
Abstract
Most hereditary tumors show aberrations in DNA repair genes or their regulators. In contrast, only a minority of sporadic tumors show alterations in these genes. As a result, genomic instability is currently considered an enhancer of tumorigenesis rather than an obligatory event in this process. However, tumor heterogeneity presents a significant technical challenge for most cancer genomics studies performed at less than 100× mean resolution depth. To address the importance of genomic instability in prostate carcinogenesis and tumor progression, we performed ultrahigh depth exome sequencing of 124 DNA damage repair/response (repairome) genes in 63 tumors and matched normal tissue samples in African Americans and Caucasians. The average sequence depth was 712-fold for DNA isolated from normal tissue and 368-fold for FFPE tumors. We identified 671 somatic mutations in tumors from African Americans and 762 somatic mutations in tumors in Caucasians. The most frequently mutated DNA repairome genes were EXO1, ATR, POLQ, NEIL3, ERCC6, BRCA2, BRCA1, XPC, JAG1, RPA1, POLE, ATM, and LIG1 in African American men, and POLQ, NEIL3, POLB, BRCA2, EXO1, ERCC6, ATR, RBBP8, BRCA1, ATM, JAG1, XPC, and POLE in Caucasians. We found that 89% of tumors had at least one mutation in nucleotide excision repair pathway genes in African Americans, whereas >40% of tumors had mutations in base excision repair pathway genes in Caucasians. We further identified a marginal increase in mutation rate in tumors in African Americans with increasing age. Tumors in Caucasians did not show a correlation with age, but a progressive increase in the mutation rate was observed at higher Gleason scores. Our data reveal significant differences in the molecular signatures in the DNA repairome in prostate cancer between African Americans and Caucasians. These data also have substantial implications regarding the well-known health disparities in prostate cancer, such as the higher mortality in African Americans than Caucasians.
Similar content being viewed by others

Introduction
Prostate cancer (PCa) is the most common type of cancer in men in USA (164,690 estimated new cases and 29,430 mortalities in 2018, according to the Surveillance, Epidemiology, and End Results database, National Cancer Institute). African American men have the highest incidence of PCa of any other racial group in the United States [1, 2] and the highest rates of aggressive disease and mortality [3, 4]. According to GLOBOCAN 2018, globally, African American men have the highest incidence rates and more aggressive types of PCa than Caucasian men. In contrast, DeSantis et al. (2019) have recently reported that overall cancer death rates declined faster in African Americans than Caucasians among both males (2.6% vs 1.6% per year) and females (1.5 vs 1.3% per year), a finding primarily driven by more significant declines related to lung cancer, colorectal cancer, and PCa (American Cancer Society) [5]. The extent of genetic instability varies among tumors of distinct racial groups, owing to genetic factors, lifestyle-related factors, and occupation [6, 7]. Genetic DNA repair alterations may thus play a crucial role in explaining the racial differences in PCa. The inherent genetic/genomic instability of prostate tumors has been studied in numerous investigations [8,9,10]. However, why some tumors are more unstable than others, or what exactly causes this instability, remains unclear.
DNA is highly vulnerable to chemical modifications, which can cause several types of DNA lesions, such as double-strand DNA breaks, base loss, or base modification. These lesions can occur endogenously or because of exposure to environmental toxins [11, 12]. Cells have thus evolved a series of mechanisms tailored to maintaining genomic integrity [13, 14]. We have previously provided evidence that three DNA repair polymerases, pol β, η, and κ, are often somatically mutated in PCa, and some of these mutations affect enzyme activity, catalytic efficiency, or the fidelity of DNA synthesis [15,16,17]. The incidence of germline mutations in DNA-repair genes among men is higher in those with metastatic PCa than localized PCa [18].
Most experts agree that genomic instability plays a role during cancer evolution, but its exact role is debated [19]. Two types of genomic instability are evident in most cancers: large-scale alterations (e.g., aneuploidy or translocations) and small-scale alterations (e.g., single-nucleotide substitutions or microsatellite instability). The extent of somatic alterations has been examined with both PCR- and non-PCR-based methods: most tumors display low rates of genomic instability [19,20,21], although cancers arising from a mutator phenotype can contain as many as 142,000 alterations per tumor cell [22].
Regardless of the method used, quantifying the somatic instability of heterogeneous tumor genomes is challenging [23, 24]. To address the importance of genomic instability in DNA repair processes for prostate carcinogenesis, we deep sequenced the exons of 124 genes involved in DNA damage repair/response in 63 PCa tumors and matched healthy tissue (peripheral blood lymphocytes or adjacent normal tissue), at very high resolution. We selected the DNA repair/damage response pathway because mutations targeting this pathway are more likely to cause a mutator phenotype than mutations in other pathways. We report that somatic alteration of DNA repair/response (repairome) genes is nearly obligatory in prostate tumors. However, somatic mutations differ among racial groups.
Results
Clinical characteristics of the samples
We sequenced DNA extracted from 63 prostate tumors and matched control tissue from the same patient; 61.9% (n = 39) of the patients were Caucasian, and 38% (n = 24) were African American. We included patients diagnosed between the ages of 46 and 89 years (mean 60 ± 6.2 years) with Gleason scores between 6 and 9, and PSA scores from 2 to 26 (Table 1).
To examine somatic mutations in DNA damage response and repair genes in PCas, we performed systematic somatic mutation analysis of DNA repair/damage response genes using SureSelect custom target enrichment, after tissue microdissection and DNA extraction. Illumina paired-end libraries were prepared from the captured target regions and sequenced on HiSeq 2000 or HiSeq 2500 Sequencing System (Illumina) with 99 bp reads. Somatic variants were called with VarScan. Six FFPE samples from African Americans and five samples from Caucasians did not pass the quality control and were excluded from the analysis. We thus analyzed somatic mutations in 19 tumors in African Americans and 33 tumors in Caucasians, in addition to matched normal tissue.
We identified 671 somatic point mutations in tumors in African Americans; 344 were predicted to generate missense mutations, 321 were predicted to generate nonsense mutations, 5 were predicted to generate stop-gain mutations, and 1 was predicted to generate stop loss codon read-through (Fig. 1a). We identified 762 somatic point mutations in the Caucasian cohort; 425 were predicted to generate missense mutations, 306 were predicted to generate nonsense mutations, 27 were predicted to generate stop codon read-through, and 4 were predicted to generate splicing (Fig. 1b).

a All somatic mutations (SNVs, non-SNVs, and stop codons) identified in PCa samples from African Americans. b All somatic mutations (SNVs, non-SNVs, and stop codons) identified in PCa samples from Caucasians. Validation status was determined by comparing tumor/normal read counts for each allele with VarScan. For validation, we had more than 50 reads with base quality ≥15 (Phred score) for both normal and tumor samples. The somatic p value significance threshold was set as <0.05 and was calculated by VarScan with Fisher’s exact test.
The most frequently mutated DNA repairome genes in African Americans tumors were EXO1 (89%), ATR (73%), POLQ (68% of PCa), NEIL3 (47%), ERCC6 (42%), BRCA2 (52.6%), BRCA1 (52%), XPC (47%), JAG1 (47%), RPA1 (42%), ATM (42%), POLE (36%), and LIG1 (26%) (Fig. 2a). We also identified mutations in the non-DNA repair genes AR (15.7% of PCa) and TMPRSS2 (42% of PCa), which were included as controls. We then focused on whether a mutator phenotype was potentially present. In this analysis, ATM and ATR were found to be associated with higher mutational burden (i.e., 50 mutations/tumor; Supplementary data 2) in all samples except one.

a Somatic mutations identified in the DNA repairome in African Americans PCa (n = 19). b Somatic mutations identified in the DNA repairome in Caucasians PCa (n = 33). Samples are ordered in columns, and mutated genes are displayed in rows. Color indicates the type of mutation: non synonymous (yellow), synonymous (blue), both (sea green), and stop-gain/loss (magenta).
In Caucasians, the most frequently mutated DNA repairome genes were POLQ (48.4%), NEIL3 (45.4%), POLB (45.4%), BRCA2 (42.4%), EXO1 (39%), ERCC6 (36%), ATR (30%), RBBP8 (24%) BRCA1, ATM, JAG1, XPC, and POLE (24%) (Fig. 2b). The non-DNA repair genes AR and TMPRSS2 were found to be mutated in 12% and 30% of PCa samples, respectively. Regarding the mutator phenotype, we identified BRCA2 and NEIL3 to be associated with higher mutational burden (i.e., 20 mutations/tumor) in tumors in Caucasians (Supplementary data 2).
Clinical implications
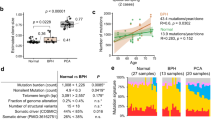
Next, we sought to identify whether age might influence the mutational rate in these two racial groups. In PCa tumors in African Americans, we found a marginal increase in the mutation rate for patients diagnosed at ages younger than 65 years (average 33 mutations) vs 65 years or older (average 41 mutations). We did not find a significant (p = 0.41) association between mutations in DNA-repair genes and age in both groups (Fig. 3a, b).

a Mutation rate in different age groups in African American PCa. b Mutation rate in different age groups in Caucasian PCa. c Comparison of number of mutations in African American PCa with Gleason score 6 and Gleason score > 6. d Comparison of number of mutations in Caucasian PCa with Gleason score 6 and Gleason score > 6.
A slight increase in the mutation rate (18 vs 24 mutations, p = 0.55) was observed with higher Gleason scores only in Caucasians (Fig. 3d). Because of the limitation of samples with a Gleason score 6 in both races, we were unable to draw conclusions regarding low vs high Gleason scores. Gleason score 7 samples were further evaluated on the basis of 4 + 3 vs 3 + 4, but we did not find a significant difference with regards to clinical parameters (data not shown). We also did not find an association between PSA and the mutation rate in either racial group (data not shown).
Recurrent somatic mutations in DNA repairome genes
Next, we focused on identifying recurrent mutations in top mutated DNA repair/response (DDR) genes. In PCa tumors in African Americans, ERCC6 in the nuclear excision repair pathway (NER) showed recurrent mutation of M1097V (methionine-1097 to valine) (Table 2). The XPC gene in the NER pathway of DNA repair was not only frequently mutated but also contained two recurrent mutations (Table 2). In the base excision repair (BER) pathway, we identified three recurrent mutations in NEIL3 (Table 2). The mismatch repair (MMR) pathway gene EXO1 had three recurrent mutations: p.H354R, p.V458M, and p.E589K (Table 2). Among these, p.E589K (glutamic acid-586 to lysine) was the most prevalent missense mutation identified in PCa in African Americans. ATR was also a top mutated gene in tumors in African Americans, with two recurrent mutations (Table 2). TMPRSS2, a non-DNA repair gene, was also frequently mutated in tumors in African Americans and contained a recurrent missense mutation (valine-160 to methionine; Table 2).
The most recurrent missense mutations in PCa tumors in Caucasians were detected in the POLB gene in the BER pathway. The somatic mutation p.E216K (glutamic acid-216 to lysine) was present in 33% of the tumors (8/24; Table 3). In the EXO1 gene in the MMR pathway, we identified two recurrent mutations (Table 3), including p.H354R (histidine-354 to arginine), which was also found in tumors in African Americans. ERCC6 in the NER pathway also had the recurrent mutation p.M1097V (methionine-1097 to valine) in PCa tumors in Caucasians (Table 3) and was also found in those in African Americans.
Somatic mutations in functional domains of DNA repairome genes
Our next aim was to analyze the distribution of these mutations at specific protein domains. Protein domains are particular sequences that have formed over evolution through duplication, recombination, or both. Domains often encode a structural entity associated with specific cellular functions. We systematically analyzed somatic mutations in the top five highly mutated DNA repair genes in both groups (tumors in African Americans and Caucasians) in terms of conserved protein domains. Here, we used mutation mapper, a publicly available visualization tool by cBioPortal (www.cbioportal.org) for cancer genomics data. We filtered out the top five mutated genes, ATR, BRCA1, NEIL3, ERCC6, and EXO1, in PCa tumors in African Americans and NEIL3, BRCA1, EXO1, ERCC6, and POLQ in PCa tumors in Caucasians. Our aim was to identify domains with mutations in all possible domains in the above-listed genes. In African Americans, in the ATR gene, the highest mutation burden was found in the FAT domain, where five somatic mutations were identified (Fig. 4a). We also identified one missense mutation in PI3K(s). In the BRCA1 gene, we identified three missense mutations in the BRCT domain. In the helix-2turn-helix domain (H2TH) of NEIL3, a DNA-binding domain (DNA glycosylase/AP lyase enzymes), we found two missense mutations. For the ERCC6 gene, we identified one silent mutation in the SNF2 domain and two silent mutations in the helicase conserved domain. For EXO1, we found two missense mutations in the XPG_N domain (Fig. 4a). In tumors in Caucasians, in BRCA1, we did not find mutations in the BRCT domain (in contrast to PCa tumors in African Americans). We identified four missense mutations in the H2TH domain and five missense mutations in the ZF-GRF domain of NEIL3. In EXO1, we identified two missense mutations in the XPG_I domain. In ERCC6, we found two missense mutations in the SNF2_N domain and four silent mutations in the helicase domain. We identified three missense mutations in the DNA_polA domain of POLQ (Fig. 4b).

a Location of mutations and protein domains encoded by the top mutated genes ATR, BRCA1, NEIL3, ERCC6, and EXO1 in African American PCa samples. b Location of mutations and protein domains encoded by the op mutated genes BRCA1, NEIL3, EXO1, ERCC6, and POLQ in Caucasian PCa samples, determined using cBioPortal/mutation-mapper. Protein domains are distinguished by color. On the graph of each gene, the y axis represents the number of mutations identified and the x axis reflects the number of amino acid residues. Somatic mutations are shown in circles; missense mutations are shown in green-circle, silent mutations are shown in purple-circle, and truncating mutations are shown in black-circle. Helic helicase, H2TH helix-2turn-helix domain, ZF-GRF zinc finger GRF domain, BRCT BRCA1.
Mutations in the MMR, BER, and NER pathways
We also examined the distribution of mutations in genes in the NER, MMR, BER, and homologous recombination (HR) pathways in prostate tumors from African Americans and Caucasians. In African Americans, the most commonly mutated gene in the NER pathway was ERCC6, followed by EXO1 in the MMR pathway, NEIL3 in the BER pathway, and BRCA1 and BRCA2 in the HR pathway (Fig. 5a). In tumors in Caucasians, mutation frequencies within NER, MMR, and BER genes were approximately equal. Among tumors from Caucasians, the most mutations were found in the NEIL3 gene in the BER pathway and BRCA1 in the HR pathway. ERCC6 in the NER pathway and EXO1 in the MMR pathway were the second most mutated genes (Fig. 5b).

a most frequently mutated genes across NER, MMR, BER, and HR pathways in African Americans (n = 19) and b most frequently mutated genes across NER, BER, MMR, and HR pathways in Caucasians (n = 33).
Mutation spectra across the DNA repairome genes in PCa
To further characterize the genetic instability, we sought to examine the T- to C-transitions in our data. We discovered markedly higher T- to C-transition frequencies (27%) in prostate tumors in African Americans than Caucasians (17.84%) (Fig. 6).

Bars show the mutational context (%) of C.A>G.T, C.G>G.C, C.T>G.A., T.A>A.T, T.C>A.G, and T.G>A.C.
Predicted effects of missense mutations
In our last objective, we attempted to identify substitutions affecting protein function through prediction methods by using sorting intolerant from tolerant (SIFT) [25] and Protein Variation Effect Analyzer (PROVEAN) [26]. SIFT predicts whether the amino acid substitution alters the protein function. Chromosome, start position, references, and variants were submitted to SIFT as input. The intolerance range of SIFT is ≤0.05, thus indicating that a variant is damaging/deleterious to the protein function, and a score > 0.05 predicts the tolerance range. In African Americans, out of 671 mutations, 278 were predicted to be deleterious by PROVEAN, and 217 were predicted to be damaging by SIFT (Supplementary data 3). In Caucasians, out of 762 mutations, 231 missense mutations were predicted to be deleterious by PROVEAN, and 207 missense mutations were predicted to be damaging by SIFT (Supplementary data 4). We did not analyze prediction in the context of protein stability that causes deleteriousness of nonsynonymous mutations; however, further studies in the area could be performed to elucidate this aspect.
Discussion
The DDR genes (DNA “repairome”) protects genome stability. To address the importance of genomic instability in human prostate carcinogenesis, we deep sequenced the exons of 124 genes involved in the DNA damage repair/response in 52 PCas and matched normal tissue samples (peripheral blood lymphocytes), at very high resolution (312–768 average read depth). We found 1433 somatic mutations in these tumors (24–38 mutations per patient, depending on race and Gleason score). We further found that all prostate tumors had somatic mutations in DDR genes, regardless of the Gleason score. Somatic mutations included indels, stop-gain, LOH, and missense substitutions. Approximately 30–40% of these somatic mutations were predicted to be damaging in different types of software analysis and thus were unlikely to be “passenger” mutations. Control non-DNA repair genes were included in the analysis: AR was the 59th highest gene hit in African Americans, and TMPRSS2 was the 14th highest gene hit in Caucasians. Thus, many DNA repair genes are somatically mutated more often than well characterized targets in prostate tumors. We also found that somatic mutations in BRCA2, NEIL3, ATM, and ATR were associated with higher mutational burden in prostate tumors. The higher rate of somatic mutations in the DNA repairome in this study compared with previous publications [8] may be the result of: (a) the much higher read depth that we used and (b) the exclusion of samples with less than 50% tumor tissue (upon biopsy) in our analysis (other studies often use a 10–20% tumor cutoff [24]. We conclude that a mutation “hit” in a DNA repair gene appears to be an obligatory event in PCa, a finding consistent with the mutator phenotype hypothesis.
Our data also show that, although DNA repair genes are often “hit” in PCa, only some of these mutations are prevalent (clonal), and in only some tumors (Supplementary data 1), during primary prostate tumor development. Furthermore, the DNA repairome somatic mutation frequency does not directly correlate with the higher Gleason score (>7). These data are somewhat but not fully consistent with the mutator phenotype hypothesis for prostate tumor progression [27]. An alternative hypothesis consistent with these data is that mutator mutations may become clonal very late in tumor evolution, after successive “bottlenecks,” such as chemo/immunotherapy, changes in the tumor microenvironment, or metastasis. Until that “final clonality” point, intratumor genetic heterogeneity may actually regulate tumor caused mortality, by keeping “aggressive” tumor cells (also sometimes called “cancer stem cells”) [28] from dominating. In this model, tumors may resemble genetic Darwinian evolution landscapes in microscale. The presence of various tumor genotypes may reduce the chances of organismal mortality until successive bottlenecks result in the selection of a highly lethal genotype.
Another major goal of this study was to examine the issue of health disparities in PCa: African Americans have the highest incidence of PCa in the world. We found that part of this difference between African Americans and Caucasians may be due to the high heterogeneity in the somatic mutations of the DNA repairome: although the top somatically mutated genes were similar in both racial groups, the exact order and frequency of specific mutations (biomarkers) differed (Fig. 4). For example, the most common somatic mutation in Caucasians, p.E216K in POLB, found in 43% of patients, was found in only three African Americans. p.E589K of EXO1 and p.Q939K of XPC were identified in 42 and 36% of African American tumors only. Moreover, African Americans had more somatic mutations in DNA repair genes per tumor than Caucasians. This finding, together with the established higher incidence and mortality observed in African Americans, suggests that somatic mutations of the DNA repairome may be important in prostate tumor progression.
We identified mutations in all major DNA repair pathways in tumors in both African Americans and Caucasians. NEIL3 and BRCA1 were the top hits in Caucasians, and ATR was the top hit in African Americans. Some patients showed only LOH, whereas others showed only missense mutations. However, most patients had both LOH and missense mutations, often in different genes. The overall picture is very complex and heterogeneous. This finding is consistent with the analysis of whole genome studies in prostate tumors [29]. However, our study has certain limitations. Because very few samples had a Gleason score 6 or lower, or one higher than 8, we could not reach conclusions in the analysis of low vs high Gleason scores. Furthermore, very few patients were <40 years or >70 years old at diagnosis, and the incidence of DNA-repair gene mutations may differ between these age groups and the rest of the population. Lastly, we did not have information on the smoking habits of these patients, which might have affected the DNA repair process or mutations. In addition, ancestry of the samples was not determined. These limitations can be addressed is future studies.
A relatively recent method of targeting tumors is synthetic lethality. This approach relies on the sensitivity of tumors with, e.g., prevalent loss-of-function BRCA1/BRCA2 mutations to PARP1 or POLQ inhibitory drugs [30]. Indeed, owing to the inhibition of both the HR and NHEJ pathways, these tumor cells cannot repair double strand breaks. Recently, this approach has been found to be efficacious even for tumors displaying LOH at BRCA1/BRCA2, but not complete loss of function (presumably because the second hit remains unknown in these tumors) or mutations in genes such as ATM, ATR, PALB2, and FANCA [31, 32]. The synthetic lethality approach has also been extended to the use of other chemotherapeutic drugs like platinum [33]. We found that 20 (39%) patients with PCa had prevalent (i.e., >50% mutant) missense or frameshift somatic mutations at both (a) BRCA1 or BRCA2, and (b) POLQ, ATM, ATR, or PARP1. Of these 20 patients, 15 had LOH at BRCA1 or BRCA2 (Supplementary data 1). Furthermore, at least two of these prevalent missense mutations in each patient (including at least one in each of the HR and NHEJ pathways) has been reported to affect protein function, survival, outcome, or response to therapy (data not shown). We conclude that these somatic mutations may be good biomarkers for selecting patients to be treated with a synthetic lethality approach. The fact that 20–25% of metastatic prostate tumors have defects in the DNA repairome, mostly in the HR pathway [33], suggests that synthetic lethality may be an attractive approach for the treatment of advanced and/ or metastatic PCa.
We identified several recurrent somatic mutations in the DNA repairome in prostate tumors. These mutations may serve as important biomarkers of tumor progression. The most common mutation in Caucasians was p.E216K in POLB, which was found in 14 tumors (43%), whereas in African Americans, p.E589K in EXO1 was found in 8 tumors (42%). Both mutations have been reported in previous studies. The p.E216K mutation in POLB does not alter polymerase activity in vitro [15], but it has cellular effects in vivo (manuscript in preparation). The p.E589K mutation in EXO1 has been reported to affect the response to cisplatin treatment in patients with head and neck tumors [34], and thus may also be functional. Furthermore, all tumors with a Gleason score >7 contained either the p.E216K POLB mutation or a prevalent missense mutation in EXO1 (p.E589K, p.V458M, or p.H354R). One of these tumors also contained a prevalent missense mutation in XPC, p.Q939K, which has been reported to affect the response to cisplatin in patients with osteosarcoma [35]. The p.H354R mutation in EXO1 has been shown to affect survival in pancreatic cancer [36]. Thus, all these somatic mutations may be important biomarkers of prostate tumor progression.
As the cost of genomic technologies (such as GWAS and NGS) drops and studies demonstrating the importance of genetic screening for disease outcome amount, genomic profiling of tumors will become part of standard cancer care. Traditional thinking dictates that germline alterations are more important for cancer prognosis, since they predate tumors, while somatic alterations are more important for therapy, since they are more likely to contribute to therapeutic resistance, metastasis, and mortality. However, more recent reports suggest that with regards to the DDR pathway, both germline and somatic mutations are of therapeutic importance. For example, 12% of metastatic PCa patients have germline alterations in HR DDR genes, with BRCA1, BRCA2, and ATM, the most commonly affected genes [37]. More specifically, germline BRCA1/BRCA2 alterations have been associated with a higher Gleason score, metastases at diagnosis, rates of overall survival, and metastasis-free survival in PCa [37] as well as response to treatment [32]. Furthermore, the importance of germline variants in the DNA repairome is not confined to the prostate, but it has been shown in other tumors, such as colorectal and breast/ovarian [38]. Thus, future translational studies for the discovery of therapeutic biomarkers in the DNA repairome should interrogate both the germline and somatic variants.
In conclusion, through deep sequencing of highly pure tumor biopsies, we found that the DNA repairome was somatically mutated in all prostate tumors tested. Highly mutated genes included EXO1, ERCC6, POLQ, NEIL3, BRCA1, BRCA2, ATM, and ATR. We further observed that the well-known difference in PCa incidence and mortality between African Americans and Caucasians may have a genetic basis, specifically involving the DNA repairome. Indeed, tumors in African Americans, compared with Caucasians, had distinct mutations in the DNA repairome, a higher rate of somatic mutations overall, and major disparities in genes such as XPC, ATR, and MBD4. If validated, these data can be used to select important biomarkers of PCa progression, mortality and racial disparities, and to guide therapeutic options.
Methods and materials
Clinical data
PCa samples were obtained from Tulane University Hospital in New Orleans, LA, under ethical approval granted by Tulane University’s local research ethics committee. Prostate samples were obtained from patients who underwent radical prostatectomy for PCa. Each sample was microscopically verified for the presence of tumor by a pathologist and those samples containing more than 50% of tumor were selected for our study. Correlative clinical data were collected and entered into databases. DNA was extracted from 63 PCa tumors and 63 matched normal tissue samples (peripheral blood lymphocytes), and only samples found to be composed of >50% tumor cells, were included the study. Targeted exome sequencing was performed for all 63 tumor samples and 63 matched normal tissue samples; 11 samples were excluded because of low read coverage or failure to pass quality control. Specimens were grouped based on self-identified race.
Microdissection
Specimens were formalin fixed, embedded in paraffin, sectioned, and transferred on microscopic slides where they were deparaffinized and stained with hematoxylin and eosin. Selected populations of carcinoma cells were microdissected and tumor DNA was then extracted from the microdissected cells using established methods [39].
Genomic DNA (gDNA) extraction
DNA from normal tissue and peripheral blood lymphocytes was extracted with a DNeasy blood and tissue kit (Qiagen Inc, Valencia, CA), as described by the manufacturer. DNA from FFPE samples was extracted with the Qiagen/heating method. Then 18 0 μL ATL buffer was added to the tube, and samples were subjected to high-heat treatment at 90 °C for 20 min to melt the paraffin. After 20 min, the samples were incubated at RT for 3 min and quickly centrifuged. Then 20 μL of proteinase K was added, and samples were briefly vortexed and incubated at 56 °C for 16 h. The tubes were quickly centrifuged, and 200 μL of buffer AL and 200 μL of ethanol were added. The mixture was added to a DNeasy Mini spin column and centrifuged for 1 min at 8000 rpm. The following steps were performed according to the manufacturer’s protocol.
SureSelect custom target enrichment library preparation
We constructed exon baits for 124 DNA damage repair/response genes (Supplementary data 1). The length of each oligonucleotide of the customized bait was 120 bp, with 5× tiling. The average number of baits per target was 630.07, and the total number of baits was 36,544. We obtained RNA probes for the baits from Agilent SureSelect with a target size of 0.429230 Mb (SureSelect ELID 0308271, Agilent, Santa Clara, CA). The gDNA (50–500 ng) was diluted with 1× low Tris-EDTA buffer (10 ng/μL) and sheared with a Bioruptor (Diagenode). To obtain a target size of 200–350 bp, seven 30 s on/off cycles were applied. Agilent’s SureSelect XT Target Enrichment protocol version 1.5 was followed for library preparation with the following modifications. In brief, sheared DNA samples were purified with Agencourt AMPure XP magnetic Beads. DNA ends were repaired with an End it DNA repair kit (Epicentre Madison, WI), and adenine bases were added to the ends of repaired fragments (Ligation kit, NEB Ipswich, MA). Samples were purified with Agencourt AMPure XP magnetic beads, and indexing-specific paired-end adapters were ligated. The ligated library was then amplified with the Herculase II Fusion DNA Polymerase system and the following program: 2 min at 98 °C, six cycles of 30 s at 98 °C, 30 s at 65 °C, and 1 min at 72 °C, and 10 min at 72 °C. The library was hybridized with no modifications, as described by the manufacturer (SureSelect Agilent, Santa Clara, CA). Subsequently, post selection was performed with Streptavidin T1, and final extension was used to add the index. For indexing, the Herculase II Fusion DNA Polymerase 2 system was used with the following program: 2 min at 98 °C, 12 cycles of 30 s at 98 °C, 30 s at 57 °C, and 1 min at 72 °C, and 10 min at 72 °C. The indexed library was purified and analyzed with a 2100 Bioanalyzer (Agilent, Santa Clara, CA).
Massively parallel exome sequencing and alignment
Samples were sequenced at the John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine (Miami, FL) and UW Biotechnology Center (Madison, WI). Flow cells were prepared, and sequencing clusters were generated according to Illumina library protocols. Sequencing was performed with 99 base paired-end sequencing on Hiseq 2000 or Hiseq 2500 genome analyzers, in accordance with the Illumina Genome Analyzer operating manual. The average sequence coverage was 712-fold for normal samples and 368-fold for tumor samples (Supplementary data 2).
Downstream analysis of exome sequencing
Downstream analysis of samples was performed at the Tulane Cancer Center at the Crusaders Next Generation Sequence Analysis Core. Before downstream analysis, quality control checks on raw sequence data were run with FastQC. To generate the prerequisite input BAM file for SAMtools mpileup function, we aligned paired-end reads to the reference human genome (GRCh37) with STAR Aligner. The BAM files were then sorted and indexed with a reference sequence.
Germline variant calling and somatic mutation detection by VarScan2
Variant calling was performed with the SAMtools mpileup function with the command samtools mpileup -f [reference sequence] [parameters] [BAM file(s)] >myData.mpileup. In this study, we present data for tumor samples and matched (normal) controls to distinguish acquired/somatic mutations (<0.01% of variants) from inherited germline variation (>99.99% of variants). As described by the developer [40], the VarScan somatic command accepts mpileup input from a normal and tumor sample (in that order); at every position meeting the minimum coverage requirement, it calls both samples independently to identify possible variants such as germline, LOH, or somatic variants. We also specified the threshold –somatic-p-value (Fisher’s exact test) of the read counts supporting reference and variant alleles in normal samples to determine the statistical significance between normal and tumor samples. The following two commands were used, as described by the developer [40]:
To run SAMtools mpileup on the BAM files for normal and tumor samples:
samtools mpileup –B –q 1 –f reference.fasta normal.bam tumor.bam >normal-tumor.mpileup
To detect somatic mutations, with VarScan run on the mpileup output file:
java –jar VarScan.jar somatic normal-tumor.mpileup output.basename –min-coverage 10 –min-var-freq 0.08 –somatic-p-value 0.05,
where (normal-tumor.mpileup) is the mpileup output file, and (output.basename) is the basename for the VarScan output files.
Functional annotation of genetic variants by ANNOVAR
We used ANNOVAR (http://www.openbioinformatics.org/annovar/) to annotate single-nucleotide variants and insertions/deletions, to examine their functional consequences on genes, or to identify variants reported in dbSNP [41].
Prediction of variant effects
We applied SIFT (http://sift-dna.org) and PROVEAN (http://provean.jcvi.org) to predict whether a given amino acid substitution affects protein function.
Statistical test
The validation status was determined by comparing tumor/normal read counts for each allele with VarScan. For validation, we had more than 50 reads with base quality ≥15 (Phred score) for both normal and tumor samples. The somatic p value significance threshold was set as <0.05 and was calculated by VarScan with Fisher’s exact test. Mutation rates in DNA-repair gene mutations and age, race, or Gleason score were evaluated with Fisher’s exact test.
References
Bostwick DG, Burke HB, Djakiew D, Euling S, Ho SM, Landolph J, et al. Human prostate cancer risk factors. Cancer. 2004;101:2371–490.
Gronberg H. Prostate cancer epidemiology. Lancet. 2003;361:859–64.
Gaines AR, Turner EL, Moorman PG, Freedland SJ, Keto CJ, McPhail ME, et al. The association between race and prostate cancer risk on initial biopsy in an equal access, multiethnic cohort. Cancer Causes Control. 2014;25:1029–35.
Powell IJ. Epidemiology and pathophysiology of prostate cancer in African-American men. J Urol. 2007;177:444–9.
DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Cancer statistics for African Americans, 2019. CA Cancer J Clin. 2019;69:211–33.
Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med. 2003;349:366–81.
Nelson WG, Demarzo AM, Yegnasubramanian S. The diet as a cause of human prostate cancer. Cancer Treat Res. 2014;159:51–68.
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9.
Fox EJ, Prindle MJ, Loeb LA. Do mutator mutations fuel tumorigenesis? Cancer Metastasis Rev. 2013;32:353–61.
Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, Ghori M, et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet. 2018;50:682–92.
Burgers PMJ, Kunkel TA. Eukaryotic DNA replication fork. Annu Rev Biochem. 2017;86:417–38.
Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85.
Caglayan M, Wilson SH. Oxidant and environmental toxicant-induced effects compromise DNA ligation during base excision DNA repair. DNA Repair. 2015;35:85–9.
Ross CA, Truant R. DNA repair: a unifying mechanism in neurodegeneration. Nature. 2017;541:34–35.
An CL, Chen D, Makridakis NM. Systematic biochemical analysis of somatic missense mutations in DNA polymerase beta found in prostate cancer reveal alteration of enzymatic function. Hum Mutat. 2011;32:415–23.
Makridakis NM, Caldas Ferraz LF, Reichardt JK. Genomic analysis of cancer tissue reveals that somatic mutations commonly occur in a specific motif. Hum Mutat. 2009;30:39–48.
Yadav S, Mukhopadhyay S, Anbalagan M, Makridakis N. Somatic mutations in catalytic core of POLK reported in prostate cancer alter translesion DNA synthesis. Hum Mutat. 2015;36:873–80.
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–53.
Marx J. Debate surges over the origins of genomic defects in cancer. Science. 2002;297:544–6.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8.
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13.
Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37:590–2.
Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci USA. 2003;100:776–81.
Shi W, Ng CKY, Lim RS, Jiang T, Kumar S, Li X, et al. Reliability of whole-exome sequencing for assessing intratumor genetic heterogeneity. Cell Rep. 2018;25:1446–57.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22.
Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–45.
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–24.
Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7:20–37.
Christenson ES, Antonarakis ES. PARP inhibitors for homologous recombination-deficient prostate cancer. Expert Opin Emerg Drugs. 2018;23:123–33.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708.
Athie A, Arce-Gallego S, Gonzalez M, Morales-Barrera R, Suarez C, Casals Galobart T, et al. Targeting DNA repair defects for precision medicine in prostate cancer. Curr Oncol Rep. 2019;21:42.
Nogueira GAS, Costa EFD, Lopes-Aguiar L, Lima TRP, Visacri MB, Pincinato EC, et al. Polymorphisms in DNA mismatch repair pathway genes predict toxicity and response to cisplatin chemoradiation in head and neck squamous cell carcinoma patients. Oncotarget. 2018;9:29538–47.
Caronia D, Patino-Garcia A, Milne RL, Zalacain-Diez M, Pita G, Alonso MR, et al. Common variations in ERCC2 are associated with response to cisplatin chemotherapy and clinical outcome in osteosarcoma patients. Pharmacogenomics J. 2009;9:347–53.
Dong X, Jiao L, Li Y, Evans DB, Wang H, Hess KR, et al. Significant associations of mismatch repair gene polymorphisms with clinical outcome of pancreatic cancer. J Clin Oncol. 2009;27:1592–9.
Schweizer MT, Antonarakis ES. Prognostic and therapeutic implications of DNA repair gene mutations in advanced prostate cancer. Clin Adv Hematol Oncol. 2017;15:785–95.
Lin PC, Yeh YM, Wu PY, Hsu KF, Chang JY, Shen MR. Germline susceptibility variants impact clinical outcome and therapeutic strategies for stage III colorectal cancer. Sci Rep. 2019;9:3931.
Akalu A, Reichardt JK. A reliable PCR amplification method for microdissected tumor cells obtained from paraffin-embedded tissue. Genet Anal. 1999;15:229–33.
Koboldt DC, Larson DE, Wilson RK. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr Protoc Bioinform. 2013;44:15.4.1–17.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Acknowledgements
This work was supported by PC094628 and NIH grant 8P20 GM103518 (NM) and Carol Lavin Bernick grant (MA).
Author information
Authors and Affiliations
Contributions
NM and SY conceived the experiments and wrote the manuscript. SY also performed exome sequencing downstream analysis. MA, MB, CW, WJ, and VKC performed exome sequencing downstream analysis. SM performed tumor DNA isolation. EF provided analytical advice, and KM provided prostate tumors and normal tissue resources.
Corresponding authors
Ethics declarations
Conflict of interest
NM is an employee of 1Globe Health Institute and declares no equity ownership.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yadav, S., Anbalagan, M., Baddoo, M. et al. Somatic mutations in the DNA repairome in prostate cancers in African Americans and Caucasians. Oncogene 39, 4299–4311 (2020). https://doi.org/10.1038/s41388-020-1280-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-1280-x
This article is cited by
-
Genetic and biological drivers of prostate cancer disparities in Black men
Nature Reviews Urology (2023)
-
A novel RBBP8(p.E281*) germline mutation is a predisposing mutation in familial hereditary cancer syndrome
Journal of Molecular Medicine (2023)
-
The potential of cell-free and exosomal microRNAs as biomarkers in liquid biopsy in patients with prostate cancer
Journal of Cancer Research and Clinical Oncology (2022)
