
Abstract
NMDA receptor antagonists have a vital role in extinction, learning, and reconsolidation processes. During the reconsolidation window, memories are activated into a labile state and can be reconsolidated in an altered form. This concept might have significant clinical implications in treating PTSD. In this pilot study we tested the potential of a single infusion of ketamine, followed by brief exposure therapy, to enhance post-retrieval extinction of PTSD trauma memories. 27 individuals diagnosed with PTSD were randomly assigned to receive either ketamine (0.5 mg/kg 40 min; N = 14) or midazolam (0.045 mg/kg; N = 13) after retrieval of the traumatic memory. 24 h following infusion, participants received a four-day trauma-focused psychotherapy. Symptoms and brain activity were assessed before treatment, at the end of treatment, and at 30-day follow-up. Amygdala activation to trauma scripts (a major biomarker of fear response) served as the main study outcome. Although PTSD symptoms improved equally in both groups, post-treatment, ketamine recipients showed a lower amygdala (−0.33, sd = 0.13, 95%HDI [−0.56,−0.04]) and hippocampus (−0.3 (sd = 0.19), 95%HDI [−0.65, 0.04]; marginal effect) reactivation to trauma memories, compared to midazolam recipients. Post-retrieval ketamine administration was also associated with decreased connectivity between the amygdala and hippocampus (−0.28, sd = 0.11, 95%HDI [−0.46, −0.11]), with no change in amygdala-vmPFC connectivity. Moreover, reduction in fractional anisotropy in bi-lateral uncinate fasciculus was seen in the Ketamine recipients compared with the midazolam recipients (right: post-treatment: −0.01108, 95% HDI [−0.0184,−0.003]; follow-up: −0.0183, 95% HDI [−0.02719,−0.0107]; left: post-treatment: −0.019, 95% HDI [−0.028,−0.011]; follow-up: −0.017, 95% HDI [−0.026,−0.007]). Taken together it is possible that ketamine may enhance post-retrieval extinction of the original trauma memories in humans. These preliminary findings show promising direction toward the capacity to rewrite human traumatic memories and modulate the fear response for at least 30 days post-extinction. When combined with psychotherapy for PTSD, further investigation of ketamine dose, timing of administration, and frequency of administration, is warranted.
Similar content being viewed by others

Introduction
Reactivation of a stored memory (i.e., bringing the previously encoded memory back into consciousness), might change its state from consolidated into a labile, in which its content or meaning may be altered. In a process known as reconsolidation, the memory is then stored again in this altered form [1]. This process is especially relevant to post-traumatic stress disorder (PTSD) as translational theories link the disorder to fear learning and updating [2, 3]. One of the signature symptom clusters in PTSD comprises overgeneralization of fear and occasional re-experiencing of the traumatic memory with its original emotional intensity and vividness. Memory retrieval, either spontaneous or via deliberate exposure [4], may open a time window in which adaptive or maladaptive memory modifications can promote either attenuation or persistence of PTSD symptoms [1, 5].
In the laboratory, fear learning is typically modeled using a paradigm in which neutral cues are paired with aversive outcomes (e.g, electric shocks) and acquire aversive value [6]. Following fear acquisition, repeated presentation of cues in the absence of outcome leads to the extinction of fear responses. But these responses usually return, either spontaneously or following further exposure to the cue or the aversive outcome [7, 8], suggesting that the original memory is not substantially altered. Extinction is likely achieved through inhibition of amygdala fear responses by the ventromedial prefrontal cortex (vmPFC) [9]. However, if extinction training is conducted within the reconsolidation window (at least 10 min but less than 6 h after reactivation [10, 11]), attenuation of fear can be long-lasting [8, 9] and likely involves modification of the original memory [12, 13]. Such post-retrieval extinction does not appear to rely on prefrontal mechanisms [14, 15], and may directly target the memory trace in the amygdala [1, 16, 17]. Similar mechanism may operate in exposure therapy when the trauma memory is recalled into a labile state. During this state the traumatic memory is processed with the therapist so it can be re-consolidated in a new form [18]. This process helps the patient to understand that they are not re-exposed to the actual event, they are in a safe environment, and that the original emotional memory-traces in the brain can be altered.
Functional connectivity between the amygdala and hippocampus may also play an important role in the processing of aversive memories [15, 19, 20]. In humans, the functional coupling of the amygdala and hippocampus was associated with the return of fear, with higher coupling after extinction that occurred outside of the reconsolidation window, compared to extinction within the reconsolidation window [15]. In rodents, increased coupling between dorsal hippocampus and amygdala basolateral nuclei was observed during non-REM sleep following a threat task [19].
Anatomical connectivity was also found to play a role in fear conditioning. In vivo magnetic resonance diffusion tensor imaging (DTI) in rodents showed alterations in fractional anisotropy (FA) in the amygdala and hippocampus following fear conditioning. In humans, the uncinate fasciculus (UNC), a white matter bundle connecting the orbito-frontal cortex with limbic structures within the anterior temporal lobe (i.e., amygdala and hippocampus) [21], was found to be related to fear conditioning. Rapid increase in right UNC FA was found to be correlated with decreased response to conditioned stimulus [22]. In contrast, reduced UNC FA was found in people with PTSD diagnosis [23] as well as subthreshold PTSD [24].
Extinction may be further enhanced with the use of ketamine, a non-competitive N-methyl-D-aspartate glutamate receptor (NMDAR) antagonist. The NMDAR has a key role in learning, extinction, and reconsolidation in both animals and humans [25,26,27]. Moreover, accumulating evidence suggests that on the molecular level, ketamine, in sub-anesthetic doses, promotes neurogenesis [28, 29], cell proliferation [30], and synaptogenesis [29, 31], all of which are important in reconsolidation processes [32, 33]. On the white matter bundle level, ketamine was found to promote a rapid (hours) increase in white matter [34], with a decline over longer periods (months) [35, 36]. However, long-term changes in white matter were tested only with chronic use of ketamine and not with sub-anesthetic dose. A recent study reported a dramatic decline in PTSD symptoms following multiple ketamine infusions, compared to midazolam [37], but the effect was only transient with a median time for loss of response of 27.5 days.
Recent studies have demonstrated the potential of post-retrieval extinction - using behavioral and pharmacological agents - to reduce fear responses. In spider phobia, repeated exposure to a spider image after fear reactivation attenuated amygdala response to that image 24 h later [38]. Administering propranolol (a β-blocker) after reactivation of fear of spiders resulted in decreased avoidance response and increased approach behavior in spider-fearful people [39]. Lastly, administration of propranolol before exposure therapy yielded a steeper decline in PTSD symptoms at follow-up [40]. Taken together, an intriguing possibility is that combining ketamine infusion with psychotherapy will have a synergistic effect [41]. The idea of combining ketamine with psychotherapy was also examined in pain and other mental disorders. Recent systematic review on ketamine-assisted psychotherapy (KAP) found heterogeneity in the use and administration of KAP, with some studies administering ketamine before, during or after the psychotherapy. Results suggest that psychotherapy might prolong the short-term effect of ketamine [42]. In PTSD, a small (n = 5 in each group), randomized control trial combining one time ketamine infusion with Trauma Interventions using Mindfulness Based Extinction and Reconsolidation (TIMBER) psychotherapy (3 session in first week, and 9 weekly sessions) found better response and prolonged effect to the TIMBER + ketamine group, compared to control [43].
In this initial investigation, we explored the ability of a single subanesthetic intravenous infusion of ketamine (0.5 mg/kg over 40 min) to enhance post-retrieval extinction of real traumatic memories. To control for the subjective effects of ketamine, we randomized individuals with PTSD to receive an infusion of either ketamine or the benzodiazepine midazolam. We have used active control in order to keep the participants blind. The use of midazolam in such studies and reasoning was reported and discussed in previous published work [37, 44]. As ketamine was found to promote reconsolidation processes, the infusion was followed by an extinction-reconsolidation focused four-day exposure-based therapy (which was initiated 24 h post-infusion while BDNF level peaks [28, 29]).
The aim of this novel pilot exploration was to identify neural biomarkers for post-retrieval extinction of the original traumatic memory for a potential translational clinical trial. Our main hypothesis focused on the amygdala; we predicted a greater reduction in amygdala reactivity to trauma cues in individuals with PTSD who received ketamine, compared to those who were administered midazolam. As the target-engagement mechanism was of reconsolidation-based extinction (and not classic extinction), we expected no change in vmPFC activation or its functional coupling with the amygdala. Moreover, we hypothesized that if ketamine produces greater post-retrieval extinction than midazolam, this effect will also be associated with a reduction in functional connectivity between the amygdala and the hippocampus. We also hypothesized that ketamine will induce alterations on white matter bundles connecting the frontal and temporal lobes. Lastly, we hypothesized that ketamine will be associated with greater reduction in PTSD symptoms than midazolam when combined with exposure treatment.
Methods and Materials
Participants
Of the 118 participants who signed informed consent and screened, 33 were eligible and 28 were randomized (see CONSORT in Supplemental). One subject voluntarily left the experiment immediately after the infusion, resulting in 27 participants who were included in the analyses (mean age = 37.8, SD = 10.7, range = 24–63; 14/13 subjects in ketamine/midazolam groups respectively; females (n = 10), males (n = 17)). All participants had chronic PTSD (more than 1-year), with the average time since trauma being 12.11(∓8.62) years in the ketamine group and 13.88(∓11.18) years in the midazolam group. Out of the 28 participants, two identified as Black, two as Hispanic, one as American Indian, one as Asian and one as Native Hawaiian. Two didn’t state their race or ethnicity. See Table 1 for sample characteristics.
The study was registered in clinicaltrials.gov (NCT02727998). Important to notice that while the primary outcome registered in the clinicaltrial.gov was PCL scores, one of the main outcomes in the NARSAD Independent Investigator Award (submitted before trial registration and data collection) was neural biomarker (see Supplemental material Original funded NARSAD grant application Aim 2). The neural mechanize is crucial toward an NIH grant application but this outcome was not registered in error.
Exclusion criteria included a diagnostic history of bipolar disorder, borderline personality disorder, obsessive-compulsive disorder, schizophrenia or schizoaffective disorder, or current psychotic features as determined by the Structured Clinical Interview for DSM-IV (SCID) [45]; dementia was also an exclusion criterion, as were current suicide risk, moderate or higher severity of substance use disorder in the 3 months prior to randomization, and history of mild-to-severe traumatic brain injury (TBI). Participants who were currently engaged in trauma focus therapy were also ineligible to participate in the study. Lastly, patients were excluded for acute medical illness. PTSD diagnosis was established using the Clinician-Administered PTSD Scale (CAPS-5) [46]. The PTSD Checklist for DSM-5 (PCL-5) was used to monitor changes in PTSD symptoms over time [47].
Randomization
Randomization was managed by the Investigational Drug Services (IDS) Pharmacy at Yale New Haven Health, which also prepared the study drugs for infusion. The entire study team was blind to the conditions. Participants were randomized in counterbalanced blocks of 10-subjects each stratified by gender.
Retrieve and reprocess exposure therapy
The therapy used in this study consisted of five sessions in total. The first session, conducted before the MRI scans, was a 2 h long meeting where the patient’s life history was taken with a focus on the index trauma. The session also included information on the disorder and the building of an exposure hierarchy, following the guidelines outlined in the prolonged exposure manual [45]. The following four sessions were conducted by an experienced therapist (O.D., I.H.R), each lasting 90–120 min. During the session patients were asked to recall and discuss their traumatic memory through imaginal exposure. The therapy also included processing of the trauma, during which the therapist and the patient discussed the implications of the trauma and ways to reframe the patient’s understanding of the event, and their behavior during the event. Additionally, during each of these four sessions, in-vivo exposure was conducted with a member of the research team, based on the exposure hierarchy constructed with the therapist. The length of psychotherapy was based on two major factors: (1) we wanted to harvest the long-term effects of a single infusion of ketamine, which diminishes after seven days (see discussion) and (2) the promising results of a one-week trauma-focused psychotherapy intervention [48] which allowed us to condense the entire study protocol to accommodate a regular working week.
Procedure
Eligible participants completed an imagery-development procedure in which they described the traumatic event associated with their PTSD (Criteria A), as well as a sad event and an event in which they felt relaxed for details on the procedure, see [49] for details. Using this information, we developed a 120 s audiotape script of each event, narrated by a male member of the research staff.
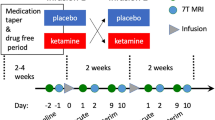
A day after psychoeducation and building an in-vivo exposure hierarchy session, participants were scanned in the MRI during recall of their traumatic, sad and relax event. The scripts were presented three times, in a fixed order, to avoid ending with the traumatic script. Immediately following script replay, infusion of either ketamine (0.5 mg/kg) or midazolam (0.045 mg/kg) began inside the MRI and lasted for 40 min. The following four days included exposure-based psychotherapy with an experienced clinician (OD, or IHR) in which trauma memory was recalled and reconsolidated after processing. The psychotherapy sessions were accompanied by in-vivo exposure, corresponding to the patients’ avoidance behavior. A week after infusion, participants went through an MRI with a similar procedure to the baseline, excluding the infusion itself. Lastly, we followed participants up for 30 and 90 days. Due to COVID restrictions, 90 days follow-up was mostly done using remote questionnaires, so MRI data is valid for the baseline, 7 and 30 days. See Fig. 1 for study procedure. The study was approved by the Yale University Institutional Review Board (IRB; 1509016530).

Days 3–6 = days 3,4,5 and 6 in which the PE sessions were conducted. Recall procedure inside the magnet included 3 rounds of each audio recall script (traumatic, sad, and relax, 120 seconds each).
MRI Scans
MRI data were collected with a Siemens 3 T Prisma scanner, using a 32-channel receiver array head coil. High-resolution structural images were acquired by Magnetization-Prepared Rapid Gradient-Echo (MPRAGE) imaging (TR = 1.9 s, TE = 2.77 ms, TI = 900 ms, flip angle = 9°, 176 sagittal slices, voxel size = 1 × 1 × 1 mm, 256 × 256 matrix in a 256 mm FOV). Functional MRI scans were acquired while the participants were listening to the narrated scripts, using a multi-band Echo-planar Imaging (EPI) sequence (multi-band factor = 4, TR = 1000 ms, TE = 30 ms, flip angle = 60°, voxel size = 2 × 2 × 2 mm, 602 mm-thick slices, in-plane resolution = 2 × 2 mm, FOV = 220 mm). For the diffusion-weighted images (DWIs) b-value was set at 1000 s/mm2 with an acquisition of a reference image (b = 0). 64 directions were scanned, with the phase-encoding gradient applied in the anterior–posterior (AP) direction.
MRI Preprocessing
Data were preprocessed with Fmriprep, version 1.5.8 [50]. For a complete preprocessing procedure please see supplement 5. For information on motion parameters and the comparison between groups, see supplement 3, Table S1.
Activation level analysis
For each of the three fMRI sessions, a first-level analysis comparing the first 60 s of the first traumatic script vs. the first 60 s of the first relax script was conducted using FSL 6.0.3 [51], through a Nipype pipeline [52]. We have regressed out DVARS, framewise displacement and the first 6 anatomical component correlations. To avoid capturing habituation effects, we chose to focus on the first 60 s of the first presentation of each script.
The resulting contrasts of parameter estimates (COPE), after z-scoring, were then used to assess specific regions of interest (ROIs) in line with our preliminary hypothesis (i.e., the amygdala, hippocampus, and ventromedial prefrontal cortex (vmPFC)). All ROIs masks were taken from neuroSynth [53]. Mask of each ROI used is presented in the relevant figure.
Each subject’s COPE was masked using the relevant ROI and averaged across all voxels of that region. The extraction of activation for specific ROIs was conducted using Nilearn [54]. Statistical analyses were conducted using python, and Bayesian comparison of groups was done using the pyMC3 package [55]. Lastly, we compared group differences in the same brain regions during the sad (vs. relax) scripts using the same method. This was done to account for an alternative explanation: that the results we see are general to all negative emotions, and not specific to trauma-related ones.
Functional Connectivity analysis
To assess connectivity between different ROIs, we used the DiFuMO atlas [56], including 256 regions, among them the amygdala, hippocampus (anterior and posterior), and ventromedial prefrontal cortex (vmPFC; and vmPFC anterior; see supplement 1 for region maps). We employed the python package Nilearn [54] to extract time series, using low-pass filtering of 0.1 Hz and high-pass filtering of 0.01 Hz, and regressing out framewise displacement, CSF, white-matter volume, 6 rotation and translations variables, and first six anatomical components based noise correction (CompCor). Then, we extracted the pearson correlation between those regions from the first 60 s of the first traumatic script. We applied pyMC3 [55] to compare changes in correlation between the relevant ROIs (amygdala, anterior and posterior hippocampus, anterior vmPFC, and vmPFC). Lastly, we conducted the same procedure on the first 60 s of the sad script (as we did in the activation analysis), in order to test whether these effects are related to negative emotions in general or to trauma-related ones specifically.
Diffusion-weighted images analysis
Diffusion-weighted images analysis was done using standard TRACULA (TRActs Constrained by UnderLying Anatomy) protocol [57, 58]. TRACULA is an automatic WM pathways reconstruction tool. It uses global probabilistic tractography [59] with anatomical priors to reconstruct major WM tracts such as the UNC [57]. TRACULA preprocessing includes image corrections for eddy currents, simple head motions, B0 distortion correction, and computation of the diffusion tensor. The affine intra-subject registration with the T1-weighted images was done using Freesurfer’s “bbregister” [60]. Inter-subject registration was done using the affine inter-subject registration to MNI152 space. The TRACULA standard procedures were then performed to fit the ball-and-stick model of diffusion to the diffusion-weighted images and for probabilistic tracking and track segmentation. For each participant, following visual inspection of reconstructed tracts, we extracted diffusion tensor mean fractional anisotropy (FA), estimated with dtifit, in bi lateral uncinate fasciculus. Motion parameter was calculated as the sum of deviations from average rotation and average translation [58] (for information on motion parameters and comparison between groups, see supplement 3, Table S1). Percent bad slices was equal to 0 and average dropout score was equal to 1 in all scans.
Statistical approach
In accordance with recent guidelines and findings [61,62,63], analysis in this manuscript was based on a Bayesian approach. DTI analysis was conducted using brms, all other analyses were conducted using PyMC3, probabilistic programming packages in R and python [55], respectively. This also promotes better measures of uncertainty in the findings, which is most critical in small sample sizes [64]. To assess the difference in reactivation (as well as functional connectivity) to the traumatic memory, we have used a Bayesian multilevel model. The model included the ROI (amygdala, hippocampus, vmPFC) activation as the dependent variable. Activation of the ROI at baseline was entered as a covariate into the model (to account for possible random differences between the groups at baseline). The model included the group (ketamine/midazolam) and the interaction of time and group; lastly, subjects were entered as random intercept (to account for the within-subject random effects). As activation at baseline was entered as covariate, we expected to see a group effect (i.e., a difference between ketamine and midazolam at the end of treatment and at follow-up). Here we report the difference between the posterior distribution of each group at T2 (end of treatment) and T3 (30 days).
Effect sizes were calculated by dividing this difference between distributions with the total unexplained variance of the model. A difference was considered robust (i.e., significant) when the 95% High Density Interval (HDI) was excluding zero (i.e. either the interval is completely below or above zero) [65]. To promote better inference, we also report the Bayes-Factor of each of the effect sizes in each analysis. Bayes Factor tests what is the probability that our hypothesis (i.e., the difference between the groups) is true, given the data, compared to the null hypothesis (Bayes Factor 1 over 0, i.e., BF10). The calculation gives the reader another aspect of the strength of the evidence. BF10 > 3 is considered robust, whereas BF10 < 1 suggests that the null hypothesis is more probable.
Assessment the changes in FA in the right and left UNC were done in R, using Bayesian multilevel regression models (BMLMs) as implemented in the ‘brms’ package [66]. HDI were calculated using the coda package [67]. Results are considered robust (i.e., significant) if HDI does not include zero. FA residuals controlling for age, gender, and motion were calculated and baseline corrected to account for initial differences in FA values. In an attempt to improve convergence while reducing overfit, specified mildly informative conservative priors were applied with priors set as a student T distribution mean of 0 and standard deviation of 1 to reduce the effects of outliers [68, 69]. All parameters indicated a good fit of the data (No divergences, Rhat < 1.01 and ESS > 1000).
Originally, based on classic power calculations (using G*power) [70] the required sample size to detect an effect size of 0.8 (commonly observed in psychotherapy) was determined to be 40 (20 for each group), with 90% power. Due to COVID restrictions, data collection was halted earlier. Using the same analysis (G*power), power was reduced to 80%. As we used a Bayesian model, we calculated the relevant power for our fMRI using simulated data. We have simulated a different number of observations per group to test our power (i.e., the proportions of a robust effect, given the sample size). Results suggest that in a sample size of 25, given an effect size of 0.6 (one-tailed hypothesis), our power is 90%. For details on the simulation analysis, see supplement 4. Full analysis scripts can be found here: https://github.com/orduek/KPE.
Results
Changes in PTSD symptoms
PCL-5 scores were recorded before treatment, at the end of treatment, and at 30 and 90 days follow-up. Although PTSD symptoms significantly and robustly improved over time [before treatment, M = 46.6, sd = 13.35 end of treatment (7 days after infusion): M = 33.8, sd = 18.37; 30-day follow-up: M = 30.79, sd = 15.79, 90-day: M = 31.04, sd = 17.55] there was no significant difference between the ketamine and midazolam groups in the rate of improvement or in the PTSD score at the end of treatment and follow-up (mean difference = 0.41, 95% HDI [−1.42, 2.31]). Comparing symptoms at the end of treatment with symptoms before treatment showed significant difference (mean difference = 12.56, 95%HDI [21.84,3.15], effect-size [95%HDI] = 0.8 [0.23,1.39], BF10 = 10.65). Comparing 30 days follow-up to baseline showed significant difference as well (mean difference = 16.1, 95%HDI [25.33, 6.48], effect-size [95%HDI] = 1.00[0.40,1.60] BF10 = 35.76). The 90 days follow-up showed stability with difference from before treatment = 15.9 95%HDI [25.74,6.35], effect-size [95%HDI] = 0.98[0.38,1.60], BF10 = 32.59; supplement 2, fig. S1.
Neural m echanism
Based on our hypotheses, the main analysis focused on activation and connectivity patterns of the amygdala. In each ROI (i.e amygdala, hippocampus, vmPFC), we contrasted activation during the traumatic script with activation during the relax script at each timepoint. Here we report the effects derived from the interaction between time and medication group.
Amygdala reactivity
Activation difference for traumatic vs. relax memories was lower in the ketamine group compared to the midazolam group at the end of treatment, with mean activation of ketamine group −0.15 (sd = 0.33; N = 14), mean activation of midazolam group 0.18 (sd = 0.3; N = 12) and mean posterior difference: −0.33, sd = 0.13, 95%HDI [−0.56,−0.04]; effect size [95%HDI] = 1.00 [0.16,1.86], BF10 = 7.45). This difference was marginally significant at the 30-day follow-up (ketamine: −0.14 (sd = 0.35; N = 12), midazolam: 0.15 (sd = 0.38; N = 10), mean difference: −0.28, sd = 0.14, 95%HDI [−0.56, 0.01], effect size [95%HDI] = 0.87 [−0.01,1.75], BF10 = 2.58; Fig. 2). There were no group differences in amygdala activation during the sad imagery scripts at any time point (supplement 2, fig. S2).

A Differences between the ketamine and midazolam groups in amygdala reactivity to trauma vs. relax scripts across the three-time points. Each dot is a participant, the horizontal line in the middle of the boxplot represents median. On the right of each panel is the posterior distribution of the difference between the groups. The black line is the 95% HDI. B Average amygdala reactivity to trauma vs. relax in the ketamine group (blue) and the midazolam group (orange) in the three-time points. Error bars represent standard error of the mean. Brain image depicts the mask of the amygdala ROI (center: MNI [−22,0,−20], [22,0,−20]).
Hippocampus reactivity
Hippocampal activation to trauma vs. relax memories was marginally lower in the ketamine (mean = −0.11, sd = 0.38; N = 14) compared to the midazolam (mean = 0.18, sd = 0.53; N = 12) group at the end of treatment, with mean difference −0.3 (sd = 0.199), 95%HDI [−0.65, 0.04], effect size [95%HDI] was 0.71 [−0.12, 1.49], BF10 = 1.77. There was no group difference in the 30-day follow-up (ketamine: −0.15 (sd = 0.38; N = 12), midazolam: 0.01 (sd = 0.42; N = 10), mean difference −0.17, sd=0.18, 95%HDI [−0.54, 0.21]), effect size [95%HDI] was 0.39 [−0.48, 1.25], BF10 = 0.65; see Fig. 3. There were no group differences in hippocampus activation of the sad imagery script at any time point (supplement 2 fig. S3).

A Differences between the ketamine and midazolam groups in hippocampus reactivity to trauma vs. relax scripts across the three time points. Each dot is a participant, the horizontal line in the middle of the boxplot represents median. On the right of each panel is the posterior distribution of the difference between the groups. The black line is the 95% HDI. B Average hippocampus reactivity to trauma vs. relax in the ketamine group (blue) and the midazolam group (orange) in the three time points. Error bars represent standard error of the mean. Brain image depicts the mask of the hippocampus ROI (center: MNI [−28,−18,−16], [28,−18,−16]).
vmPFC reactivity
We found no group differences in vmPFC activation to traumatic imagery scripts (compared to relax) at any time point. There was no group difference in vmPFC activation during the sad imagery scripts compared to the relax ones at any of the three-time points. For a full description of the vmPFC activation results see supplement 2 fig. S4, S5.
Amygdala-vmPFC functional connectivity
Functional connectivity (FC) between the amygdala and vmPFC was not significantly different between the groups at any time point. For details, please see supplement 2 fig. S6.
Amygdala - Hippocampus functional connectivity
Functional connectivity (FC) between the amygdala and posterior hippocampus at the end of treatment was significantly lower in the ketamine group (mean 0.15, sd = 0.32; N = 14) than in the midazolam group (mean 0.43, sd = 0.18; N = 12), with mean difference: −0.285, sd = 0.11, 95%HDI [−0.50, −0.05], effect size [95%HDI] = 1.06 [0.18,1.92], BF10 = 7.59. At 30-days follow-up we found no difference between the ketamine (mean FC: 0.37, sd = 0.26; N = 12) and midazolam (mean FC: 0.29, sd = 0.35; N = 10) groups (mean difference: 0.08, sd = 0.15, 95%HDI [−0.17, 0.32], effect size [95%HDI] = 0.43 [−1.14.,0.62], BF10 = 0.55). A similar analysis of the connectivity between the amygdala and anterior hippocampus revealed no group differences (supplement 2). There were also no group differences in amygdala-posterior hippocampus functional connectivity during the sad imagery script at any of the three-time points (supplement 2 fig. S7).
Alterations in White matter
A robust interaction between session (baseline, post-treatment, 30-day follow-up) and drug on FA in the right UNC (post-treatment interaction coefficient: −0.01, 95% HDI [−0.02, −0.002]; follow-up interaction coefficient: −0.01, 95% HDI [−0.013,−0.003]), suggests a differential response to drug across sessions. In order to investigate the interaction, each session was analyzed separately. Ketamine showed a robust reduction in FA in the right UNC compared with midazolam group that lasts for at least 30 days (post-treatment: −0.011, 95% HDI [−0.018,−0.003]; follow-up: −0.018, 95% HDI [−0.027,−0.01]). Figure 4a shows the right UNC WMI corrected for age, gender and motion across time. Analyzing the left UNC, yielded similar results, with a robust interaction between session and drug on FA in the left UNC (post-treatment interaction coefficient: −0.019, 95% HDI [−0.035, −0.004]; follow-up interaction coefficient: −0.017, 95% HDI [−0.032,−0.002]). In order to investigate the interaction, each session was analyzed separately. Ketamine showed a robust reduction in FA in the right UNC compared with midazolam group that lasts for at least 30 days (post-treatment: −0.019, 95% HDI [−0.028,−0.011], BF10 = 9.45; follow-up: −0.017, 95% HDI [−0.026,−0.009], BF10 = 137). Figure 4b shows the left UNC WMI corrected for age, gender and motion across time.

Differences between the ketamine and midazolam groups in the (A) right and (B) left UNC FA adjusted for age, gender, motion and baseline corrected. Each dot is a participant, boxplots include the median, two hinges and two whiskers, and the density distribution of participants. C An illustration of the UNC.
Discussion
In this work, we provided preliminary evidence that a single infusion of 0.5 mg/kg ketamine for 40 min, when combined with exposure and processing of the traumatic memory, lead to alterations in neural circuit functioning, although it did not produce reduction in PTSD symptoms greater than midazolam. These changes were more consistent with modification of the original memory (i.e., post-retrieval extinction) than with enhancement of classic extinction. The ketamine group showed lower amygdala reactivity to recalled subjective traumatic events compared to the midazolam group, with no change in connectivity between the amygdala and vmPFC. Moreover, connectivity between the amygdala and posterior-hippocampus was reduced in the ketamine, but not the midazolam group, at the end of treatment. Last, ketamine, but not midazolam, was also associated with long-term reduction in FA in the UNC. The combined results suggest a potential, larger reduction in the level of neuronal reactivity associated with the original trauma memory in the ketamine group compared to midazolam.
The amygdala is a key node in neural networks engaged in extinction and post-retrieval extinction processes [9, 15, 71]. Some studies argue that diminished amygdala activation that is independent of vmPFC inhibitory function, is associated with post-retrieval extinction, which is in line with the classic reconsolidation findings, blocking protein synthesis in the basolateral amygdala (BLA) [1]. Other studies have found that extinction learning involves an inhibitory signal from the prefrontal cortex, mainly the vmPFC, to the amygdala and hippocampus [9]. Taken together, these findings suggest that extinction learning should present with higher connectivity between vmPFC and amygdala and higher activation of the vmPFC. Reconsolidation-based extinction (i.e. post-retrieval extinction), on the other hand, should present with lower activation in the amygdala during retrieval, regardless of vmPFC-amygdala connectivity. Our findings of diminished activation of the amygdala and lack of enhanced connectivity between vmPFC and amygdala, in response to trauma recall in the ketamine group, are consistent with the post-retrieval extinction theory [14]. Interestingly, a recent study reported an association between decreased PTSD symptoms and increased amygdala-vmPFC connectivity during emotional face viewing, especially in participants who received multiple infusions of ketamine [72]. Our results suggest a possible path of combining ketamine with psychotherapy and assessing their effect on the neural mechanisms underlying real autobiographical memories. While ketamine may have a general effect on regulating emotions, our theory proposes that combining it with the retrieval of the traumatic memory may lead to modification of the original traumatic memory through a process called post-retrieval extinction. However, further research is needed to confirm this theory.
NMDARs are required for the transition of memory from a fixed to a labile state. For example, an NMDAR antagonist, but not an AMPAR antagonist, blocked the reconsolidation process [27]. NMDAR activation may be upstream of the stimulation of protein synthesis in the mechanistic cascade responsible for making memories labile, as the addition of anisomycin does not augment the interference with reconsolidation produced by an NMDAR antagonist [1, 27]. Midazolam, a benzodiazepine positive allosteric modulator of GABAA receptors, has also been reported to interfere with the reconsolidation of fear [73, 74]. While some studies have found that benzodiazepine impaired extinction learning [75, 76], others have shown that if benzodiazepine is administered after memory reactivation (i.e., within the reconsolidation window), it blocks the reconsolidation of fear and as such promotes extinction of fear response [73, 74]. As midazolam’s half-life is relatively short (2.3 h) [77], it is highly unlikely that it will interfere with post-retrieval extinction which occurred 24 h post-infusion. Based on the current study procedures, we cannot rule out that some reconsolidation blockade might have occurred in both agents as the infusion was conducted following baseline memory activation task in the MRI. It is thus possible that the effect shown at the end of treatment derives from both the elevation of activation in the midazolam group and the reduced activation in the ketamine group.
Studies in animals show that 90 min following acute subanesthetic dose, ketamine causes reduction in synaptic density (PSD-95) due to the blockade of NMDA receptors [78]. However, 4 h post-injection there is a significant increase in WMI [34]. By 24 h following acute subanesthetic dose, evidence show ketamine promotes neuroplasticity by enhancing glutamate release, raising BDNF levels, activating mTORC1 signaling, producing cortical synaptogenesis, and stimulating hippocampal neurogenesis [28, 29], all of which are important to the reconsolidation processes [32, 33]. Supporting these findings, ketamine-related increase in hippocampal BDNF gene expression reduces reactivation of fear memories in rats [79]. However, the well-known antidepressant [80, 81] and anxiolytic [82] effects of ketamine emerge only 24 h to several days after ketamine administration. This enhanced neural plasticity and synaptic reorganization may be the main mechanism of action underlying our results, as traumatic memories were retrieved during the plastic stage, 24 h post-infusion, and reprocessed in a safe environment. This procedure was repeated for a total of 4 post-infusion daily exposures to further enhance the reconsolidation of the altered memory trace, as BDNF levels only return to baseline after 7 days [83]. In contrast, midazolam’s effect on memory reconsolidation (via GABA-A receptor signaling) appears to last only up to 60 min [84] post-administration.
It was suggested that ketamine does not simply increase connectivity, rather, it promotes the reorganization of the connections [85, 86]. Phoumthipphavong et al. (2016) checking for the effect of a single sub-anesthetic dose on mice dendritic architecture, found increase in synaptic density as well as retraction of distal spines. Reduction in FA in the UNC, represents a similar process i.e., reduced white matter integrity in long range axon bundles. Studies suggest that memories are stored in neuronal ensembles [87, 88]. These ensembles trigger the emotional response. Repeated activation of the memory in a safe environment, where the response is not initiated, alters the behavioral response and thus the ensemble that triggers the memory. As the response, the long range connections over the bundle (UNC) diminishes. It is possible that the interaction of psychotherapy and ketamine yields this reorganization of synapses, but our current design does not allow us to conclude such a thing yet. Further exploration of dosage, frequency and timing of ketamine when combined with psychotherapy is needed.
Our results also show reduced amygdala-hippocampus connectivity following ketamine infusion. This reduction was only apparent 7 days after infusion and not in the follow-up session (30 days), suggesting a transient effect. This finding is consistent with prior research demonstrating a role for amygdala-hippocampus interaction in the acquisition and modification of fear memories [15]. For example, the strength of amygdala-hippocampus connectivity is associated with the ability to encode long-term memories [20, 89,90,91,92,93]. Coupling between the amygdala and dorsal hippocampus during non-REM sleep increases following threat conditioning in rodents [19]. The role of the hippocampus in contextual fear memory was recently highlighted in a study that disrupted the reconsolidation of fear by inhibiting protein synthesis in hippocampal ensemble [94].
In healthy individuals, higher amygdala-hippocampus connectivity may be associated with enhanced memory under stress, but not neutral, conditions [92]. Although initial evidence suggested that anterior (rather than posterior) hippocampus was associated with emotional memories [95], posterior hippocampal connectivity was perturbed in PTSD (compared to healthy controls and Anxiety) in both resting-state and task-based (emotional faces) measures [96]. Moreover, individuals with PTSD had a smaller posterior hippocampus [97], and impaired posterior-anterior hippocampal connectivity [98]. The decreased connectivity between posterior-hippocampus and amygdala found in this investigation may serve as a potential biomarker of reduced fear memory trace following an NMDA enhanced post-retrieval extinction. Results may also point to the role of the posterior-hippocampus in real (context-dependent) traumatic memories.
The facilitation of post-retrieval extinction of trauma memories in this study raises the possibility that behavioral interventions may enhance the reported efficacy of ketamine in the treatment of PTSD [44]. PTSD is a highly disabling disorder with few effective pharmacotherapy options [99]. It is speculated that PTSD derives from overgeneralization of fear responses and excessive or maladaptive reconsolidation of the traumatic memories. As such, reconsolidation-based treatments can be clinically relevant for treating such a disorder. Although some evidence-based treatments for PTSD are highly effective, they reach 50% remission at best [100]. Using ketamine as a facilitator is consistent with a recent study conducted in participants with harmful drinking patterns [101]. While ketamine alone was ineffective in reducing drinking, when administered in conjunction with alcohol cues, ketamine interfered with the reconsolidation of alcohol-reward-related memories and significantly reduced drinking.
Although neural activity indicated a greater reduction in levels of distress associated with the traumatic memory in the ketamine group, we did not find any differences in PTSD symptoms between the experimental groups. A high level of heterogeneity characterizes PTSD symptomatology and thus presents a challenge for detecting nuances in treatment response, more so, in small samples. In this context, other studies which tested the effectiveness of exposure therapy on PTSD or fear of flying reported similar outcome differences in physiological fear response between the experimental groups at the end of the intervention with no difference in the self-report measure of symptoms [102, 103]. It is possible that a subjective account of psychological distress, measured by verbal account of psychiatric symptoms, is less sensitive to change than objective neurobiological measures. This hypothesis is in line with the NIMH Research Domain Criteria (RDoC) initiative to further establish biomarkers associated with mental disorders.
This study has some limitations. First, the number of participants in each group is relatively small, and larger studies will need to replicate the findings. This concern is partially mitigated by the longitudinal aspect of the study, as well as the findings, which are consistent with other animal and human studies. Another potential limitation is the use of an active psychotropic drug, midazolam. We used midazolam as a comparator to control for the subjective effects of ketamine and to preserve the blind conditions. This limits our inferences, however, to the relative effects of similarly tolerable doses of ketamine and midazolam. In order to fully unfold the effect of ketamine, a control group receiving ketamine without recall of trauma is needed. Lastly, it is important to note that since all participants received psychotherapy, it is difficult to determine the specific effect of ketamine alone and the effect of the combination of ketamine and psychotherapy.
While preliminary, the present pilot study finds that one-time ketamine infusion might enhance post-retrieval extinction of traumatic memories. This is apparent in the effects found in the ketamine vs. the midazolam group: diminished amygdala reactivity to recall of traumatic events, reduced WMI in the UNC, and attenuation of connectivity between amygdala and hippocampus. These findings complement previous findings in both animal models and humans, on a wider spectrum of psychiatric disorders and both appetitive and aversive memories. As the enhancement of post-retrieval extinction presented here was demonstrated using real-life traumatic events, the applicability of this procedure is high and it might serve as a potential novel future intervention for PTSD and anxiety disorders.
Data availability
Data and analysis can be found in the Github repository.
Code availability
All analysis scripts can be found here https://github.com/orduek/KPE.
References
Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 2000;406:722–6.
Lis S, Thome J, Kleindienst N, Mueller-Engelmann M, Steil R, Priebe K, et al. Generalization of fear in post-traumatic stress disorder. Psychophysiology 2020;57:e13422.
Kaczkurkin AN, Burton PC, Chazin SM, Manbeck AB, Espensen-Sturges T, Cooper SE, et al. Neural substrates of overgeneralized conditioned fear in PTSD. Am J Psychiatry. 2017;174:125–34.
Duek O, Spiller TR, Rubenstein A, Pietrzak RH, Harpaz-Rotem I. Exploration of a novel model of intrusive symptoms in posttraumatic stress disorder among US veterans. JAMA Netw Open. 2022;5:e223555–e223555.
Lee JLC, Nader K, Schiller D. An update on memory reconsolidation updating. Trends Cogn Sci. 2017;21:531–45.
Rescorla RA. Probability of shock in the presence and absence of CS in fear conditioning. J Comp Physiol Psychol. 1968;66:1.
Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96.
Bouton ME. Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull. 1993;114:80–99.
Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: Role of the amygdala and vmPFC. Neuron 2004;43:897–905.
Schiller D, Monfils M-H, Raio CM, Johnson DC, Ledoux JE, Phelps EA. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature 2010;463:49–53.
Monfils M-H, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science 2009;324:951–5.
Gershman SJ, Monfils M-H, Norman KA, Niv Y. Correction: The computational nature of memory modification. Elife. 2017;6:e23763.
Bang JW, Shibata K, Frank SM, Walsh EG, Greenlee MW, Watanabe T, et al. Consolidation and reconsolidation share behavioral and neurochemical mechanisms. Nat Hum Behav. 2018;2:507–13.
Schiller D, Kanen JW, LeDoux JE, Monfils M-H, Phelps EA. Extinction during reconsolidation of threat memory diminishes prefrontal cortex involvement. Proc Natl Acad Sci USA. 2013;110:20040–5.
Agren T, Engman J, Frick A, Björkstrand J, Larsson E-M, Furmark T, et al. Disruption of reconsolidation erases a fear memory trace in the human amygdala. Science 2012;337:1550–2.
Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron 2002;36:527–38.
Feng P, Zheng Y, Feng T. Resting-state functional connectivity between amygdala and the ventromedial prefrontal cortex following fear reminder predicts fear extinction. Soc Cogn Affect Neurosci. 2016;11:991–1001.
Foa E, Hembree E, Rothbaum BO. Prolonged exposure therapy for PTSD: Emotional processing of traumatic experiences therapist guide. Oxford University Press, USA; 2007.
Girardeau G, Inema I, Buzsáki G. Reactivations of emotional memory in the hippocampus-amygdala system during sleep. Nat Neurosci. 2017;20:1634–42.
Hermans EJ, Kanen JW, Tambini A, Fernández G, Davachi L, Phelps EA. Persistence of Amygdala-hippocampal connectivity and multi-voxel correlation structures during awake rest after fear learning predicts long-term expression of fear. Cereb Cortex. 2017;27:3028–41.
Von Der Heide RJ, Skipper LM, Klobusicky E, Olson IR. Dissecting the uncinate fasciculus: Disorders, controversies and a hypothesis. Brain. 2013;136:1692–707.
Hölzel BK, Brunsch V, Gard T, Greve DN, Koch K, Sorg C, et al. Mindfulness-based stress reduction, fear conditioning, and the uncinate fasciculus: A pilot study. Front Behav Neurosci. 2016;10:124.
Koch SBJ, van Zuiden M, Nawijn L, Frijling JL, Veltman DJ, Olff M. Decreased uncinate fasciculus tract integrity in male and female patients with PTSD: A diffusion tensor imaging study. J Psychiatry Neurosci. 2017;42:331–42.
Costanzo ME, Jovanovic T, Pham D, Leaman S, Highland KB, Norrholm SD, et al. White matter microstructure of the uncinate fasciculus is associated with subthreshold posttraumatic stress disorder symptoms and fear potentiated startle during early extinction in recently deployed Service Members. Neurosci Lett. 2016;618:66–71.
Milton AL, Lee JLC, Butler VJ, Gardner R, Everitt BJ. Intra-amygdala and systemic antagonism of NMDA receptors prevents the reconsolidation of drug-associated memory and impairs subsequently both novel and previously acquired drug-seeking behaviors. J Neurosci. 2008;28:8230–7.
Milton AL, Schramm MJW, Wawrzynski JR, Gore F, Oikonomou-Mpegeti F, Wang NQ, et al. Antagonism at NMDA receptors, but not β-adrenergic receptors, disrupts the reconsolidation of pavlovian conditioned approach and instrumental transfer for ethanol-associated conditioned stimuli. Psychopharmacology 2012;219:751–61.
Mamou CB, Gamache K, Nader K. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci. 2006;9:1237–9.
Clarke M, Razmjou S, Prowse N, Dwyer Z, Litteljohn D, Pentz R, et al. Ketamine modulates hippocampal neurogenesis and pro-inflammatory cytokines but not stressor induced neurochemical changes. Neuropharmacology 2017;112:210–20.
Duman RS, Li N, Liu R-J, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012;62:35–41.
Bai X, Yan Y, Canfield S, Muravyeva MY, Kikuchi C, Zaja I, et al. Ketamine enhances human neural stem cell proliferation and induces neuronal apoptosis via reactive oxygen species-mediated mitochondrial pathway. Anesth Analg. 2013;116:869–80.
Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010;329:959–64.
Barker JM, Boonstra R, Wojtowicz JM. From pattern to purpose: How comparative studies contribute to understanding the function of adult neurogenesis. Eur J Neurosci. 2011;34:963–77.
Suárez-Pereira I, Carrión ÁM. Updating stored memory requires adult hippocampal neurogenesis. Sci Rep. 2015;5:13993.
Sydnor VJ, Lyall AE, Cetin-Karayumak S, Cheung JC, Felicione JM, Akeju O, et al. Studying pre-treatment and ketamine-induced changes in white matter microstructure in the context of ketamine’s antidepressant effects. Transl Psychiatry. 2020;10:432.
Liao Y, Tang J, Ma M, Wu Z, Yang M, Wang X, et al. Frontal white matter abnormalities following chronic ketamine use: a diffusion tensor imaging study. Brain 2010;133:2115–22.
Young JT, Vlasova RM, Howell BR, Knickmeyer RC, Morin E, Kuitchoua KI, et al. General anaesthesia during infancy reduces white matter micro-organisation in developing rhesus monkeys. Br J Anaesth. 2021;126:845–53.
Feder A, Costi S, Rutter SB, Collins AB, Govindarajulu U, Jha MK, et al. A randomized controlled trial of repeated ketamine administration for chronic posttraumatic stress disorder. Am J Psychiatry. 2021;178:193–202.
Björkstrand J, Agren T, Åhs F, Frick A, Larsson E-M, Hjorth O, et al. Disrupting reconsolidation attenuates long-term fear memory in the human amygdala and facilitates approach behavior. Curr Biol. 2016;26:2690–5.
Soeter M, Kindt M. An Abrupt Transformation of phobic behavior after a post-retrieval amnesic agent. Biol Psychiatry. 2015;78:880–6.
Brunet A, Saumier D, Liu A, Streiner DL, Tremblay J, Pitman RK. Reduction of PTSD symptoms with pre-reactivation propranolol therapy: A randomized controlled trial. Am J Psychiatry. 2018;175:427–33.
Stein MB, Simon NM. Ketamine for PTSD: Well, Isn’t that special. AJP 2021;178:116–8.
Drozdz SJ, Goel A, McGarr MW, Katz J, Ritvo P, Mattina GF, et al. Ketamine assisted psychotherapy: A systematic narrative review of the literature. J Pain Res. 2022;15:1691–706.
Pradhan BK, Wainer IW, Moaddel R, Torjman MC, Goldberg M, Sabia M, et al. Trauma Interventions using Mindfulness Based Extinction and Reconsolidation (TIMBER) psychotherapy prolong the therapeutic effects of single ketamine infusion on post-traumatic stress disorder and comorbid depression: a pilot randomized, placebo-controlled, crossover clinical trial. Asia Pac J Clin Trials: Nerv Syst Dis. 2017;2:80.
Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry. 2014;71:681–8.
First MB, Gibbon M. The structured clinical interview for DSM-IV axis I disorders (SCID-I) and the structured clinical interview for DSM-IV axis II disorders (SCID-II). Compr Handb Psychol Assess. 2004;2:134–43.
Weathers FW, Bovin MJ, Lee DJ, Sloan DM, Schnurr PP, Kaloupek DG, et al. The Clinician-Administered PTSD Scale for DSM-5 (CAPS-5): Development and initial psychometric evaluation in military veterans. Psychol Assess. 2018;30:383–95.
Blevins CA, Weathers FW, Davis MT, Witte TK, Domino JL. The Posttraumatic Stress Disorder Checklist for DSM-5 (PCL-5): Development and Initial Psychometric Evaluation. J Trauma Stress. 2015;28:489–98.
Ehlers A, Hackmann A, Grey N, Wild J, Liness S, Albert I, et al. A randomized controlled trial of 7-day intensive and standard weekly cognitive therapy for PTSD and emotion-focused supportive therapy. Am J Psychiatry. 2014;171:294–304.
Sinha R, Fox HC, Hong KA, Bergquist K, Bhagwagar Z, Siedlarz KM. Enhanced negative emotion and alcohol craving, and altered physiological responses following stress and cue exposure in alcohol dependent individuals. Neuropsychopharmacology 2009;34:1198–208.
Esteban O, Markiewicz CJ, Blair RW, Moodie CA, Isik AI, Erramuzpe A, et al. fMRIPrep: a robust preprocessing pipeline for functional MRI. Nat Methods. 2019;16:111–6.
Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TEJ, Johansen-Berg H, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 2004;23:S208–19.
Gorgolewski K, Burns CD, Madison C, Clark D, Halchenko YO, Waskom ML, et al. Nipype: a flexible, lightweight and extensible neuroimaging data processing framework in python. Front Neuroinform. 2011;5:13.
Yarkoni T, Poldrack RA, Nichols TE, Van Essen DC, Wager TD. Large-scale automated synthesis of human functional neuroimaging data. Nat Methods. 2011;8:665–70.
Abraham A, Pedregosa F, Eickenberg M, Gervais P, Mueller A, Kossaifi J, et al. Machine learning for neuroimaging with scikit-learn. Front Neuroinform. 2014;8:14.
Salvatier J, Wiecki TV, Fonnesbeck C. Probabilistic programming in Python using PyMC3. PeerJ Comput Sci. 2016;2:e55.
Dadi K, Varoquaux G, Machlouzarides-Shalit A, Gorgolewski KJ, Wassermann D, Thirion B, et al. Fine-grain atlases of functional modes for fMRI analysis. Neuroimage. 2020;221:117126.
Yendiki A, Panneck P, Srinivasan P, Stevens A, Zöllei L, Augustinack J, et al. Automated probabilistic reconstruction of white-matter pathways in health and disease using an atlas of the underlying anatomy. Front Neuroinform. 2011;5:23.
Yendiki A, Koldewyn K, Kakunoori S, Kanwisher N, Fischl B. Spurious group differences due to head motion in a diffusion MRI study. Neuroimage 2014;88:79–90.
Jbabdi S, Woolrich MW, Andersson JLR, Behrens TEJ. A Bayesian framework for global tractography. Neuroimage 2007;37:116–29.
Greve DN, Fischl B. Accurate and robust brain image alignment using boundary-based registration. Neuroimage 2009;48:63–72.
Cumming G. The New statistics: Why and how. Psychol Sci. 2014;25:7–29.
Wasserstein RL, Lazar NA. The ASA statement on p-Values: Context, process, and purpose. Am Stat. 2016;70:129–33.
Greenland S, Senn SJ, Rothman KJ, Carlin JB, Poole C, Goodman SN, et al. Statistical tests, P values, confidence intervals, and power: A guide to misinterpretations. Eur J Epidemiol. 2016;31:337–50.
Kruschke JK. Bayesian estimation supersedes the t test. J Exp Psychol Gen. 2013;142:573–603.
Kruschke JK. Rejecting or accepting parameter values in Bayesian estimation. Adv Methods Pract Psychol. Sci. 2018;1:270–80.
Bürkner P-C. brms: An R package for Bayesian multilevel models using Stan. J Stat Softw. 2017;80:1–28.
Plummer M, Best N, Cowles K, Vines K. CODA: Convergence diagnosis and output analysis for MCMC. R N. 2006;6:7–11.
Geweke J. Bayesian treatment of the independent student-t linear model. J Appl Econ. 1993;8:S19–S40.
McElreath R Statistical Rethinking: A Bayesian Course with Examples in R and STAN. CRC Press; 2020.
Faul F, Erdfelder E, Lang A-G, Buchner A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–91.
Feinstein JS, Adolphs R, Damasio A, Tranel D. The human amygdala and the induction and experience of fear. Curr Biol. 2011;21:34–8.
Norbury A, Rutter SB, Collins AB, Costi S, Jha MK, Horn SR, et al. Neuroimaging correlates and predictors of response to repeated-dose intravenous ketamine in PTSD: preliminary evidence. Neuropsychopharmacology 2021;46:2266–77.
Ferrer Monti RI, Alfei JM, Mugnaini M, Bueno AM, Beckers T, Urcelay GP, et al. A comparison of behavioral and pharmacological interventions to attenuate reactivated fear memories. Learn Mem. 2017;24:369–74.
Ortiz V, Giachero M, Espejo PJ, Molina VA, Martijena ID. The effect of Midazolam and Propranolol on fear memory reconsolidation in ethanol-withdrawn rats: influence of d-cycloserine. Int J Neuropsychopharmacol. 2015;18:1–11
Rothbaum BO, Price M, Jovanovic T, Norrholm SD, Gerardi M, Dunlop B, et al. A randomized, double-blind evaluation of D-cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan War veterans. Am J Psychiatry. 2014;171:640–8.
Hart G, Harris JA, Westbrook RF. Systemic or intra-amygdala injection of a benzodiazepine (midazolam) impairs extinction but spares re-extinction of conditioned fear responses. Learn Mem. 2009;16:53–61.
Heizmann P, Eckert M, Ziegler WH. Pharmacokinetics and bioavailability of midazolam in man. Br J Clin Pharm. 1983;16:43S–49S.
de Bartolomeis A, Sarappa C, Buonaguro EF, Marmo F, Eramo A, Tomasetti C, et al. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:1–12.
Duclot F, Perez-Taboada I, Wright KN, Kabbaj M. Prediction of individual differences in fear response by novelty seeking, and disruption of contextual fear memory reconsolidation by ketamine. Neuropharmacology 2016;109:293–305.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4.
Krystal JH, Abdallah CG, Sanacora G, Charney DS, Duman RS. Ketamine: A Paradigm Shift for Depression Research and Treatment. Neuron 2019;101:774–8.
Glue P, Neehoff SM, Medlicott NJ, Gray A, Kibby G, McNaughton N. Safety and efficacy of maintenance ketamine treatment in patients with treatment-refractory generalised anxiety and social anxiety disorders. J Psychopharmacol. 2018;32:663–7.
Kim J-W, Autry AE, Na ES, Adachi M, Björkholm C, Kavalali ET, et al. Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat Neurosci. 2021;24:1100–9.
Bustos SG, Maldonado H, Molina VA. Midazolam disrupts fear memory reconsolidation. Neuroscience 2006;139:831–42.
Phoumthipphavong V, Barthas F, Hassett S, Kwan AC Longitudinal Effects of Ketamine on Dendritic Architecture In Vivo in the Mouse Medial Frontal Cortex. eNeuro 2016;3:ENEURO.0133-15.2016.
Savalia NK, Shao L-X, Kwan AC A Dendrite-Focused Framework for Understanding the Actions of Ketamine and Psychedelics. Trends Neurosci. 2020. 21 December 2020. https://doi.org/10.1016/j.tins.2020.11.008.
Rogerson T, Cai DJ, Frank A, Sano Y, Shobe J, Lopez-Aranda MF, et al. Synaptic tagging during memory allocation. Nat Rev Neurosci. 2014;15:157–69.
Rogerson T, Jayaprakash B, Cai DJ, Sano Y, Lee Y-S, Zhou Y, et al. Molecular and Cellular Mechanisms for Trapping and Activating Emotional Memories. PLoS One. 2016;11:e0161655.
Dolcos F, LaBar KS, Cabeza R. Remembering one year later: role of the amygdala and the medial temporal lobe memory system in retrieving emotional memories. Proc Natl Acad Sci USA. 2005;102:2626–31.
Goldfarb EV. Enhancing memory with stress: Progress, challenges, and opportunities. Brain Cogn. 2019;133:94–105.
Shields GS, McCullough AM, Ritchey M, Ranganath C, Yonelinas AP. Stress and the medial temporal lobe at rest: Functional connectivity is associated with both memory and cortisol. Psychoneuroendocrinology 2019;106:138–46.
de Voogd LD, Klumpers F, Fernández G, Hermans EJ. Intrinsic functional connectivity between amygdala and hippocampus during rest predicts enhanced memory under stress. Psychoneuroendocrinology 2017;75:192–202.
Richardson MP, Strange BA, Dolan RJ. Encoding of emotional memories depends on amygdala and hippocampus and their interactions. Nat Neurosci. 2004;7:278–85.
Ressler RL, Goode TD, Kim S, Ramanathan KR, Maren S. Covert capture and attenuation of a hippocampus-dependent fear memory. Nat Neurosci. 2021;24:677–84.
Dolcos F, LaBar KS, Cabeza R. Interaction between the amygdala and the medial temporal lobe memory system predicts better memory for emotional events. Neuron 2004;42:855–63.
Chen AC, Etkin A. Hippocampal network connectivity and activation differentiates post-traumatic stress disorder from generalized anxiety disorder. Neuropsychopharmacology 2013;38:1889–98.
Bonne O, Vythilingam M, Inagaki M, Wood S, Neumeister A, Nugent AC, et al. Reduced posterior hippocampal volume in posttraumatic stress disorder. J Clin Psychiatry. 2008;69:1087–91.
Lazarov A, Zhu X, Suarez-Jimenez B, Rutherford BR, Neria Y. Resting-state functional connectivity of anterior and posterior hippocampus in posttraumatic stress disorder. J Psychiatr Res. 2017;94:15–22.
Krystal JH, Davis LL, Neylan TC, A Raskind M, Schnurr PP, Stein MB, et al. It Is Time to Address the Crisis in the Pharmacotherapy of Posttraumatic Stress Disorder: A Consensus Statement of the PTSD Psychopharmacology Working Group. Biol Psychiatry. 2017;82:e51–e59.
Schottenbauer MA, Glass CR, Arnkoff DB, Tendick V, Gray SH. Nonresponse and dropout rates in outcome studies on PTSD: review and methodological considerations. Psychiatry 2008;71:134–68.
Das RK, Gale G, Walsh K, Hennessy VE, Iskandar G, Mordecai LA, et al. Ketamine can reduce harmful drinking by pharmacologically rewriting drinking memories. Nat Commun. 2019;10:5187.
Vermes JS, Ayres R, Goés AS, Real ND, Araújo ÁC, Schiller D, et al. Targeting the reconsolidation of traumatic memories with a brief 2-session imaginal exposure intervention in post-traumatic stress disorder. J Affect Disord. 2020;276:487–94.
Maples-Keller JL, Price M, Jovanovic T, Norrholm SD, Odenat L, Post L, et al. Targeting memory reconsolidation to prevent the return of fear in patients with fear of flying. Depress Anxiety. 2017;34:610–20.
Acknowledgements
The main source of funding for this work was provided by: Independent Investigator Grant from the Brain and Behavior Research Foundation (IHR); by the Clinical Neurosciences Division of the National Center for PTSD (IHR); a donation from the American Brain Society (IHR), and the Yale Center for Clinical Investigation (YCCI) supported by CTSA Grant from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH). The contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH. We thank Professor Daniela Schiller for very helpful comments on this manuscript. An earlier version of the manuscript is published in the preprint server of medrXiv and can be accessed here: https://doi.org/10.1101/2021.07.07.21260166.
Author information
Authors and Affiliations
Contributions
I.H.R. designed the study. O.D., B.K., S.A., C.G., M.M., and I.H.R. collected the data. O.D., N.K., and Y.L. analyzed the data, I.L. and I.H.R. contributed to the data analyses. J.K. has contributed to the interpretation of the results. O.D., N.K., I.L., and I.H.R. wrote the first draft of the manuscript. All authors contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.K. registered patents: Glutamate Modulating Agents in the Treatment of Mental Disorders. US Patent No. 8,778,979 B2 Patent Issue Date: July 15, 2014. US Patent Application No. 15/695,164: Filing Date: 09/05/2017; Intranasal Administration of Ketamine to Treat Depression United States Patent Number: 9592207, Issue date: 3/14/2017. Licensed to Janssen Research & Development. Other interests J.K: Consultant (the Individual Consultant Agreements listed below are less than $5,000 per year): Aptinyx, Inc.; Atai Life Sciences; AstraZeneca Pharmaceuticals; Biogen, Idec, MA; Biomedisyn Corporation; Bionomics, Limited (Australia); Boehringer Ingelheim International; Cadent Therapeutics, Inc.; Clexio Bioscience, Ltd.; COMPASS Pathways, Limited, United Kingdom; Concert Pharmaceuticals, Inc.; Epiodyne, Inc.; EpiVario, Inc.; Greenwich Biosciences, Inc.; Heptares Therapeutics, Limited (UK); Janssen Research & Development; Jazz Pharmaceuticals, Inc.; Otsuka America Pharmaceutical, Inc.; Perception Neuroscience Holdings, Inc.; Spring Care, Inc.; Sunovion; Pharmaceuticals, Inc.; Takeda Industries; Taisho Pharmaceutical Co., Ltd. Board of Directors: Freedom Biosciences, Inc. Stock: Biohaven Pharmaceuticals; Sage Pharmaceuticals.; Spring Care, Inc. Stock Options: Biohaven Pharmaceuticals Medical Sciences; EpiVario, Inc.; RBNC Therapeutics, Inc.; Terran Biosciences, Inc.; Tempero Bio, Inc. Patents and Inventions: Seibyl JP, Krystal JH, Charney DS. Dopamine and noradrenergic reuptake inhibitors in treatment of schizophrenia. US Patent #:5,447,948.September 5, 1995; Zarate, C, Charney, DS, Manji, HK, Mathew, Sanjay J, Krystal, JH, Yale University “Methods for Treating Suicidal Ideation”, Patent Application No. 15/379,013 filed on December 14, 2016 by Yale University Office of Cooperative Research; Arias A, Petrakis I, Krystal JH. – Composition and methods to treat addiction. Provisional Use Patent Application no.61/973/961. April 2, 2014. Filed by Yale University Office of Cooperative Research; Chekroud, A., Gueorguieva, R., & Krystal, JH. “Treatment Selection for Major Depressive Disorder” [filing date 3rd June 2016, USPTO docket number Y0087.70116US00]. Provisional patent submission by Yale University; Gihyun, Yoon, Petrakis I, Krystal JH – Compounds, Compositions and Methods for Treating or Preventing Depression and Other Diseases. U. S. Provisional Patent Application No. 62/444,552, filed on January 10, 2017 by Yale University Office of Cooperative Research OCR 7088 US01; Abdallah, C, Krystal, JH, Duman, R, Sanacora, G. Combination Therapy for Treating or Preventing Depression or Other Mood Diseases. U.S. Provisional Patent Application No. 62/719,935 filed on August 20, 2018 by Yale University Office of Cooperative Research OCR 7451 US01; John Krystal, Godfrey Pearlson, Stephanie O’Malley, Marc Potenza, Fabrizio Gasparini, Baltazar Gomez-Mancilla, Vincent Malaterre. Mavoglurant in treating gambling and gaming disorders. U.S. Provisional Patent Application No. 63/125,181filed on December 14, 2020 by Yale University Office of Cooperative Research OCR 8065 US00 NON Federal Research Support: AstraZeneca Pharmaceuticals provides the drug, Saracatinib, for research related to NIAAA grant “Center for Translational Neuroscience of Alcoholism [CTNA-4]; Novartis provides the drug, Mavoglurant, for research related to NIAAA grant “Center for Translational Neuroscience of Alcoholism [CTNA-4]; Cerevel provides the drug PF-06412562 for A Translational and Neurocomputational Evaluation of a D1R Partial Agonist for Schizophrenia (1 U01 MH121766-01). Scientific Advisory Board: Biohaven Pharmaceuticals; BioXcel Therapeutics, Inc. (Clinical Advisory Board); Cadent Therapeutics, Inc. (Clinical Advisory Board); Cerevel Therapeutics, LLC; EpiVario, Inc.; Eisai, Inc.; Jazz Pharmaceuticals, Inc.; Lohocla Research Corporation; Novartis Pharmaceuticals Corporation; PsychoGenics, Inc.; RBNC Therapeutics, Inc.; Tempero Bio, Inc.; Terran Biosciences, Inc.; Income Greater than $10,000: Editor - Biological Psychiatry. OD is consultant for Madrigal Mental Care. All remaining authors have nothing to disclose. I.H.R receives funding from Boehringer Ingelheim Pharma GmbH & Co. KG through Investigator-Initiated Research into “Subtyping PTSD - Precision Psychiatry”.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Duek, O., Korem, N., Li, Y. et al. Long term structural and functional neural changes following a single infusion of Ketamine in PTSD. Neuropsychopharmacol. 48, 1648–1658 (2023). https://doi.org/10.1038/s41386-023-01606-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-023-01606-3
This article is cited by
-
Effects of a dissociative drug on fronto-limbic resting-state functional connectivity in individuals with posttraumatic stress disorder: a randomized controlled pilot study
Psychopharmacology (2024)
-
Neural patterns differentiate traumatic from sad autobiographical memories in PTSD
Nature Neuroscience (2023)
