since TAMs carry out a pro-tumorigenic role via triggering T-cell dysfunction [9]. B7 superfamily 1 (B7S1) and CD39 expressed on TAMs have been shown to be involved in the direct suppression of T cell activity [10, 11]. Cytokines that TAMs secrete are involved in inhibiting T-cell activity as well. For example, IL-10 secreted by TAMs enhances N-glycan branching, which in turn curtails the sensitivity of T cells to the antigen [12]. Moreover, TAMs recruit CCR6-positive regulatory T cells (Treg) to render the TME into an immunosuppressive milieu in colorectal cancer models, and the infiltration of Tregs driven by TAMs has further been studied in ovarian cancer and laryngeal squamous cell carcinoma [13,14,15]. As the role of TAMs in TME is appreciated to be suppressing antitumor immunity, inhibition of TAM infiltration into TME via colony-stimulating factor 1 receptor (CSF1R) blockade treatment resulted in a significant reduction of tumor growth accompanying increase of T cell infiltration [16].
Since efficient activation of T cells is largely dependent on APCs, antigen-presenting capacities of APCs within TME has long been under extensive investigation. However, the roles of TAMs as APC within TME have been less appreciated compared to those of DCs. TAMs within TME adopt an M2-like phenotype, which express low level of MHCII, thereby exhibiting limited antigen-presenting capacity. Furthermore, low expression of CCR7, critical for LN migration, is another factor that limits their ability as APC, rendering macrophages less effective compared to DCs. A study using ascites-derived DCs or macrophages from human ovarian cancer patients showed a superior capacity of DCs activating CD8 T cells [17]. Inhibition of signal regulatory protein α (SIRPα) with CD47 blockade efficiently enabled DC-mediated cross-priming of CD8 T cells, whereas macrophages were incapable to do so [18]. In contrary to these reports, CD169+ macrophages in the LN have been shown to play critical roles in exerting antitumor immunity by cross-presenting dead cell-associated antigens to CD8 T cells [19]. Further studies using in vitro models demonstrated that macrophages can cross-present antigens to prime CD8 T cell response [20, 21]. Moreover, a recent study revealed the capability of CD206+ macrophage subset to cross-present tumor antigens [22]. Therefore, further studies on the unique potential of TAMs in priming T cells within the TME are needed, which will not only expand our understanding on their unappreciated functional diversities but also potentiate therapies targeting APCs to enhance antitumor immunity.
Dendritic cells
DC is another quintessential APCs of the immune system, especially for the activation of antitumor T cells [23,24,25,26]. There are three types of DCs found in the TME, and can be distinguished as plasmacytoid DCs (pDCs), conventional DCs (cDC1 and cDC2), and monocyte-derived DCs (moDCs) based on their phenotypical and functional properties [27]. Owing to their responsibility for the initiation of the “cancer-immunity cycle”, cDC1 (CD8α+CD103+BATF3+ CLEC9A+XCR1+) is identified as a critical APC subset for tumor antigen drainage and robust T cell activation [28, 29]. cDC1s generally appear to be more dependent on OXPHOS, displaying higher mitochondrial mass and ΔΨm than cDC2s [30]. Upon immunogenic activation, cDC1s elevate glycolysis and lactic fermentation [31]. Indeed, interruption of glycolysis impairs not only the maturation, but also the immunogenicity and T cell stimulatory capacity of DCs [30]. In line with their dependency on glycolysis, glucose deprivation impairs DC-mediated immune response by affecting the motility of splenic CD11c+ cDCs and oligomerization of CCR7, whose level correlates with T cell infiltration and patient survival [28, 32]. However, mitochondrial respiration appears to be as important as glycolysis induction for proper cDC1 activation. Splenic cDC1s in aged mice that have mitochondrial dysfunction (decrease in basal OCR and increase in proton leakage and ROS) displayed reduced endocytic activity and antigen presentation capacity [33]. In addition, bone marrow-derived cDC1-like cells have been shown to upregulate de novo fatty acid synthesis (FAS) to accumulate phospholipids upon stimulation [34]. As cDC1s are a crucial subset for effective antitumor immunity, clarifying how these metabolic pathways are intertwined in regulating the activity of cDC1s within TME will be important.
cDC2, another subset of conventional DCs, can be identified by high expression of CD11b, CD1c (human), and SIRPa (CD172a). In contrast to cDC1, ROS strongly skews the differentiation toward cDC2. Likewise, in cDC1s, upregulation of glycolysis and FAS are required for the activation and maturation of cDC2s [35]. This reflects the reason why DCs with high lipid content are more potent in priming T cells [36]. In addition to the essential roles of cDC1s in the induction and maintenance of antitumor immunity, cDC2s can effectively elicit intratumoral CD4 T cell responses and induce the polarization of diverse subsets of CD4 Th cells [37, 38]. Upon Treg depletion, cDC2 can migrate to the draining LN and improve CD4 T cell differentiation in vivo. Similarly, a high level of cDC2s is strongly associated with longer progression-free survival and higher infiltration of CD4 T cells in both melanoma and human head and neck squamous cell carcinoma (HNSCC) [38].
Although pDCs are mainly known for antiviral immunity, they are a potential APC subset in TME as well [39, 40]. pDCs are derived from either myeloid common DC progenitor (CDP) or IL-7R+ lymphoid progenitor cells, but single-cell RNA-seq (scRNA-seq) demonstrated that only myeloid-derived pDCs show similar potential to process and present antigens as cDCs [41]. Reduced expression of co-stimulatory molecules and type I interferon (IFN) upon 2-deoxyglucose (2-DG) treatment demonstrated the importance of glycolysis during activation and maturation of pDCs. However, the analysis of the whole transcriptome of human pDC upon toll-like receptor (TLR) 7/8 stimulation proved the induction of glutaminolysis and OXPHOS [35]. Further studies are needed to demonstrate the metabolic dependency of pDC differentiation. The role of pDCs in anti-cancer immunity remains controversial [41]. Although pDC infiltration is associated with poor outcomes in human breast cancer, pDC can induce tumor regression through type I IFN-mediated mechanism following intratumoral injection of TLR7 ligand in orthotopic murine mammary tumor model [39].
In addition to the DC subsets listed above, different types of DCs from human peripheral blood and tumors as well as from murine tumor models have been recently identified through scRNA-seq. For instance, AXL+SIGLEC6+ cells (AS DCs), a new subdivision between cDC-like and pDC-like cells have been demonstrated to potently activate T cells [42]. cDC2A and cDC2B, two principal cDC2 lineages, identified by combining RNA-seq and chromatin accessibility analyses with genetic reporter expression, have distinct pro- and anti-inflammatory potential and are characterized by distinct metabolic states [43]. Mature DCs enriched in immunoregulatory molecules (mregDCs), co-expressing immunoregulatory genes (Cd274, Pdcd1lg2, and Cd200) and maturation genes (Cd40, Ccr7, and Il12b), can uptake tumor antigens, upregulate IL-12 in an IFNγ-dependent manner, and initiate effector T cells response [44]. DC3s, identified as the CD88−CD1c+CD163+ subset, share similar secretory profiles of immune modulators with monocytes and cDCs. Of note, infiltration of DC3s positively correlates with the expansion and abundance of resident memory T cells in human breast cancers [45].
B cells
B cells are another type of APCs that participate in antitumor immunity by activating T cells via MHC-loaded tumor antigens and by secreting tumor-reactive antibodies. Within TME, B cells are frequently found in TLS, where multiple types of immune cells cluster to form a structure resembling secondary lymphoid organs. Mature TLS contain an organized B cell zone surrounded by T cells exerting a humoral response of antitumor immunity. Therefore, the presence of TLS has been correlated with a better prognosis of tumor patients and better response to immunotherapies in different types of cancers [46, 47]. Furthermore, patients with colocalization of CD20+ B cells with CD8 T cells showed longer survival than the patients with CD8 T cells alone in ovarian cancer [48]. In contrast, infiltration of B cells into TME has been reported to promote the progression of tumors as well. Together with earlier studies showing a better response to therapy upon B cell depletion, the population called “regulatory B cell (Breg)” secreting immunosuppressive cytokines (IL-10 and IL-35) is appreciated to be responsible for pro-tumorigenic activity of B cells [49, 50]. Nevertheless, as a rising number of studies indicate a positive prognostic value of B cells, the distinct roles of heterogenic B cell subsets should be precisely evaluated for a better therapeutic application.
Metabolism of APCs in TME
Tumors can manipulate TME to support their proliferation, and promote metastatic behavior and therapeutic resistance, in part through inducing APC tolerization by enriching TME with immunosuppressive factors that can suppress anti-tumoral activities of APCs.
One common feature of most solid tumors is dysfunctional vascularization which causes severe hypoxia [51]. Hypoxia induces dual consequences on the migration of DCs based on the duration [52, 53]. Short-time hypoxia results in a better migration of moDCs, while long-term hypoxia inhibits the migration of DCs [52]. The hypoxia-inducible factor 1 (HIF1α) expression is inhibited by direct binding of long non-coding RNA Dpf3 (lncRNA-Dpf3), and this further impairs glycolysis and inhibits CCR7-mediated DC migration [53]. Availability of oxygen affects the feature of TAMs as well. The expression of regulated in development and DNA damage response 1 (REDD1) is upregulated in TAMs under hypoxia to support angiogenesis and pro-tumoral phenotype, and fine-tunes the M2 phenotype of TAMs [54, 55]. Association between hypoxia and M2 TAMs is further appreciated by their distribution within the TME. TAMs adopting M2-like feature distribute in the hypoxic region, while M1 TAMs infiltrate into the normoxic region [56]. Although the cue for this distribution seems not yet clear, TIE2 expression on TAMs has been shown to play a role in the guidance to the vasculatures [57]. TAMs with stabilized HIF1α under hypoxia can induce the expression of vascular endothelial growth factor (VEGF) and the differentiation into M2 TAMs [58]. Furthermore, TAMs further enhance the hypoxia of tumors forming a feed-forwarding loop to modulate tumor metabolism, which subsequently results in therapeutic inefficiency [59]. Hypoxia might have adverse effects on proper B cell activities within TME as well. Forced stabilization of HIF1α by depleting Von Hippel-Landau tumor suppressor protein (pVHL) in B cells resulted in the reduction of clonal expansion, recall response, and high-affinity antibody production via perturbation of mTORC1 activity [60]. Furthermore, stabilization of HIF1α has been shown to drive IL-10 expression in B cells in encephalomyelitis [61]. Although not many studies are done on metabolic aspects of B cells within TME, these studies show the potential involvement of hypoxia in hampering antitumor immunity exerted by B cells via limiting humoral response and driving IL-10 secreting regulatory B cell development.
Fatty acid (FA) enriched in the TME has been shown to contribute pro-tumorigenic phenotypes of TAMs. CD36 has been demonstrated as the receptor responsible for the uptake of lipids in TAMs, and these TAMs utilized FAO as an energy source. Enhanced FAO facilitates the generation of pro-tumoral TAMs via JAK1-STAT6 signaling, which is activated by a high level of oxidative stress due to increased ROS production [62]. Recently, tumor-derived glucosylceramide has been demonstrated to induce M2 polarization of macrophages via triggering ER stress [63]. Furthermore, TAMs accumulated with lipids via caspase-1 activation have been shown to exhibit pro-tumoral properties [64]. The lipid metabolism of TAMs has further been featured in several studies. M2 TAMs in ovarian cancer have been shown to adopt deregulated peroxisome proliferator-activated receptor (PPAR) signaling due to the accumulation of oxidized low-density lipoprotein (LDL) in the TME [62]. Perturbation of lipid uptake sufficiently ameliorated pro-tumorigenic potential of TAMs, showing a critical role of lipid metabolism in pro-tumoral TAMs [65]. Prominent use of lipid metabolism by M2 macrophages also reflects their survival advantage over M1 macrophages within lipid-rich TME. Glutamine metabolism is another way that TAMs depend on to produce energy. Depletion of glutamine synthetase (GS) polarize TAMs into M1-like TAMs, which consequently led to the inhibition of metastasis [66]. Furthermore, M2 skewing of macrophages by high level of α-ketoglutarate (αKG) via glutaminolysis suggested therapeutic potential of targeting glutamine metabolism of macrophages within TME [67].
The role of lipid, particularly triglycerides (TG), in DCs has also been reported [68]. Upregulation of scavenger receptor A expressed on DCs leads to the uptake of lipids from TME [68, 69]. In addition to the uptake of lipids, alteration of metabolic pathway can promote the synthesis and accumulation of FAs within DCs. Dysfunctional DCs prefer to use FAs as the carbon source via augmenting FAO instead of glycolysis [70]. In ovarian cancer, DCs exhibit ER stress response and drive IRE1a/XBP1 activation, leading to the synthesis and accumulation of FAs and TG [71]. In melanoma, tumor cells activate WNT5a/β-catenin-PPARγ signaling, which in turn upregulates the expression of carnitine palmitoyltransferase-1a (CPT1A) to promote FAO [72]. FA-carrying tumor-derived exosomes (TDEs) drive DCs to activate PPARα signaling pathway and promote FAO by increasing intracellular lipids [73]. Several studies have shown that abnormal accumulation of lipids in DCs is one of the major factors impairing antigen cross-presentation [74]. Oxidized lipids, especially electrophilic oxidatively truncated lipids (ox-tr-LB), which covalently adduct with heat shock protein 70 (Hsp70), mainly cause defective trafficking of peptide-MHCI (pMHCI) complexes from phagosome/lysosome to the cell surface [75]. Furthermore, lipid accumulation in DCs can downregulate CD86 and upregulate tolerogenic cytokine IL-10 [76]. These studies demonstrate the importance of lipid metabolism regulating the functionality of DCs within TME.
Metabolites that can act as immune signaling molecules in APCs
Metabolites produced by tumors such as lactate, succinate, and αKG are utilized not only for feeding metabolic pathways, but also function as signaling molecules to the neighboring cells (Table 1). The immunoregulatory function of these metabolites are accentuated as a modulator of TME, which are now drawing attention as therapeutic targets to awake antitumor immunity.
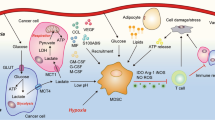
Low extracellular pH reduces glucose consumption and lactate production, increases mitochondrial respiration, and inhibits mTORC1 activity. These environmental factors govern the differentiation of monocytes into DCs [77]. Furthermore, lactate produced by highly glycolytic tumor cells are considered an immunomodulatory molecule [78], inducing metabolic polarization of macrophages into M2 TAMs by triggering the expression of M2-associated genes (e.g., arginase 1 (Arg1) and VegfA) in a HIF1α-dependent manner [58]. An investigation of receptors responsible for lactate sensing demonstrated that the odorant receptor OLFR78, a type of G protein-coupled receptor (GPCR), as a responsible sensor for lactate uptake by TAMs in TME [79]. GPCR-mediated inducible cyclic AMP early repressor (ICER) has further been shown to dictate lactate as a pro-tumorigenic signal altering TAMs to obtain M2-like features [80]. Macrophages seem to engage lactate as an epigenetic modulator as well. Although histone lactylation has only been demonstrated in a bacterial infection model, preferential lactylation in macrophages in the resolving phase rather than the pro-inflammatory phase suggests possible roles of lactate in sculpting pro-tumorigenic function of TAMs [81]. Lactate can also limit antigen-presenting capacity of DCs by stimulating G protein-coupled receptor 81 (GPR81) expression, which downregulates cell surface expression of MHCII and decreases cAMP, IL-6, and IL-12 [82]. Acidification of TME by lactate limits antigen uptake and destabilizes the antigen-MHCI complex [83]. Furthermore, low pH reduces the antigen binding capacity of mannose receptor (MR), such as DEC205, expressed on a variety of APCs, including DCs, by inducing conformational change (Fig. 1) [83, 84].

Low pH resulting from lactate from tumor cells leads to the downregulation of cAMP, IL-6, and IL-12 by stimulating GPR81. Furthermore, lactate limits the APC function of DCs by suppressing MHCII-mediated antigen presentation and inducing a conformational change of mannose receptor (MR), which is responsible for antigen (Ag) binding. A hypoxic environment limits the migration of DCs to the LN, and enrichment of IDO and adenosine induce DCs to express immunosuppressive cytokines. Fatty acid oxidation (FAO) fueled by lipids in TME is prominently used by dysfunctional DCs that are impaired with antigen cross-presentation. Lipid uptake by scavenger receptor A (SR-A) results in the accumulation of lipids within DC, which consequently affects peptide-MHCI (pMHCI) trafficking by forming an adduct with heat shock protein 70 (HSP70).
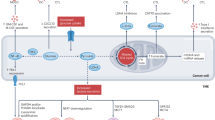
Metabolites from the tricarboxylic acid (TCA) cycle not only serve as metabolic precursors, but also actively play a role as messengers between tumor cells and immune cells. Tumors harboring mutations on TCA cycle enzymes, such as succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase (IDH), reshape TME, which in turn affect immunosurveillance. Succinate, as a ligand of succinate receptor 1 (SUCNR1), conveys signal to phosphoinositide 3-kinase (PI3K)-HIF1α cascade within TAMs and promotes the migration of TAMs into TME [85]. 2-hydroxyglutarate (2-HG), an analogous metabolite produced by mutant IDH, has been suggested to lead dysfunctionality of TAMs via altering tryptophan metabolism [86]. Alteration of tryptophan metabolism is one of the strategies that tumor utilizes to suppress antitumor immunity. IDO, a rate-limiting enzyme of tryptophan catabolism, is found upregulated in a wide range of cells, including tumor cells and immune subsets. Reduced availability of tryptophan due to high levels of IDO within TME induces T cell dysfunction within TME. Likewise, in tumors with mutant IDH, attenuation of Bar-adapter encoding gene 1 (BIN1) expression in human cancers has also been demonstrated to alter tryptophan metabolism by promoting IDO activity [87]. Pro-tumoral macrophage is another compartment that express a high level of IDO. IDO that is highly expressed in TAMs further contribute to perturb antitumor immunity by depleting tryptophan but enriching metabolic product, kynurenine (KYN), which functions as the ligand for aryl hydrocarbon receptor (AhR). KYN suppresses the nuclear factor kappa light chain of activated B cells (NF-κB) pathway involving kruppel-like factor 4 (KLF4), and leads to M2 polarization and T cell dysfunction via CD39 expression [11, 88]. As the role of KYN has been implicated in the formation of immunosuppressive TME, its depletion via the administration of kynureninase reversed immunosuppressive TME. However, whether this involves the modulation of the activity of TAMs is not clear (Fig. 2) [89]. Accumulation of IDO will also render DCs to further contribute to the alteration of TME into immunosuppressive TME [90]. Depletion of tryptophan within TME markedly decreases Ag uptake and the expression of CD40 and CD80 on DCs. In addition, DCs under tryptophan-deprived conditions significantly increase inhibitory receptors (ILT3 and ILT4), which leads to the induction of Tregs [91]. KYN has been evidenced to be strongly correlated with cDC2 differentiation. IDO1-expressing cDC1s induce regulatory cDC2 through the secretion of L-KYN. This, in turn, recruits the AhR-activated cDC2 subset into a tolerogenic pool. Since the importance of KLF4 in regulating functionality and fate specification of cDC2s have been reported, whether tolerance of cDC2 induced by KYN within TME involves KLF4 should be further investigated [92]. Furthermore, IL-12 plus GM-CSF-treated tumor-bearing mice induce long-term maintenance of IDO+ DC in the tumor-draining LNs (TDLNs) through transient induction of IFNγ. In vitro system modeling demonstrated that IDO expression is maintained by a positive feedback loop between IDO-KYN and AhR-IDO. This renders DCs to be tolerogenic, inhibiting T-cell proliferation as well as inducing Treg differentiation [93]. Tregs generated in the TDLN can further upregulate IDO expression in DCs via CTLA4/B7 interaction [94]. In addition to IDO, adenosine accumulated within hypoxic TME stimulates A2B adenosine receptor, skewing DC differentiation into a distinct subset that secretes high levels of immune suppressors, pro-inflammatory and tolerogenic factors such as IL-6, IL-8, IL-10, and TGFβ [95]. Taken together, TME enriched with metabolites secreted by tumor cells actively modulate metabolic phenotypes and functionalities of APCs into pro-tumorigenic APCs.

Lactate transported by MCT1 and GPCRs (e.g., OLFR78 and GPR132) shapes pro-tumoral properties of TAMs by upregulating M2-associated genes. Enrichment of TCA cycle metabolites affects the metabolic properties of TAMs. SUCNR1-mediated succinate uptake activates PI3K-HIF1α signaling to induce polarization of TAMs. Furthermore, accumulation of kynurenine within TAMs generated from tryptophan and 2-HG (generated by mutant IDH on tumor cells) by sequential activities of IDO and TDO, binds to aryl hydrocarbon receptor (AhR), and suppresses NF-κB signaling. Glutaminolysis produce α-ketoglutarate (αKG), supplying intermediate for the TCA cycle, and further alters the epigenome of macrophages. Increased dependency on fatty acid oxidation (FAO) or ER stress induced by lipids enriched in TME leads macrophages to adopt pro-tumoral characteristics.
The impact of tumor aggressiveness on the function of APCs
The dysfunction of APCs is largely driven by the metabolic rewiring of tumor cells. However, whether the metabolic features of APCs are altered gradually depending on the progression of tumor or how the tumor aggressiveness and properties affect APCs is unclear. Below, we introduce studies regarding how APCs are affected by two indicators of tumor aggressiveness; cancer stem cell (CSC) and epithelial-to-mesenchymal transition (EMT).
CSC has been described to be responsible for treatment resistance, tumor metastasis, and tumor recurrence. Of note, the maintenance of CSC highly relies on the ADP-ribosylation factor 1 (Arf1)-mediated lipolysis pathway that provides energy from FA. Disruption of lipid metabolism by Arf1 ablation enhanced antitumor immunity exerted by DC via triggering ER stress and secretion of damage-associated molecular patterns (DAMPs) [96]. This indirectly shows how CSC can escape antitumor immunity. Furthermore, accumulation of ROS, which correlates with tumor aggressiveness, due to oxidative stress or hypoxia within TME leads to the upregulation of heme oxygenase 1 (HO-1) expression in DCs and TAMs [97, 98]. The induction of HO-1 in APCs profoundly contributes to the shaping of immunosuppressive TME. HO-1 expression in DCs inhibits lipopolysaccharide (LPS)-induced phenotypic maturation and production of pro-inflammatory cytokines, which hinders T-cell proliferation, and increases IL-10 production [99]. Moreover, an enhanced beneficial effect of antitumor vaccine by ablation of HO-1 in TAMs demonstrates how the abundance of ROS and aggressiveness of tumor can affect the function of APCs [100].
EMT is another feature that is associated with tumor aggressiveness in many cancers. Compared to differentiated tumors that still retain epithelial features, tumor cells undergoing mesenchymal transition gradually rewire their metabolism to support their energy demand. TGFβ, a well-known inducer of EMT, drives the upregulation of glycolysis via inducing the expression of glycolytic enzymes in glioblastoma cells [101]. Additionally, transcription factors (Snail, Slug, Twist, and Zeb1) involved in the process of EMT commonly upregulate glycolysis, while they downregulate mitochondrial respiration [102,103,104,105]. For example, SNAIL enhanced glycolytic flux by increasing glucose uptake and downregulating the expression of fructose-bisphosphatase 1 (FBP1) via suppressing promoter activity [104]. In a murine pancreatic cancer model, the tumor deficient of Zeb1 lacked a glycolytic switch upon the blockage of OXPHOS with oligomycin treatment [106]. An increase of glycolysis in tumors undergoing EMT will shape the TME into a lactate-rich environment, which will further perturb proper antitumor immunity. In line with this, Zeb1 induction in breast cancer has been demonstrated to be critical for pro-tumoral macrophage polarization [107]. This intrigues the question of whether the ablation of lactate signaling in TAMs will alleviate the aggressiveness of tumor. Furthermore, The Cancer Genome Atlas (TCGA) analysis showed that EMT-high tumors are prone to be enriched with TAMs, revealing the association between the metabolic shift of aggressive tumors with pro-tumoral features of macrophages [108]. However, the “cause and consequence” relationship between the aggressiveness of tumor and macrophage phenotype is yet to be clarified, as TAMs can promote EMT via secreting TGFβ [109]. In addition to TGFβ as an EMT-inducer secreted by TAMs, co-culture of TAMs with HNSCC cells demonstrated that epidermal growth factor (EGF) from TAMs also triggers EMT via conveying its signal to activate extracellular signal-regulated protein kinase 1/2 (ERK1/2) [110]. Besides, tumor-associated dendritic cells (TADC) has been studied to render colon cancer cells to obtain EMT features and CSC properties by enhancing CD133 and aldehyde dehydrogenase (ALDH) via secreting C-X-C motif ligand 1 (CXCL1) [111]. These studies imply that the association of tumor aggressiveness and APC function is not a uni-directional event, but rather intertwined.
Therapeutic approaches to restore the competent function of APCs
Based on recent observations on the dynamic interaction between tumor cells and TAMs through metabolites and nutrients within TME, an extensive amount of effort has been devoted to target metabolic features of TAMs to repolarize into M1 macrophages. TAMs expressing high levels of ARG1 render TME into an immunosuppressive environment by depleting arginine, which is critical for T cells and NK cells. Treatment of CB-1158, an ARG1 inhibitor under clinical trials, has been shown to restore the antitumor immunity exerted by T cells, both in vitro and in vivo syngeneic tumor model, and increase the population of pro-inflammatory macrophages [112]. Activation of PI3Kγ alters macrophages into immunosuppressive phenotype via activation of NF-κB-C/EBPβ. TAMs exhibited pro-inflammatory features upon the treatment of PI3Kγ blockade, which further led to enhanced antitumor immunity via altering T cell content [113]. Recently, the initiative role of protein kinase RNA-like ER kinase (PERK) promoting M2 polarization of TAM via upregulation of serine metabolism has been demonstrated. In this study, PERK inhibition with GSK2656157 not only delayed tumor progression, but also induced higher expansion of effector T cells [114]. Owing to its critical role in the M2 polarization of macrophages, targeting glutamine metabolism seems like a promising way to enhance antitumor immunity [115]. Inhibition of GS repolarized M2 macrophages into M1 macrophages, which further enhanced lymphocyte recruitment with less suppressive activity [66]. Furthermore, AMP-activated protein kinase (AMPK) activation by metformin treatment blocked M2 skewing of macrophages, which prevented metastasis of Lewis lung adenocarcinoma [116]. Targeting lactate signaling in TAMs can be another option to control tumor progression. G protein-coupled receptor 132 (GPR132) expressed on macrophages senses lactate within TME and leads macrophages to adopt M2-like phenotypes to promote breast cancer metastasis [117]. Treatment of rosiglitazone, an agonist of PPARγ, impeded tumor progression via inhibiting GPR132 expression on TAMs [118]. Furthermore, as the role of tryptophan metabolism has been demonstrated to be critical in modulating antitumor immunity, targeting tryptophan catabolism at different levels showed better antitumor immunity. Direct degradation of kynurenine with the administration of PEGlyated kynureninase substantially improved antitumor immunity against different types of cancers when combined with checkpoint inhibitors [89]. Furthermore, IDO-specific peptide (IDO vaccine) successfully reshaped immunosuppressive TME into immune-supportive TME, enriched with less M2 macrophages but with higher amount of M1 macrophages [119]. Nowadays, clinical trials targeting the tryptophan-kynurenine-AhR axis (e.g., AhR inhibitor) are under investigation in conjunction with checkpoint blockades [120]. Although studies on immunometabolism revealed targetable vulnerabilities which largely widened therapeutic options, targeting a single metabolic pathway of immune cells will lead to another round of metabolic rewiring of tumors as they retain high plasticity. Therefore, exploitation of metabolic targets that can improve antitumor immunity and perturb tumor metabolism simultaneously would be encouraged.
DCs (especially cDC1s) are central inducers of the immune response, which can further influence responsiveness to cancer therapies. Conventional cancer therapies, such as radiotherapy and chemotherapy, influence and require DC functions to improve therapeutic efficacy. Chemotherapy can trigger immunogenic tumor cell death, which results in releasing of stimulatory factors, such as apoptotic tumor cells, ATP and high-mobility-group box 1 (HMGB1). In turn, these factors play a role as alarmins that activate and mobilize DCs, which enhance the cross-presentation of tumor-associated antigens (TAAs) to elicit antitumor CD8 T cell responses [121]. Immunogenic chemotherapies, such as anthracycline- or oxaliplatin-treatment lead tumor cells to release ATP, which then recruits DCs and activates NOD-like receptor family, pyrin domain containing-3 protein (NLRP3) inflammasome, allowing the secretion of IL-1β [122]. In addition, there are several therapeutic approaches directly targeting DCs using DC-specific antibodies to deliver antigen/adjuvant or nanoparticles and adoptive transfer of autologous, antigen-loaded and activated DCs: DC activating factors, DC mobilizing agents, antigen-presenting, and antigen carriers [123]. The followings are the strategies targeting DCs to enhance antitumor immunity. (1) Providing exogenous activation signals, particularly derivatives of Agonists for TLRs or STING, can drive immunogenic DC activation. For example, treatment of tumor-bearing mice with NS-398 (COX-2 inhibitor), to reduce prostaglandin synthesis of tumor cells, can enhance the transcription factor Zbtb46, induce cDC lineage maturation, and enhance the activity of DCs [124,125,126]. Specifically, the lifespan of TLR-activated BMDCs is extended upon inhibition of mTOR due to reduced NO production as well as improved mitochondrial function [127]. (2) Vaccines and DC-specific antibodies to deliver antigen/adjuvant to increase antigen-specific T cell responses. DC vaccines include TAA-derived peptides, whole tumor lysates, and recombinant TAA-expressing viruses. It has been reported that DCs exhibit both accuracy and therapeutic efficacy after exposure to TAAs oxidized with mannan [123, 128, 129]. Furthermore, sarcosine-treated DC vaccines increase the migration ability of DCs, which is associated with the upregulation of COX1 and PIK3CG in B16F10 melanoma and glioma [130]. Dasatinib (tyrosine kinase inhibitor)-stimulated DC vaccines downregulate the IDO expression level and IDO-mediated tryptophan metabolism via inhibiting c-KIT [131]. Yellow fever vaccine (YF-17D) can enhance the activity of general control nonderepressible 2 kinase (GCN2), a sensor of amino acid starvation, thereby augmenting antigen presentation capacity of DCs, which leads to the modulation of an adaptive immune response [132]. Since vaccines have shown limited efficacy to date, combining adjuvants (GM-CSF or TLR Agonists) with vaccines are on the rise for in vivo provision [133]. For example, using a combination of OK432 (Picibanil), TLR7/8 ligand (CL097) and reduced PGE2 is manifested, as this combination stimulated the maturation of moDCs and increased the expression of co-stimulatory molecules and IL-12p70 production [134]. (3) Targeting metabolism in situ reprograms tumour-infiltrating DCs (TIDCs). For instance, inhibition of FAS using 5-(tetradecyloxy)-2-furoic acid (TOFA) or cerulenin prevents the accumulation of lipids, which in turn restores immunostimulatory activity and tumor control of TIDC [68, 135]. A rising number of preclinical studies regarding how to restore antitumor immunity by harnessing the metabolic properties of APCs will contribute to broaden the therapeutic options available to combat tumor (Table 2).
Conclusion
Successful clearance of cancer largely relies on the proper activation of APCs. Although immunotherapy has revolutionized and fueled cancer therapies, a significant percentage of cancer patients do not benefit from cancer immunotherapies, partly due to low T cell infiltration or low tumor mutation burdens. Mounting evidence shows not only immunotherapy but also radiotherapy or chemotherapy requires the proper function of APCs to improve therapeutic efficacy. While tolerance and dysfunction of immune cells within TME are major hurdles that has to be overcome to fully harness the potential of APCs in cancer immunotherapy, immunostimulatory APCs are being recognized as a targetable source to elicit favorable adaptive immune responses. As expanded upon in previous sections, metabolic alteration of APCs is not a passive feature appearing during the immune response, but rather an active process that APCs engage to bolster adaptive immunity via regulating the functional status of T cells. Improved knowledge of how APCs are regulated in TME allowed therapeutic exploitation in clinical settings. However, direct targeting of metabolic pathways of APCs as a strategy to treat cancers has been marginally successful. Failure of targeting APC metabolism in clinical trials might be due to several reasons: insufficiency of targeting a single metabolic pathway to overcome the metabolic thresholds of APCs within TME, which is more harsh than preclinical models, and plasticity of tumor cells that can further rewire their metabolism. Therefore, targeting metabolism in combination with other therapies, including checkpoint inhibitors, will be a promising avenue. Moreover, investigating further on how multiple metabolic pathways in APCs are intertwined to interpret different metabolic signals within TME will help figure out a better target and refine the strategies to target the metabolism of APCs for reverting the dysfunctionality of APCs. A better understanding of the metabolic features of APCs in TME will change the landscape of cancer therapies that can improve the survival of patients with better outcomes.
Data availability
This manuscript does not contain original experimental data.
References
Gaudino SJ, Kumar P. Cross-talk between antigen presenting cells and T cells impacts intestinal homeostasis, bacterial infections, and tumorigenesis. Front Immunol. 2019;10:360.
Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 2014;74:705–15.
Geeraerts X, Fernández-Garcia J, Hartmann FJ, de Goede KE, Martens L, Elkrim Y, et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep. 2021;37:110171.
Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184:792–809.e23.
Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun. 2021;12:1151.
Liu L, Lu Y, Martinez J, Bi Y, Lian G, Wang T, et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc Natl Acad Sci USA. 2016;113:1564–9.
Das Gupta K, Shakespear MR, Curson JEB, Murthy AMV, Iyer A, Hodson MP, et al. Class IIa histone deacetylases drive toll-like receptor-inducible glycolysis and macrophage inflammatory responses via pyruvate kinase M2. Cell Rep. 2020;30:2712–28.e8.
Koo SJ, Szczesny B, Wan X, Putluri N, Garg NJ. Pentose phosphate shunt modulates reactive oxygen species and nitric oxide production controlling Trypanosoma cruzi in macrophages. Front Immunol. 2018;9:202.
Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. 2012;7:e50946.
Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T cells. Immunity. 2018;48:773–86.e5.
Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci. 2019;22:729–40.
Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits CD8(+) T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity. 2018;48:299–312.e5.
Sun W, Wei FQ, Li WJ, Wei JW, Zhong H, Wen YH, et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br J Cancer. 2017;117:1631–43.
Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE. 2011;6:e19495.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–59.
Tang-Huau TL, Gueguen P, Goudot C, Durand M, Bohec M, Baulande S, et al. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat Commun. 2018;9:2570.
Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity. 2017;47:363–73.e5.
Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34:85–95.
Barrio MM, Abes R, Colombo M, Pizzurro G, Boix C, Roberti MP, et al. Human macrophages and dendritic cells can equally present MART-1 antigen to CD8(+) T cells after phagocytosis of gamma-irradiated melanoma cells. PLoS ONE. 2012;7:e40311.
Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci USA. 2013;110:11103–8.
Modak M, Mattes AK, Reiss D, Skronska-Wasek W, Langlois R, Sabarth N, et al. CD206+ tumor-associated macrophages cross-present tumor antigen and drive antitumor immunity. JCI Insight. 2022;7:e155022.
Hildner K, Edelson Brian T, Purtha Whitney E, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–100.
Cheng W-C, Tsui Y-C, Ragusa S, Koelzer VH, Mina M, Franco F, et al. Uncoupling protein 2 reprograms the tumor microenvironment to support the anti-tumor immune cycle. Nat Immunol. 2019;20:206–17.
Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Disco. 2016;6:71–9.
Møller SH, Wang L, Ho PC. Metabolic programming in dendritic cells tailors immune responses and homeostasis. Cell Mol Immunol. 2022;19:370–83.
DeVito NC, Plebanek MP, Theivanthiran B, Hanks BA. Role of tumor-mediated dendritic cell tolerization in immune evasion. Front Immunol. 2019;10:2876.
Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30:324–36.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Wculek SK, Khouili SC, Priego E, Heras-Murillo I, Sancho D. Metabolic control of dendritic cell functions: digesting information. Front Immunol. 2019;10:775.
Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–9.
Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44:924–38.
Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol. 2015;195:2624–32.
Stüve P, Minarrieta L, Erdmann H, Arnold-Schrauf C, Swallow M, Guderian M, et al. De novo fatty acid synthesis during mycobacterial infection is a prerequisite for the function of highly proliferative T cells, but not for dendritic cells or macrophages. Front Immunol. 2018;9:495.
Basit F, Mathan T, Sancho D, de Vries IJM. Human dendritic cell subsets undergo distinct metabolic reprogramming for immune response. Front Immunol. 2018;9:2489.
Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. 2012;143:1061–72.
Sittig SP, Bakdash G, Weiden J, Sköld AE, Tel J, Figdor CG, et al. A comparative study of the T cell stimulatory and polarizing capacity of human primary blood dendritic cell subsets. Mediators Inflamm. 2016;2016:3605643.
Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell. 2019;177:556–71.e16.
Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013;73:4629–40.
Rodrigues PF, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, Tussiwand R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. 2018;19:711–22.
Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res. 2004;10:7466–74.
Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356:eaah4573.
Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallée V-P, Mendoza A, et al. Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell. 2019;179:846–63.e24.
Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. 2020;580:257–62.
Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo-Koussou Y, Hidalgo S, et al. Transcriptional and functional analysis of CD1c+ human dendritic cells identifies a CD163+ subset priming CD8+CD103+ T cells. Immunity. 2020;53:335–52.e8.
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–5.
Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol. 2019;70:58–65.
Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. 2012;18:3281–92.
Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Disco. 2016;6:247–55.
Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. PD-1hi identifies a novel regulatory B-cell population in human hepatoma that promotes disease progression. Cancer Disco. 2016;6:546–59.
Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92.
Filippi I, Morena E, Aldinucci C, Carraro F, Sozzani S, Naldini A. Short-term hypoxia enhances the migratory capability of dendritic cell through HIF-1α and PI3K/Akt pathway. J Cell Physiol. 2014;229:2067–76.
Liu J, Zhang X, Chen K, Cheng Y, Liu S, Xia M, et al. CCR7 chemokine receptor-inducible lnc-Dpf3 restrains dendritic cell migration by inhibiting HIF-1α-mediated glycolysis. Immunity. 2019;50:600–15.e15.
Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74:24–30.
Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016;24:701–15.
Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. 2014;5:5350–68.
Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19:512–26.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63.
Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019;79:795–806.
Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;537:234–8.
Meng X, Grötsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX, et al. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat Commun. 2018;9:251.
Su P, Wang Q, Bi E, Ma X, Liu L, Yang M, et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 2020;80:1438–50.
Di Conza G, Tsai CH, Gallart-Ayala H, Yu YR, Franco F, Zaffalon L, et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat Immunol. 2021;22:1403–15.
Niu Z, Shi Q, Zhang W, Shu Y, Yang N, Chen B, et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat Commun. 2017;8:766.
Park J, Lee SE, Hur J, Hong EB, Choi JI, Yang JM, et al. M-CSF from cancer cells induces fatty acid synthase and PPARβ/δ activation in tumor myeloid cells, leading to tumor progression. Cell Rep. 2015;10:1614–25.
Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20:1654–66.
Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985–94.
Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16:880–6.
Arai R, Soda S, Okutomi T, Morita H, Ohmi F, Funakoshi T, et al. Lipid accumulation in peripheral blood dendritic cells and anticancer immunity in patients with lung cancer. J Immunol Res. 2018;2018:5708239.
Dong H, Bullock TNJ. Metabolic influences that regulate dendritic cell function in tumors. Front Immunol. 2014;5:24.
Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2015;161:1527–38.
Zhao F, Xiao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, et al. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. 2018;48:147–60.e7.
Yin X, Zeng W, Wu B, Wang L, Wang Z, Tian H, et al. PPARα inhibition overcomes tumor-derived exosomal lipid-induced dendritic cell dysfunction. Cell Rep. 2020;33:108278.
Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of dendritic cells in tumor microenvironment: for immunotherapy. Front Immunol. 2021;12:613492.
Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. 2017;8:2122.
Gardner JK, Mamotte CDS, Patel P, Yeoh TL, Jackaman C, Nelson DJ. Mesothelioma tumor cells modulate dendritic cell lipid content, phenotype and function. PLoS ONE. 2015;10:e0123563.
Erra Díaz F, Ochoa V, Merlotti A, Dantas E, Mazzitelli I, Gonzalez Polo V, et al. Extracellular acidosis and mTOR inhibition drive the differentiation of human monocyte-derived dendritic cells. Cell Rep. 2020;31:107613.
Wang Z-H, Peng W-B, Zhang P, Yang X-P, Zhou Q. Lactate in the tumour microenvironment: From immune modulation to therapy. eBioMedicine. 2021;73:103627.
Vadevoo SMP, Gunassekaran GR, Lee C, Lee N, Lee J, Chae S, et al. The macrophage odorant receptor Olfr78 mediates the lactate-induced M2 phenotype of tumor-associated macrophages. Proc Natl Acad Sci USA. 2021;118:e2102434118.
Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. 2018;19:1319–29.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–80.
Brown TP, Bhattacharjee P, Ramachandran S, Sivaprakasam S, Ristic B, Sikder MOF, et al. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene. 2020;39:3292–304.
Burgdorf S, Porubsky S, Marx A, Popovic ZV. Cancer acidity and hypertonicity contribute to dysfunction of tumor-associated dendritic cells: potential impact on antigen cross-presentation machinery. Cancers. 2020;12:2403.
Cao L, Shi X, Chang H, Zhang Q, He Y. pH-Dependent recognition of apoptotic and necrotic cells by the human dendritic cell receptor DEC205. Proc Natl Acad Sci USA. 2015;112:7237–42.
Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. 2020;77:213–27.e5.
Friedrich M, Sankowski R, Bunse L, Kilian M, Green E, Ramallo Guevara C, et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer. 2021;2:723–40.
Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–9.
Venkateswaran N, Lafita-Navarro MC, Hao YH, Kilgore JA, Perez-Castro L, Braverman J, et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019;33:1236–51.
Triplett TA, Garrison KC, Marshall N, Donkor M, Blazeck J, Lamb C, et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat Biotechnol. 2018;36:758–64.
Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Disco. 2014;4:879–88.
Brenk M, Scheler M, Koch S, Neumann J, Takikawa O, Häcker G, et al. Tryptophan deprivation induces inhibitory receptors ILT3 and ILT4 on dendritic cells favoring the induction of human CD4+CD25+ Foxp3+ T regulatory cells. J Immunol. 2009;183:145–54.
Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. 2015;42:916–28.
Li Q, Harden JL, Anderson CD, Egilmez NK. Tolerogenic phenotype of IFN-γ-induced IDO+ dendritic cells is maintained via an autocrine IDO-kynurenine/AhR-IDO loop. J Immunol. 2016;197:962–70.
Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–12.
Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. 2008;112:1822–31.
Wang G, Xu J, Zhao J, Yin W, Liu D, Chen W, et al. Arf1-mediated lipid metabolism sustains cancer cells and its ablation induces anti-tumor immune responses in mice. Nat Commun. 2020;11:220.
Luu Hoang KN, Anstee JE, Arnold JN. The diverse roles of heme oxygenase-1 in tumor progression. Front Immunol. 2021;12:658315.
Oshi M, Gandhi S, Yan L, Tokumaru Y, Wu R, Yamada A, et al. Abundance of reactive oxygen species (ROS) is associated with tumor aggressiveness, immune response, and worse survival in breast cancer. Breast Cancer Res Treat. 2022;194:231–41.
Chauveau C, Rémy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. 2005;106:1694–702.
Alaluf E, Vokaer B, Detavernier A, Azouz A, Splittgerber M, Carrette A, et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight. 2020;5:e133929.
Rodríguez-García A, Samsó P, Fontova P, Simon-Molas H, Manzano A, Castaño E, et al. TGF-β1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017;284:3437–54.
Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316–31.
Røsland GV, Dyrstad SE, Tusubira D, Helwa R, Tan TZ, Lotsberg ML, et al. Epithelial to mesenchymal transition (EMT) is associated with attenuation of succinate dehydrogenase (SDH) in breast cancer through reduced expression of SDHC. Cancer Metab. 2019;7:6.
Yu J, Li J, Chen Y, Cao W, Lu Y, Yang J, et al. Snail enhances glycolysis in the epithelial-mesenchymal transition process by targeting FBP1 in gastric cancer. Cell Physiol Biochem. 2017;43:31–8.
Zhou Y, Lin F, Wan T, Chen A, Wang H, Jiang B, et al. ZEB1 enhances Warburg effect to facilitate tumorigenesis and metastasis of HCC by transcriptionally activating PFKM. Theranostics. 2021;11:5926–38.
Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19:518–29.
Jiang H, Wei H, Wang H, Wang Z, Li J, Ou Y, et al. Zeb1-induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis. 2022;13:206.
Tiwari JK, Negi S, Kashyap M, Nizamuddin S, Singh A, Khattri A. Pan-cancer analysis shows enrichment of macrophages, overexpression of checkpoint molecules, inhibitory cytokines, and immune exhaustion signatures in EMT-high tumors. Front Oncol. 2021;11:793881.
Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352:160–8.
Gao L, Zhang W, Zhong WQ, Liu ZJ, Li HM, Yu ZL, et al. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol Rep. 2018;40:2558–72.
Hsu YL, Chen YJ, Chang WA, Jian SF, Fan HL, Wang JY, et al. Interaction between tumor-associated dendritic cells and colon cancer cells contributes to tumor progression via CXCL1. Int J Mol Sci. 2018;19:2427.
Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:101.
Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–42.
Raines LN, Zhao H, Wang Y, Chen HY, Gallart-Ayala H, Hsueh PC, et al. PERK is a critical metabolic hub for immunosuppressive function in macrophages. Nat Immunol. 2022;23:431–45.
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–30.
Ding L, Liang G, Yao Z, Zhang J, Liu R, Chen H, et al. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget. 2015;6:36441–55.
Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA. 2017;114:580–5.
Cheng WY, Huynh H, Chen P, Peña-Llopis S, Wan Y. Macrophage PPARγ inhibits Gpr132 to mediate the anti-tumor effects of rosiglitazone. Elife. 2016;5:e18501.
Nandre R, Verma V, Gaur P, Patil V, Yang X, Ramlaoui Z, et al. IDO vaccine ablates immune-suppressive myeloid populations and enhances antitumor effects independent of tumor cell IDO status. Cancer Immunol Res. 2022;10:571–80.
Labadie BW, Bao R, Luke JJ, Reimagining IDO. Pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin Cancer Res. 2019;25:1462–71.
Sánchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Pérez-Gracia JL, Sánchez-Arráez A, et al. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol. 2017;28:xii44–55.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
Fuertes MB, Woo S-R, Burnett B, Fu Y-X, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34:67–73.
Pandey VK, Amin PJ, Shankar BS. COX-2 inhibitor prevents tumor induced down regulation of classical DC lineage specific transcription factor Zbtb46 resulting in immunocompetent DC and decreased tumor burden. Immunol Lett. 2017;184:23–33.
Chi H, Li C, Zhao FS, Zhang L, Ng TB, Jin G, et al. Anti-tumor activity of Toll-like receptor 7 agonists. Front Pharm. 2017;8:304.
Amiel E, Everts B, Fritz D, Beauchamp S, Ge B, Pearce EL, et al. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J Immunol. 2014;193:2821–30.
Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–60.
Zhang J, Wang Y, Wu Y, Ding ZY, Luo XM, Zhong WN, et al. Mannan-modified adenovirus encoding VEGFR-2 as a vaccine to induce anti-tumor immunity. J Cancer Res Clin Oncol. 2014;140:701–12.
Dastmalchi F, Karachi A, Yang C, Azari H, Sayour EJ, Dechkovskaia A, et al. Sarcosine promotes trafficking of dendritic cells and improves efficacy of anti-tumor dendritic cell vaccines via CXC chemokine family signaling. J Immunother Cancer. 2019;7:321.
Chu CL, Lee YP, Pang CY, Lin HR, Chen CS, You RI. Tyrosine kinase inhibitors modulate dendritic cell activity via confining c-Kit signaling and tryptophan metabolism. Int Immunopharmacol. 2020;82:106357.
Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science. 2014;343:313–7.
Moyer TJ, Zmolek AC, Irvine DJ. Beyond antigens and adjuvants: formulating future vaccines. J Clin Invest. 2016;126:799–808.
Yi DH, Stetter N, Jakobsen K, Jonsson R, Appel S. 3-Day monocyte-derived dendritic cells stimulated with a combination of OK432, TLR7/8 ligand, and prostaglandin E(2) are a promising alternative for cancer immunotherapy. Cancer Immunol Immunother. 2018;67:1611–20.
Jiang L, Fang X, Wang H, Li D, Wang X. Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells. Front Immunol. 2018;9:2927.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5.
Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257–70.
Acknowledgements
JP was supported by the National Research Foundation of Korea (NRF-2021R1A6A3A14039520). P-CH was supported in part by the SNSF project grants (31003A_182470), Ludwig Cancer Research, and the European Research Council Starting Grant (802773-MitoGuide).
Author information
Authors and Affiliations
Contributions
JP, LW, and P-CH wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
P-CH is a scientific advisor for Elixiron Immunotherapeutics and a co-founder of Pilatus Biosciences.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, J., Wang, L. & Ho, PC. Metabolic guidance and stress in tumors modulate antigen-presenting cells. Oncogenesis 11, 62 (2022). https://doi.org/10.1038/s41389-022-00438-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-022-00438-y
