
Abstract
The formation and extinction of fear memories represent two forms of learning that each engage the hippocampus and amygdala. How cell populations in these areas contribute to fear relapse, however, remains unclear. Here, we demonstrate that, in male mice, cells active during fear conditioning in the dentate gyrus of hippocampus exhibit decreased activity during extinction and are re-engaged after contextual fear relapse. In vivo calcium imaging reveals that relapse drives population dynamics in the basolateral amygdala to revert to a network state similar to the state present during fear conditioning. Finally, we find that optogenetic inactivation of neuronal ensembles active during fear conditioning in either the hippocampus or amygdala is sufficient to disrupt fear expression after relapse, while optogenetic stimulation of these same ensembles after extinction is insufficient to artificially mimic fear relapse. These results suggest that fear relapse triggers a partial re-emergence of the original fear memory representation, providing new insight into the neural substrates of fear relapse.
Similar content being viewed by others

Introduction
The biological capacity to produce adaptive behavioral responses in actively changing environments is critical to an animal’s survival. Contextual fear conditioning (CFC) is a form of learning whereby an animal learns to associate a conditioned stimulus (i.e., a context) with an unconditioned aversive stimulus (e.g., foot shocks) to produce a conditioned response to the conditioned stimulus (e.g., freezing). Conditioned responses can be mitigated through extinction learning via repeated exposure to the conditioned context in the absence of the foot shock. However, while extinction learning can be effective at attenuating fear, animals are susceptible to fear relapse under several conditions, including exposure to stressors, the passage of time, and re-exposure to the unconditioned stimulus [1,2,3,4,5,6]. This observation in rodents shares numerous similarities to clinical observations: exposure therapy – a clinical analog to extinction learning – can be effective at reducing fear in subsets of patients with anxiety disorders or post-traumatic stress disorder. However, many patients are still susceptible to fear relapse following successful exposure therapy [7,8,9]. Despite an extensive body of literature investigating the neural substrates of fear and extinction learning [10,11,12,13,14,15], the neural substrates underlying fear relapse are comparatively less understood.
Fear reinstatement is a form of fear relapse whereby extinguished fear of a conditioned stimulus (CS) returns following re-exposure to the unconditioned stimulus (US) alone [3]. Fear reinstatement has been discussed as being a non-associative learning phenomenon driven by a changing US representation strength across conditioning and extinction [16]. It has also separately been proposed that the CS mediates reinstatement during recall after US presentation, by retrieving the CS-US memory [2, 17, 18]. The neural circuitry underlying fear relapse has also been studied in recent years. It has been demonstrated, for example, that pharmacological activation of noradrenergic activity is sufficient to drive fear relapse after extinction [19], that dopamine-1 receptor blockade in the infralimbic cortex prevents fear reinstatement [20], and that activity of the bed nucleus of the stria terminalis is necessary for fear reinstatement [21]. However, how brain regions central to emotional memory processing – such as the amygdala and hippocampus – contribute to fear relapse has been understudied. Nonetheless, it has been shown that pharmacological inactivation of either the prelimbic cortex, dorsal CA1, or ventral hippocampus disrupts fear relapse [22], and blockade of either mRNA or protein synthesis in the CA1 of the hippocampus prevents fear relapse [23]. More broadly, similar sets of brain regions, including the amygdala and hippocampus, are involved in the relapse of both fear and drug intake [24]. However, whether and how discrete neuronal populations active during fear conditioning contribute to fear reinstatement remains incompletely understood.
Previous studies have demonstrated that cells in the dorsal dentate gyrus of the hippocampus (DG), in the CA1 of the hippocampus, and in the basolateral amygdala (BLA) that are active during fear conditioning (hereafter referred to as the DG, CA1, and BLA fear ensembles) are preferentially reactivated during fear memory recall [25,26,27,28], and are necessary and sufficient for the expression of defensive behaviors such as freezing [29,30,31]. Additionally, recent evidence has indicated that extinction learning is mediated by interactions between local BLA interneurons and a BLA fear ensemble [32], while a new set of cells simultaneously emerges in both the hippocampus [33,34,35] and BLA [36], possibly to encode extinction learning. It has also been shown that, after fear extinction, activation of the brain-wide neuronal ensemble active during fear conditioning is capable of driving freezing, suggesting that there is a latent representation of the fear memory that can be activated exogenously after extinction [37]. Whether fear reinstatement re-engages the original memory-encoding neuronal population or gives rise to a new representation, however, remains unclear.
Methods and materials
Subjects
Wildtype male C57BL/6 mice (6–8 weeks of age; Charles River Labs) were housed in groups of 4–5 mice per cage. The animal facilities (vivarium and behavioral testing rooms) were maintained on a 12:12-h light cycle (lights on at 0700). Mice were placed on a diet containing 40 mg/kg doxycycline (Dox) for a minimum of 2 days before receiving surgery with access to food and water ad libitum. Mice recovered for at least 10 days after surgery. Dox-containing diet was replaced with standard mouse chow (ad libitum) 48 h prior to behavioral tagging to open a time window of activity-dependent labeling [25].
All procedures relating to mouse care and treatment conformed to the institutional and National Institutes of Health guidelines for the Care and Use of Laboratory Animals. No statistical methods were used to predetermine sample size; however, sample sizes were chosen based on sample sizes in previous studies [25]. Data collection and analysis were not performed blind to the conditions of the experiments.
Activity-dependent viral constructs
pAAV9-cFos-tTA (titer of ~1.5 × 1013 GC/mL), pAAV9-TRE-eYFP (titer of ~1 × 1013 GC/mL), pAAV9 -TRE-ArchT-eYFP (titer of ~1 × 1013 GC/mL), and pAAV9-TRE-ChR2-eYFP (titer of ~1 × 1013 GC/mL) were constructed as previously described [38]. pAAV9-c-Fos-tTA was combined with pAAV9-TRE-eYFP or pAAV9-TRE-ArchT-eYFP or pAAV9-TRE-ChR2-eYFP prior to injection at a 1:1 ratio. See Supplementary Methods for Stereotaxic Surgery methods.
Optogenetic methods
Optic fiber implants were plugged into a patch cord connected to a 520 nm green laser diode or 473 nm blue laser diode controlled by automated software (Doric Lenses). Laser diode output was tested at the beginning of every experiment to ensure that at least 10 mW of power was delivered at the end of the optic fiber tip (Doric lenses). For the inhibition experiments in Fig. 3 and Supplementary Fig. 3, mice began the stimulation trial with a 2-min light-off epoch, followed by 2-min optical stimulation (15 ms pulse width, 20 Hz), and then repeated, such that the mice underwent a light-OFF/ON/OFF/ON pattern for a total of 8-min. For the excitation experiments in Fig. 4, mice underwent stimulation (15 ms pulse width, 20 Hz) for the full 60-s behavioral session.
Behavioral tagging
Dox diet was replaced with standard lab chow (ad libitum) 48-h prior to behavioral tagging. Fear conditioning: Mice were placed into a conditioning chamber and received fear conditioning (see Supplementary Methods) over a 500-s training session (including exposure to four 1.5 mA foot shocks). Following behavioral tagging, the male mouse was returned to their home cage with access to Dox diet [25].
Behavior
All behavior assays were conducted during the light cycle of the day (0730–1930). Mice were handled for 1–2 days, 2 min per day, before all behavioral experiments, and were run by cage. The entire behavioral schedule includes fear conditioning, extinction, reinstatement, and recall, as well as subsequent immunohistochemical methods (See supplementary methods).
In vivo calcium imaging
Mice were injected with AAV9-Syn-GCaMP6f into either the right CA1 or right BLA. Two to four weeks later, mice were implanted with a gradient index (GRIN) lens above the prior injection site. For CA1 implants, overlying cortex was aspirated, as previously described [39]. A miniaturized microscope (Inscopix) was used to collect Ca2+ imaging videos in mice undergoing the fear reinstatement schedule. Videos were captured using nVista (Inscopix) at 20 Hz in a 720 × 540 pixel field of view (1.1 microns/pixel). Ca2+ imaging videos were cropped, spatially bandpass filtered, and motion corrected offline using Inscopix Data Processing Software v1.1. A ΔF/F movie was computed using the mean fluorescence of the movie as the baseline and PCA/ICA was used for automated segmentation of cell masks [40]. PCA/ICA putative cell masks were each manually inspected to verify that cells were accurately captured with high fidelity. Cells across imaging sessions were aligned and registered to each other using the automated CellReg software in Matlab [41]. Population vectors (PVs) were computed for the entirety of the CFC session by taking the average Ca2+ transient rate for each cell while the mouse was in the fear conditioning chamber. See Supplementary Methods for further calcium imaging ensemble characterization.
Cell counting
To calculate the percentage of reactivated cells we counted the number of eYFP-positive cells, cFos-positive cells, and both eYFP- and cFos-positive (Overlapped) cells. Reactivation was calculated as (Overlap/eYFP*100). Overlap was compared across groups using unpaired t-test (two-groups) and one-way ANOVA (more than two groups). Relative expression of eYFP and cFos across groups was calculated as (#eYFP cells/Area) and (#cFos cell/Area) in arbitrary area units. See Supplementary Methods for Cell Counting criteria.
Data analysis
Data were analyzed using Inscopix nVista in conjunction with custom-made R, Python, and Matlab scripts. Data were analyzed using paired t-tests (two factors) or with one-way and repeated measures ANOVAs (more than two factors), Wilcoxon signed-rank tests, and Mann–Whitney U tests (two-tailed, corrected for multiple comparisons using false discovery rate adjustments). Post-hoc analyses (Tukey’s) were used to characterize treatment and interaction effects, when statistically significant (alpha set at p < 0.05, two-tailed).
Results
Hippocampal cells active during fear conditioning are less active after fear extinction and re-engaged after fear reinstatement
We first developed a 5-day behavioral protocol for fear reinstatement, a model of fear relapse in rodents [3]. Mice underwent CFC on day 1, followed by two subsequent extinction (EXT) sessions on days 2 and 3. 24 h after the second EXT session, mice received an immediate shock (IS) in a novel context to reinstate the original fear memory. The following day, mice were placed in a post-reinstatement recall test (IS-Recall) to measure the return of fear (Fig. 1a, bottom behavioral schedule; see Methods). Reinstatement led to an increase in freezing in the original conditioned context (Supplementary Fig. 1a–e) and mice froze more in the original conditioned context over a novel context after reinstatement (Supplementary Fig. 1h, i).

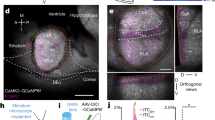
a Behavioral design for fear reinstatement. Mice underwent fear conditioning, and were then sacrificed at different points in the behavioral schedule and had tissue stained for c-Fos. b Schematic of viral strategy. A viral cocktail of AAV9-c-Fos-tTA and AAV9-TRE-eYFP was infused into the DG and BLA, and in a separate cohort of mice in the CA1, for activity-dependent induction of eYFP. c Representative microscope image for the injection sites. d Example confocal images of BLA sections. Images from left to right: virus-labeled cells (eYFP), c-Fos+ cells (cFos), merged green and red channels (Merge). Yellow arrows designate double-positive cells. e Same as (d) but for DG sections. f Same as (d) but for CA1 sections. g Quantitative analysis of overlap between FC-tagged BLA cells and c-Fos+ BLA cells in each group. The amount of overlap between FC-tagged (eYFP+) cells and c-Fos+ cells remained unchanged across all behavioral groups. (n = 8–10 mice per group; F3,32 = 1.607; one-way ANOVA). Counts were calculated as % of eYFP+ cells that were also c-Fos+. Each data point represents one mouse. h Same as quantification in (g), but for DG. Overlap between FC-tagged cells and c-Fos+ cells was high during recall after fear conditioning (FC-Recall) and significantly decreased following EXT (EXT-Recall). While overlap remained low during the reinstating shock (IS), it significantly increased during Recall after reinstatement (IS-Recall). (n = 8–10 mice per group; F3,31 = 7.703, p = 0.0006; one-way ANOVA followed by pairwise t-tests corrected with Benjamini–Hochberg method to correct for multiple comparisons; **P < 0.01, ns P > 0.05). i Same as (g), but for CA1. The amount of overlap between FC-tagged (eYFP+) cells and c-Fos+ cells remained unchanged across all behavioral groups. (n = 5–8 mice per group; F3,21 = 0.98, p = 0.42; one-way ANOVA).
Next, we determined if the cells active during fear conditioning were preferentially reactivated after mice underwent extinction and subsequent reinstatement. To that end, we tagged cells active during fear conditioning by injecting an activity-dependent viral cocktail of AAV9-c-Fos-tTA and AAV9-TRE-eYFP in the BLA, DG, and CA1 of adult male mice (Fig. 1b, c). This virus enabled expression of eYFP in cells sufficiently active to express the immediate early gene c-Fos, which is under the repressive control of the antibiotic doxycycline (DOX) [26]. We then measured immunoreactive c-Fos and calculated overlap between the set of cells active during CFC (eYFP-expressing cells) and cells active during different stages of the behavioral schedule (c-Fos-expressing cells) (Fig. 1d–f).
Previous reports have shown that the number of BLA cells active during both fear conditioning and fear memory recall correlates with freezing levels [26]. Thus, we reasoned that if reinstatement re-engages the fear ensemble, the set of cells active during fear conditioning would be active again following reinstatement. Surprisingly, we found that cells active during CFC were no differently reactivated throughout the behavioral schedule (Fig. 1g). Similarly, overlap in the CA1 remained consistent throughout the behavioral schedule (Fig. 1i). In the DG, however, we observed significant overlap between the cells active during CFC and those active during fear memory recall, as previously reported [25] (Fig. 1g). In support of the notion that the dorsal DG processes changes in environmental contingencies [42], this overlap substantially decreased after EXT. While overlap remained low after IS, it significantly increased when mice were given the IS and were placed back into the original conditioned context the following day, suggesting that fear reinstatement may re-engage the set of cells originally active during fear conditioning in the DG (Fig. 1g). A similar analysis where the number of overlapping cells was normalized to the area of interest (rather than normalized to the number of FC-tagged cells) yielded similar results (Supplementary Fig. 1j–l). Overall expression of eYFP and cFos was stable across groups in the DG and CA1, and while eYFP expression was stable across groups in the BLA, mice had significantly higher levels of cFos during post-reinstatement recall (Supplementary Fig. 1j–r).
Population activity during fear conditioning correlates with activity after fear reinstatement but not after extinction learning
Whereas our c-Fos-based labeling system allowed comparisons between activity of cells across two discrete timepoints with high spatial resolution, it was incapable of measuring activity at finer timescales due to the slow kinetics of immediate-early gene expression relative to real-time neural activity. To overcome this weakness, we next utilized an in vivo calcium (Ca2+) imaging approach to record real-time neuronal activity in the BLA or CA1 in freely moving mice during exposures to both a conditioned context and a neutral context where no shocks were delivered (Fig. 2a–d, Supplementary Table 1). While overlaps showed that the DG fear ensemble was dis-engaged after extinction and re-engaged after fear reinstatement (Fig. 1h), we were unable to perform calcium imaging in the DG due to the necessary removal of the overlying CA1, which we speculate would have disrupted the integrity of the hippocampal network. Since CA1 is the major output structure of the hippocampus, we instead recorded CA1 calcium dynamics as a real-time readout of the output of dorsal hippocampus throughout the reinstatement protocol. We tracked these cells longitudinally over the course of the reinstatement schedule in order to determine whether similar activity patterns were expressed during fear conditioning and reinstatement [41] (Supplementary Fig. 2a–d). There were no differences in overlaps of BLA cells detected with calcium imaging for each session pair (Supplementary Fig. 2e, f), consistent with our cFos immunostaining results (Fig. 1f). However, we hypothesized that the moment-by-moment activity patterns of the recorded neuronal populations might correlate with an initial population state reflecting fear acquisition during CFC. To define this initial population state, we constructed Ca2+ transient rate population vectors from the CFC session for each mouse. Then, to compare extinction and post-reinstatement recall states to CFC, we correlated population vectors from EXT and Recall (in 30 s non-overlapping time windows) to the CFC population vector. We found that over EXT, the population states in the BLA and CA1 followed a trajectory that gradually deviated from its CFC state, supporting the idea of a network-wide transformation over extinction [33, 36, 43, 44] (Fig. 2e, top left). However, during Recall, the BLA population in each mouse rebounded towards the CFC network state to an extent greater than expected by chance (Fig. 2e, top right). This rebound effect was absent in a neutral context (Fig. 2e, bottom), demonstrating that the conditioned context drove these dynamics primarily in BLA, and in CA1 of some mice (Fig. 2f, top right). Additionally, a regression continuity analysis [45] (see Methods) detected a significant change point in activity patterns at the EXT2-Recall border, suggesting that the network drastically shifted to a different state during that transition (Fig. 2e, f, left). No such divergence was detected across EXT1 to EXT2 in BLA, indicating a relatively steady transition across the two EXT sessions.

a Behavioral schedule for Ca2+ imaging cohort. b Left, freezing time course of fear reinstatement paradigm (n = 12 mice). Mice froze more in the shock context during Recall compared to the neutral context (repeated-measures ANOVA, F1,11 = 16.4, P = 0.0019). Right, pooled freezing. EXT1 vs. EXT2, Wilcoxon signed-rank test, P = 0.0015; EXT1 vs neutral EXT1, P = 0.017; EXT2 vs. Recall, P = 0.012. Data are means ± standard error of the mean. c Left, example field of view in BLA-implanted mouse, depicted as maximum projection of CFC imaging session. Blue outlines indicate cell masks. Scale bar = 100 microns. Right, fluorescence traces of 10 example cells. d Same as (b), but for CA1. e Left, Pearson correlation coefficients between population vectors (PVs) during CFC and EXT/Recall PVs (n = 6 BLA mice), for both the shock context (top) and the neutral context (bottom). Each data point represents a measure of population similarity to CFC (60 s time bins). Two piecewise regressions were fit each to EXT and Recall. P-values above indicate regression discontinuity analysis results on discontinuities between population vector correlations across sessions (EXT1 to EXT2 and EXT2 to Recall). Right, bar plots of population vector similarity (predicted and empirical). Each data point represents a mouse’s predicted r value (from the EXT regression) for Recall compared to the actual observed r value during Recall (Wilcoxon signed-rank test, P = 0.028). f Same as (e), but for CA1.
Using an algorithm for extracting co-active neurons from simultaneously recorded cells [46], we also identified neuronal ensembles during individual sessions prior to Recall (CFC, EXT1, and EXT2). We hypothesized that the ensembles identified during CFC might predict freezing behavior during Recall, while ensembles identified during late extinction sessions would not. On the Recall session, we correlated the activity of these ensembles to freezing and found that the activity of BLA ensembles extracted during CFC and EXT1 reliably predicted relapse freezing, but later ensembles extracted during EXT2 did not (Supplementary Fig. 2g). No CA1 ensembles from any session predicted freezing nor did BLA or CA1 ensembles predict freezing in the neutral context (Supplementary Fig. 2g, h). This suggests that BLA activity patterns contributed to expression of fear relapse, but only before extinction training modified these patterns. Overall, these data indicated that context-specific reinstated fear was associated with the emergence of network states in the BLA that resembled network states during fear conditioning, suggesting that a relapsed fear memory may be represented by a similar trace as the original fear memory.
Optogenetic inhibition of the BLA DG, or CA1 fear ensemble disrupts freezing after fear reinstatement
Next, we sought to determine whether the activity of cells active during fear conditioning was necessary for expression of reinstated fear. To do this, we bilaterally injected mice in either the BLA, DG, or CA1 with a virus cocktail of AAV9-c-Fos-tTA and AAV9-TRE-ArchT-eYFP to drive expression of the light-sensitive protein archaerhodopsin (ArchT) in cells active during CFC, and subsequently implanted optic fibers above the injection sites (Fig. 3a, b). Mice then underwent two EXT sessions, the reinstating shock, and recall the following day (Fig. 3c). Mice in all three experimental groups (i.e., BLA, DG, CA1) showed significant suppression of freezing during optical inhibition (Fig. 3d, f, h). In the BLA and DG, this manipulation was reversible, as freezing increased again in the following light-off epoch (Fig. 3d, f). In the CA1, however, freezing did not increase again once the light stimulation ended (Fig. 3h). eYFP controls did not show this decrease in freezing during optical inhibition, confirming that the behavioral effect was dependent on expression of ArchT (Fig. 3e, g, i). Difference scores comparing freezing during light on vs light off epochs confirmed that mice which received inhibition of the fear ensemble in either the BLA, DG, or CA1 froze significantly less during light-on periods compared to light-off periods, and control groups froze no differently across the two light conditions (Fig. 3j).

a Schematic of viral strategy. A virus cocktail of AAV9-c-Fos-tTA and AAV9-TRE-ArchT-eYFP was infused into either the BLA, DG, or CA1 for activity-dependent expression of ArchT-eYFP. b Representative microscope images of injection sites for the BLA, DG, and CA1 groups of mice. c Reinstatement behavioral schedule. Mice had the fear ensemble labeled in either the BLA, DG, or CA1 and inhibited during Recall after Shock Reinstatement. d–i Line graphs: 2-min light OFF and ON epochs during Recall for the three experimental ArchT groups (BLA ArchT, DG ArchT, CA1 ArchT) and the three no-opsin control groups (BLA eYFP, DG eYFP, CA1 eYFP). Bar graphs: Quantification of average freezing between light OFF (white bars) vs. light ON (green bars) epochs for each group. d BLA ArchT Recall: mice froze significantly less during ON epochs than OFF epochs (Main effect of Light: F(1,8) = 38.71, ***P = 0.0002; two-way repeated measures ANOVA of Light (OFF vs ON) & Epoch (1 vs 2); n = 9 mice). e BLA eYFP Recall: mice froze no differently during ON epochs than OFF epochs (No main effect of Light: F(1,7) = 0.18, n.s., P = 0.6857; two-way repeated measures ANOVA of Light (OFF vs ON) & Epoch (1 vs 2); n = 8 mice). f DG ArchT Recall: mice froze significantly less during ON epochs than OFF epochs (Main effect of Light: F(1,11) = 10.16, **P = 0.0086; two-way repeated measures ANOVA of Light (OFF vs ON) & Epoch (1 vs 2); n = 12 mice). g DG eYFP Recall: mice froze no differently during ON epochs than OFF epochs (No main effect of Light: F(1,10) = 0.88, n.s., P = 0.3710; two-way repeated measures ANOVA of Light (OFF vs ON) & Epoch (1 vs 2); n = 11 mice). h CA1 ArchT Recall: mice froze significantly less during ON epochs than OFF epochs (Main effect of Light: F(1,8) = 6.46, *P = 0.0346; main effect of Epoch: F(1,8) = 11.00, *P = 0.0106; two-way repeated measures ANOVA of Light (OFF vs ON) & Epoch (1 vs 2); n = 9 mice). i CA1 eYFP Recall: mice froze no differently during ON epochs than OFF epochs (No main effect of Light: F(1,5) = 0.03, n.s., P = 0.8688; two-way repeated measures ANOVA of Light (OFF vs ON) & Epoch (1 vs 2); n = 6 mice). j Summary graph of freezing difference scores across all groups in Fig. 3. While mice in all three experimental groups (BLA, DG, CA1; dark blue bars) show significantly less freezing during light ON epochs, all eYFP control groups (BLA, DG, CA1; light blue bars) show no difference in freezing between light ON and light OFF epochs (from left to right: n = 18 scores from 9 mice, 25 scores from 13 mice, 18 scores from 9 mice, 16 scores from 8 mice, 22 scores from 12 mice, 12 scores from 6 mice; BLA ArchT, t17 = −4.277, ***P = 0.0005; DG ArchT, t24 = −2.781, *P = 0.0104; CA1 ArchT, t17 = −2.9033, **P = 0.0099; BLA eYFP, t15 = 0.3915, n.s., P = 0.7010; DG eYFP, t21 = −0.05076, n.s., P = 0.9600; CA1 eYFP, t11 = −0.1253, n.s., P = 0.9026; one-sample t-tests, µ0 = 0). The BLA ArchT group showed a significantly more negative difference score than the BLA eYFP group, and the DG ArchT group showed a modestly more negative difference score than the DG eYFP group (three pairwise t-tests, corrected with FDR correction; BLA ArchT vs BLA eYFP, DG ArchT vs DG eYFP, CA1 ArchT vs CA1 eYFP). BLA: t32 = −3.1676, *P = 0.0101; DG: t45 = −2.1679, #P = 0.0532; CA1: t28 = −1.3427, n.s., P = 0.1902.
Previous literature has shown that optogenetic inhibition of a hippocampal fear ensemble during fear recall after fear conditioning can disrupt freezing [29]. Since inhibition of the fear ensemble after reinstatement disrupted post-reinstatement freezing (Fig. 3), we next inhibited the DG or BLA fear ensemble during an extinction recall session, to test whether fear ensemble inhibition could drive down freezing at this stage after extinction, or whether the reinstatement process re-engaged the fear ensemble to drive fear expression post-reinstatement. After our partial extinction protocol, freezing levels were significantly reduced relative to pre-extinction levels; however, moderate levels of freezing were still present (Supplementary Fig. 3c). In contrast, after a full extinction protocol, freezing levels were reduced to near-zero levels (Supplementary Fig. 1f, g). Thus, we reasoned that the moderate levels of freezing present after partial extinction could be reduced further, and if the original fear ensemble was still driving freezing after partial extinction, we would observe a further reduction in light-induced freezing. We observed that inhibition of the DG fear ensemble led to a mild, non-significant reduction in freezing (Supplementary Fig. 3a–e, g), while inhibition of the BLA fear ensemble did not disrupt freezing (Supplementary Fig. 3f). These results suggest that extinction differentially modified the BLA and DG fear ensembles, such that BLA ensemble inhibition did not disrupt freezing during extinction, while DG ensemble activity may have still been involved in contextual fear expression following partial extinction. Importantly, this result does not rule out the possibility of a floor effect, where freezing was too low for an optogenetic manipulation to have revealed a mitigation of freezing.
Optogenetic excitation of the BLA, DG, or CA1 fear ensemble is not sufficient to mimic reinstatement
Since optogenetic inhibition of the fear ensemble of either the BLA, DG, or CA1 disrupted freezing during recall after reinstatement, we finally tested whether we could artificially drive the activity of this ensemble in replacement of the immediate shock to test if we could mimic reinstatement. In separate groups of mice, we selectively expressed ChR2 in the fear ensemble in either the BLA, DG, or CA1, and implanted optic fibers bilaterally above the injection sites (Fig. 4a). Mice underwent FC and EXT, were placed in a novel environment, and rather than receiving the reinstating shock, mice had the tagged fear ensemble stimulated for 60 s. The next day, they were placed back in the original conditioned context to assess whether the stimulation could mimic reinstatement (Fig. 4b; Supplementary Fig. 4a–c). Mice that had the BLA or CA1 fear ensemble stimulated froze no more than eYFP controls, while mice that had the DG fear ensemble stimulated showed modest, non-significant increases in freezing relative to the eYFP controls (Fig. 4c–e). These results indicated that despite a crucial role for the BLA, DG, and CA1 fear ensembles in fear learning, activity of these populations was not sufficient to drive fear reinstatement.

a Schematic of viral strategy. A viral cocktail of AAV9-c-Fos-tTA and AAV9- TRE-ChR2-eYFP was infused into the BLA, DG, or CA1 for activity-dependent induction of ChR2-eYFP. Control mice received infusion of AAV9-c-Fos-tTA and AAV9-TRE-eYFP. b Behavioral schedule to test if stimulation of a fear ensemble in a novel environment can mimic reinstatement. c Top: Freezing across Recall session after BLA fear ensemble stimulation for ChR2 and eYFP groups. Bottom: Comparison of average freezing during Recall session after BLA fear ensemble stimulation, for ChR2 and eYFP groups (t14 = 0.8265, n.s., P = 0.4224; unpaired t-test; n = 8 mice in each group). d Top: Freezing across Recall session after DG fear ensemble stimulation for ChR2 and eYFP groups. Bottom: Comparison of average freezing during Recall session after DG fear ensemble stimulation, for ChR2 and eYFP groups (t14 = 1.9134, n.s., P = 0.06597; unpaired t-test; ChR2, n = 14 mice, eYFP, n = 16 mice). e Top: Freezing across Recall session after CA1 fear ensemble stimulation for ChR2 and eYFP groups. Bottom: Comparison of average freezing during Recall session after CA1 fear ensemble stimulation, for ChR2 and eYFP groups (t14 = 1.0314, n.s., P = 0.3198; unpaired t-test; n = 8 mice in each group).
Discussion
The dynamic nature of fear memory expression constitutes a difficult problem for mitigating fear in the clinic: patients with fear-related disorders who have undergone successful treatment are still prone to relapse, and the underlying causal mechanisms facilitating fear reinstatement are largely unknown. A commonly held view is that fear extinction is not an unlearning of the original trauma; rather, a second memory develops that suppresses the original aversive memory. This raises an important notion about the nature of the ensemble regulating fear expression post-reinstatement. One idea is that the original ensemble driving fear expression and a new ensemble driving fear suppression actively compete to influence behavioral output. Under this framework, fear relapse could be the result of the fear ensemble dominating in activity. Alternatively, fear relapse might be driven by recruitment of a new, discrete cellular population that does not involve the original fear ensemble. A likely scenario is a mixture of the two, where fear relapse materializes from a partial re-emergence of the original ensemble in parallel with recruitment of new neuronal connections [47], which our c-Fos labeling, calcium imaging, and optogenetics evidence collectively support. In our study, c-Fos overlaps in the DG and calcium imaging in the BLA demonstrated that the fear ensemble becomes less active across extinction and is partly re-engaged after fear reinstatement, while optogenetic inhibition of the fear ensemble in the BLA, DG, and CA1 after fear reinstatement illustrated that once the fear ensemble is re-engaged after reinstatement, its activity is necessary to drive post-reinstatement freezing.
Calcium imaging and c-Fos labeling during the fear reinstatement schedule enabled us to capture network dynamics from the hippocampus and amygdala over multiple timescales, shedding light on the activity of these regions over fear reinstatement. Consistent with prior reports of BLA cell populations up- and down-regulating their activity during extinction learning [36, 43], we observed decorrelation of the BLA population vector from the initial fear-encoding state over repeated exposures to the conditioned context. Similarly, we observed a decrease in DG fear ensemble reactivation across extinction learning. This time-dependent transformation has previously been depicted in numerous brain regions as “representational drift” [48,49,50,51,52,53], but these studies all described population states that monotonically drifted away from a reference session. In the present study, the BLA representation (Fig. 2e) and DG representation (Fig. 1h) exhibited similar dynamics, but in contrast to past work, they both regressed their neural trajectory back towards the initial representation after fear reinstatement. While the decrease in overlap across extinction we observed might be attributed simply to the passage of time, our calcium imaging results suggest that this decorrelation is driven by extinction rather than time, since the decorrelation in the BLA was only observed in the shock context and not a neutral context (Fig. 2e). This re-engagement of the fear ensemble leads us to believe that fear reinstatement may be restoring a remote memory trace similar to how optogenetic activation can artificially induce memory retrieval [25, 30, 54]. Notably, this decorrelation and regression back towards the initial representation was not detected using c-Fos imaging in the BLA. One possibility is that after our partial extinction protocol – where moderate levels of freezing are still present – both fear and extinction cells are sufficiently active to express immediate early genes [33, 34], which might mask changes in ensemble overlap. In contrast, calcium imaging offered a sensitive enough readout of the population activity to detect changes in fear ensemble activity in real-time across extinction and reinstatement.
Interestingly, the neural patterns associated with fear expression are still retrievable after putative circuit remodeling over extinction learning and fear reinstatement [14, 32, 44, 55]. Our ability to observe and manipulate the original fear ensemble after reinstatement is suggestive of a latent representation of the original memory that persists and coexists with the newly formed extinction memory [34, 56], and partially re-emerges after fear reinstatement. However, this new extinction memory may also facilitate local synaptic remodeling that modifies the original fear memory, which may explain why inhibition of the fear ensemble in DG and BLA after partial extinction did not fully eliminate freezing (Supplementary Fig. 3d–g) [32, 57]. The re-emergence of the original fear memory may also depend on strict endogenous plasticity mechanisms, which can explain why we failed to optically induce relapse through broad stimulation of the BLA, DG, or CA1 fear ensembles after extinction (Fig. 4). These results suggest that the natural endogenous fear reinstatement process might require certain temporal activity patterns for modifying the original fear ensemble that could not be artificially produced through blanketed BLA or hippocampal stimulation alone. For example, recent work showed that sequential activity from triplets of BLA neurons preceded fear learning [58] and similar patterns may be required for fear reinstatement. Fear reinstatement may then be recruiting a subset of the original fear ensemble while forming new synaptic linkages with a novel cell population. Others have shown that unique memories reside in patterns of connectivity between memory-encoding – or engram – cells in the hippocampal system [59,60,61]. In the context of our study, reinstatement could be modifying these functional linkages to engage a new set of engram cells, possibly those that are highly excitable at the time of the experience [62,63,64], forming a reinstatement ensemble that is similar, but not identical, to the original fear ensemble. In accordance with this idea, post-reinstatement recall activates a large proportion, but not all, of the original fear ensemble (Fig. 1g–i).
Notably, past studies that have promoted learning via artificial stimulation of neural ensembles have done so by inducing associations between conditioned and unconditioned stimuli [25, 31, 65]. Here, however, fear ensemble stimulation during reinstatement attempted to re-activate the latent fear memory representation (Fig. 4). Since the fear ensemble stimulation alone was insufficient to drive the re-emergence of context-specific fear, this suggests that fear reinstatement is mediated by an alternate mechanism that requires the presentation of the unconditioned stimulus and cannot be mimicked by optogenetic stimulation alone. Consistent with this, overlaps in the DG were not high during the reinstating shock but they increased the following day during post-reinstatement fear recall (Fig. 1h), suggesting that the fear ensemble is not highly engaged during the reinstating shock to drive fear reinstatement. Moreover, Yoshii et al. demonstrated that chemogenetic activation of a brain-wide fear ensemble after extinction was sufficient to increase freezing in the conditioned context; however, this increase in freezing was transient such that freezing returned to low levels during memory recall the following day [37]. This is further evidence suggesting that while the fear ensemble partly drives freezing following fear reinstatement, increasing its activity is insufficient to recapitulate the necessary plasticity that drives reinstatement. It is possible, for example, that the nociceptive sensory inputs experienced during the immediate shock provide the necessary input to drive fear relapse [65], after which fear expression is mediated, in part, by the original fear-encoding ensemble (Fig. 3). A notable limitation of our study was, since mice received the reinstating shock in a novel context, the contribution of context alone in driving fear relapse could not be disentangled from context and shock. Future studies investigating the role that sensory stimuli play for fear relapse would provide insight into such a possibility.
While our optogenetic and histological overlap experiments suggest a re-emergence of the fear ensemble in the DG, we were unable to perform calcium imaging in DG due to the technical limitations of accessing this region without significant damage to overlying hippocampal subareas. Nonetheless, we report that CA1 exhibits only marginally significant (p = 0.054, Fig. 2f) reversion of calcium dynamics to a fear conditioning-like state after reinstatement. This may be due to the functional differences between DG and CA1, with CA1 possibly responding to more contextual features of the experience [66] or being overall more subject to plasticity than DG. As others have found, day-to-day dynamics of spatial activity in DG are more stable than in CA1 [67]. Until methods are developed that allow imaging of DG while still preserving superficial hippocampal areas, it remains unknown whether the real-time network activity of DG exhibits relapse-induced reactivation of fear ensembles. However, recent work agrees with our prediction that extinction suppresses fear-related activity in the DG while those activity patterns are retrieved during spontaneous recovery, another form of fear relapse [34].
Further work exploring the competing interactions of cellular networks across fear learning and fear suppression could provide important insight into how the brain competes for the expression of fear throughout fear extinction and relapse. Our study utilizing solely male mice leaves unanswered whether female mice would differ in behavioral expression of fear relapse, and whether the underlying neural mechanisms vary. For example, while it’s been shown that male and female mice learn similarly under contextual and cued fear conditioning paradigms [68, 69], those studies have shown that female mice exhibit higher likelihoods of spontaneous recovery, while another has shown no differences in the rates of spontaneous recovery [34]. Moreover, fear retrieval has been shown to differentially recruit hippocampal and amygdalar circuits [70]. Ultimately, a deeper understanding of how fear memories are modified by time and experience may help guide development of treatments for trauma-related disorders, and these findings point to hippocampal- and BLA-mediated engrams as key nodes contributing to the re-emergence of a contextual fear memory.
References
Goode, TD, et al. 10 - Neural Circuits for Fear Relapse. In Neurobiology of Abnormal Emotion and Motivated Behaviors (Sangha, S and Foti, D eds), 2018. pp. 182–202, Academic Press.
Bouton ME, Bolles RC. Role of conditioned contextual stimuli in reinstatement of extinguished fear. J Exp Psychol Anim Behav Process. 1979;5:368–78.
Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96.
Halladay LR, et al. Reinstatement of extinguished fear by an unextinguished conditional stimulus. Front Behav Neurosci. 2012;6:18.
Goode TD, Maren S. Animal models of fear relapse. ILAR J. 2014;55:246–58.
Vervliet B, et al. Fear extinction and relapse: state of the art. Annu Rev Clin Psychol. 2013;9:215–48.
Kearns MC, et al. Early interventions for PTSD: a review. Depress Anxiety. 2012;29:833–42.
LaBar KS, Phelps EA. Reinstatement of conditioned fear in humans is context dependent and impaired in amnesia. Behav Neurosci. 2005;119:677–86.
Haaker J, et al. A review on human reinstatement studies: an overview and methodological challenges. Learn Mem. 2014;21:424–40.
Maren S. Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci. 2001;24:897–931.
Maren S, Holmes A. Stress and fear extinction. Neuropsychopharmacology. 2016;41:58–79.
Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–51.
Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72.
Bocchio M, et al. Synaptic plasticity, engrams, and network oscillations in amygdala circuits for storage and retrieval of emotional memories. Neuron. 2017;94:731–43.
LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–84.
Rescorla RA, Cunningham CL. Erasure of reinstated fear. Anim Learn Behav. 1977;5:386–94.
Bouton ME. Context, time, and memory retrieval in the interference paradigms of pavlovian learning. Psychological Bull. 1993;114:80–99.
Westbrook FR, et al. Reinstatement of fear to an extinguished conditioned stimulus: Two roles for context. J Exp Psychol-Anim Behav Process. 2002;28:97–110.
Giustino TF, et al. Locus coeruleus toggles reciprocal prefrontal firing to reinstate fear. Proc Natl Acad Sci USA. 2019;116:8570–5.
Hitora-Imamura N, et al. Prefrontal dopamine regulates fear reinstatement through the downregulation of extinction circuits. Elife. 2015;4:e08274.
Goode TD, et al. Reversible inactivation of the bed nucleus of the stria terminalis prevents reinstatement but not renewal of extinguished fear. eNeuro. 2015;2:ENEURO.0037-15.2015.
Fu J, et al. Region-specific roles of the prelimbic cortex, the dorsal CA1, the ventral DG and ventral CA1 of the hippocampus in the fear return evoked by a sub-conditioning procedure in rats. Neurobiol Learn Mem. 2016;128:80–91.
Cammarota M, et al. Inhibition of mRNA and protein synthesis in the CA1 region of the dorsal hippocampus blocks reinstallment of an extinguished conditioned fear response. J Neurosci. 2003;23:737–41.
Goode TD, Maren S. Common neurocircuitry mediating drug and fear relapse in preclinical models. Psychopharmacol (Berl). 2019;236:415–37.
Ramirez S, et al. Creating a false memory in the hippocampus. Science. 2013;341:387–91.
Reijmers LG, et al. Localization of a stable neural correlate of associative memory. Science. 2007;317:1230–3.
Nakazawa Y, et al. Memory retrieval along the proximodistal axis of CA1. Hippocampus. 2016;26:1140–8.
Ghandour K, et al. Orchestrated ensemble activities constitute a hippocampal memory engram. Nat Commun. 2019;10:2637.
Denny CA, et al. Hippocampal memory traces are differentially modulated by experience, time, and adult neurogenesis. Neuron. 2014;83:189–201.
Redondo RL, et al. Bidirectional switch of the valence associated with a hippocampal contextual memory engram. Nature. 2014;513:426–30.
Ohkawa N, et al. Artificial association of pre-stored information to generate a qualitatively new memory. Cell Rep. 2015;11:261–9.
Davis P, et al. Cellular and oscillatory substrates of fear extinction learning. Nat Neurosci. 2017;20:1624–33.
Tronson NC, et al. Segregated populations of hippocampal principal CA1 neurons mediating conditioning and extinction of contextual fear. J Neurosci. 2009;29:3387–94.
Lacagnina AF, et al. Distinct hippocampal engrams control extinction and relapse of fear memory. Nat Neurosci. 2019;22:753–61.
Khalaf O, et al. Reactivation of recall-induced neurons contributes to remote fear memory attenuation. Science. 2018;360:1239–42.
Herry C, et al. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–6.
Yoshii T, et al. Pharmacogenetic reactivation of the original engram evokes an extinguished fear memory. Neuropharmacology. 2017;113:1–9.
Ramirez S, et al. Activating positive memory engrams suppresses depression-like behaviour. Nature. 2015;522:335–9.
Resendez SL, et al. Visualization of cortical, subcortical and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat Protoc. 2016;11:566–97.
Mukamel EA, et al. Automated analysis of cellular signals from large-scale calcium imaging data. Neuron. 2009;63:747–60.
Sheintuch L, et al. Tracking the same neurons across multiple days in Ca(2+) imaging data. Cell Rep. 2017;21:1102–15.
Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19.
Grewe BF, et al. Neural ensemble dynamics underlying a long-term associative memory. Nature. 2017;543:670–5.
Hartley ND, et al. Dynamic remodeling of a basolateral-to-central amygdala glutamatergic circuit across fear states. Nat Neurosci. 2019;22:2000–12.
Thistlethwaite DL, Campbell DT. Regression-discontinuity analysis - an alternative to the ex-post-facto experiment. J Educ Psychol. 1960;51:309–17.
Lopes-dos-Santos V, et al. Detecting cell assemblies in large neuronal populations. J Neurosci Methods. 2013;220:149–66.
Clem RL, Schiller D. New learning and unlearning: strangers or accomplices in threat memory attenuation? Trends Neurosci. 2016;39:340–51.
Mau W, et al. The same hippocampal CA1 population simultaneously codes temporal information over multiple timescales. Curr Biol. 2018;28:1499–508 e4.
Rubin A, et al. Hippocampal ensemble dynamics timestamp events in long-term memory. Elife. 2015;4:e12247.
Mankin EA, et al. Neuronal code for extended time in the hippocampus. Proc Natl Acad Sci USA. 2012;109:19462–7.
Rule ME, et al. Causes and consequences of representational drift. Curr Opin Neurobiol. 2019;58:141–7.
Driscoll LN, et al. Dynamic reorganization of neuronal activity patterns in parietal cortex. Cell. 2017;170:986–99 e16.
Mau, W et al. The brain in motion: How ensemble fluidity drives memory-updating and flexibility. Elife. 2020;9.
Liu X, et al. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012;484:381–5.
Maren S. Out with the old and in with the new: Synaptic mechanisms of extinction in the amygdala. Brain Res. 2015;1621:231–8.
Maren S. Seeking a spotless mind: extinction, deconsolidation, and erasure of fear memory. Neuron. 2011;70:830–45.
Trouche S, et al. Fear extinction causes target-specific remodeling of perisomatic inhibitory synapses. Neuron. 2013;80:1054–65.
Reitich-Stolero T, Paz R. Affective memory rehearsal with temporal sequences in amygdala neurons. Nat Neurosci. 2019;22:2050–9.
Ryan TJ, et al. Memory. Engram cells retain memory under retrograde amnesia. Science. 2015;348:1007–13.
Abdou K, et al. Synapse-specific representation of the identity of overlapping memory engrams. Science. 2018;360:1227–31.
Tonegawa S, et al. The role of engram cells in the systems consolidation of memory. Nat Rev Neurosci. 2018;19:485–98.
Yiu AP, et al. Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron. 2014;83:722–35.
Rashid AJ, et al. Competition between engrams influences fear memory formation and recall. Science. 2016;353:383–7.
Cai DJ, et al. A shared neural ensemble links distinct contextual memories encoded close in time. Nature. 2016;534:115–8.
Vetere G, et al. Memory formation in the absence of experience. Nat Neurosci. 2019;22:933–40.
Tanaka KZ, et al. The hippocampal engram maps experience but not place. Science. 2018;361:392–7.
Hainmueller T, Bartos M. Parallel emergence of stable and dynamic memory engrams in the hippocampus. Nature. 2018;558:292–6.
Matsuda S, et al. Sex differences in fear extinction and involvements of extracellular signal-regulated kinase (ERK). Neurobiol Learn Mem. 2015;123:117–24.
Fenton GE, et al. Sex differences in learned fear expression and extinction involve altered gamma oscillations in medial prefrontal cortex. Neurobiol Learn Mem. 2016;135:66–72.
Keiser AA, et al. Sex differences in context fear generalization and recruitment of hippocampus and amygdala during retrieval. Neuropsychopharmacology. 2017;42:397–407.
Acknowledgements
We thank Dr. Joshua Sanes and his lab at the Center for Brain Science, Harvard University, for providing laboratory space within which the initial experiments were conducted, the Center for Brain Science Neuroengineering core for providing technical support, and the Society of Fellows at Harvard University for their support. We also thank Dr. Susumu Tonegawa and his lab for providing the activity-dependent virus cocktail, Dr. Chris MacDonald for consultation on behavioral schedules, and Drs. Leon Reijmers and Patrick Davis, for their help with formulating this project and for their feedback throughout. We also thank Vardhan Dani and Inscopix for their technical assistance as well as Helen Fawcett and the NSF Neurophotonics Research Traineeship Program.
Funding
This work was supported by an NIH Early Independence Award (DP5 OD023106-01), an NIH Transformative R01 Award, a Young Investigator Grant from the Brain and Behavior Research Foundation, a Ludwig Family Foundation grant, and the McKnight Foundation Memory and Cognitive Disorders award.
Author information
Authors and Affiliations
Contributions
YZ and SR conceived the study. YZ, WM, and SR designed experiments and YZ, WM, AM, and SR analyzed data. YZ, WM, CC, AM, EO, ED, SLG, EM, and MS conducted experiments. YZ, WM, and SR wrote the manuscript; all authors edited and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zaki, Y., Mau, W., Cincotta, C. et al. Hippocampus and amygdala fear memory engrams re-emerge after contextual fear relapse. Neuropsychopharmacol. 47, 1992–2001 (2022). https://doi.org/10.1038/s41386-022-01407-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01407-0
This article is cited by
-
Memory Trace for Fear Extinction: Fragile yet Reinforceable
Neuroscience Bulletin (2023)
-
Repeated Use of Morphine Induces Anxiety by Affecting a Proinflammatory Cytokine Signaling Pathway in the Prefrontal Cortex in Rats
Molecular Neurobiology (2023)
-
Reactivating hippocampal-mediated memories during reconsolidation to disrupt fear
Nature Communications (2022)
