
Abstract
The anterior cingulate cortex (ACC) is implicated in many pathologies, including depression, anxiety, substance-use disorders, and pain. There is also evidence from brain imaging that the ACC is hyperactive during periods of opioid withdrawal. However, there are limited data contributing to our understanding of ACC function at the cellular level during opioid withdrawal. Here, we address this issue by performing ex vivo electrophysiological analysis of thick-tufted, putative dopamine D2 receptor expressing, layer V pyramidal neurons in the ACC (ACC L5 PyNs) in a mouse model of spontaneous opioid withdrawal. We found that escalating doses of morphine (20, 40, 60, 80, and 100 mg/kg, i.p. on days 1–5, respectively) injected twice daily into male C57BL/6 mice evoked withdrawal behaviors and an associated withdrawal-induced mechanical hypersensitivity. Brain slices prepared 24 h following the last morphine injection showed increases in ACC L5 thick-tufted PyN-intrinsic membrane excitability, increases in membrane resistance, reductions in the rheobase, and reductions in HCN channel-mediated currents (IH). We did not observe changes in intrinsic or synaptic properties on thin-tufted, dopamine D1-receptor-expressing ACC L5 PyNs recorded from male Drd1a-tdTomato transgenic mice. In addition, we found that chemogenetic inhibition of the ACC blocked opioid-induced withdrawal and withdrawal-induced mechanical hypersensitivity. These results demonstrate that spontaneous opioid withdrawal alters neuronal properties within the ACC and that ACC activity is necessary to control behaviors associated with opioid withdrawal and withdrawal-induced mechanical hypersensitivity. The ability of the ACC to regulate both withdrawal behaviors and withdrawal-induced mechanical hypersensitivity suggests overlapping mechanisms between two seemingly distinguishable behaviors. This commonality potentially suggests that the ACC is a locus for multiple withdrawal symptoms.
Similar content being viewed by others


Introduction
Opioid-use disorder (OUD) is characterized. by chronic relapses driven, in part, by the desire to avoid withdrawal symptoms [1,2,3]. Withdrawal symptoms are caused by the cessation of opioid use in opioid-dependent patients. In the absence of opioids, the central nervous system becomes hyper-excitable [4]. This is due, in part, to compensatory changes put in place to overcome opioid-induced decreases in neuronal excitability. Evidence suggests that these compensatory changes take place within spinal and supraspinal regions that are heavily regulated by opioid-receptor activation, including brain regions involved in modulating autonomic activity and pain [5,6,7,8]. Therefore, opioid withdrawal is a multifaceted syndrome, resulting in severe flu-like symptoms, anxiety/restlessness, and hyperalgesia [9,10,11], which is why identifying a locus where these symptoms integrate may be crucial for understanding and perhaps treating opioid dependence.
The anterior cingulate cortex (ACC) is a brain region capable of integrating a broad range of neuronal signals spanning multiple physiological networks. For example, the ACC is implicated in many cognitive functions, including arousal, reward predictions, error processing, attention, pain, and decision-making [12,13,14,15,16,17,18]. However, the characteristic neuronal signatures that contribute to ACC activity during opioid withdrawal remain unknown. The ACC consists of layers I, II–III, and V–VI in both humans and rodents (layer IV is not present) [19, 20]. Layer I contains interneurons, layers II–III contain predominantly pyramidal neurons (PyNs), and layers V–VI contain both PyNs and interneurons, with the PyNs in layers V-VI serving as the major projection neurons of the ACC. These projection neurons target key brain regions involved in reward and motivation, arousal, and pain modulation [21, 22]. Evidence suggests that the neuronal excitability in the ACC is altered during opioid withdrawal, which may influence downstream signaling. Using blood-oxygenated-level-dependent (BOLD) functional MRI (fMRI), Lowe et al. identified increases in ACC activation upon naloxone-precipitated withdrawal in morphine-dependent mice [23]. Similarly, in humans, naloxone-precipitated withdrawal increased neuronal activity within the ACC [24]. It was shown, in rats, that ACC glutamate levels are elevated following naloxone-precipitated opioid withdrawal [25]. In humans, glutamate levels in the ACC are positively correlated with the number of previous experiences of opioid withdrawal [26], and during early withdrawal from other drugs of abuse, including alcohol, ACC glutamate levels are increased [27, 28]. Based on evidence that the ACC may play a key role in the opioid withdrawal syndrome, we aimed to examine key properties that regulate neuronal excitability within the ACC, including synaptic transmission and the intrinsic excitability of PyNs in layer V (L5) of the ACC in a mouse model of spontaneous opioid withdrawal.
Materials and methods
All experiments were done in accordance with procedures approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee.
Results
Escalating morphine administration evokes spontaneous opioid withdrawal and withdrawal-induced mechanical hypersensitivity
In order to study the effects of spontaneous withdrawal, we induced tolerance in mice by administering escalating doses of morphine (i.p.) over 5 days (Fig. 1A). Twenty-four hours following the last morphine injection, spontaneous withdrawal was measured. First, we observed a significant decrease in total locomotor activity in morphine- vs. saline-treated mice (t(9) = 6.530, p = 0.0001; Student’s unpaired t-test) (Fig. 1B). Second, we observed a statistically significant increase in the withdrawal score in mice that received morphine compared with saline controls (t(30) = 11.14, p < 0.0001; Student’s unpaired t-test) (Fig. 1C). This consisted of a statistically significant increase in the tremors and shakes (t(30) = 9.604, p < 0.0001; Student’s unpaired t-test) (Fig. 1D), number of jumps (t(30) = 4.531, p < 0.0001; Student’s unpaired t-test) (Fig. 1E), and paw flutters (t(30) = 5.882, p < 0.0001; Student’s unpaired t-test) (Fig. 1F). In addition, there was a statistically significant increase in piloerection (elevation of hair follicles due to contraction of arrectores pilorum muscles) in mice treated with morphine vs. saline (presence of piloerection: saline: 0 of 16 mice; morphine: 14 of 16 mice) (t(30) = 10.25, p < 0.0001; Student’s unpaired t-test) (data not shown). No significant difference was observed in grooming behavior (Fig. 1G) (t(30) = 1.271, p = 0.2135; Student’s unpaired t-test). These results corroborate findings from previous reports that showed that mice exposed to escalating doses of morphine experience spontaneous withdrawal-like symptoms [29,30,31,32].

A Experimental timeline. Escalating morphine was administered twice daily over a course of 5 days. Withdrawal monitoring was performed in an open field arena 24 h following the last dose. Von Frey assessments were performed at withdrawal days 1, 4, and 7, corresponding to experimental days 6, 9, and 12. B (Left) Representative traces showing mouse locomotor behavior over the course of the 30 min withdrawal monitoring. (Right) Summary graph showing that the total distance traveled was significantly decreased in morphine-treated mice (saline: n = 5; morphine: n = 6). C Summary graph showing the calculated withdrawal score in saline- vs. morphine-treated mice (saline: n = 16; morphine: n = 16) with each component broken down into individual summary graphs, including (D) tremors and shakes, (E) jumps, (F) paw flutters, and (G) grooming sessions. H Summary graph showing mechanical hypersensitivity on withdrawal days 1, 4, and 7 in saline vs. morphine-treated mice (saline: n = 10; morphine: n = 10). *p < 0.05, **p < 0.01, ***p < 0.001.
Given that pain thresholds are reduced during opioid withdrawal [33], we measured mechanical hypersensitivity in mice using an electronic von Frey apparatus. We found that on withdrawal days 1 and 4, morphine-treated mice had a reduced pain threshold compared with saline controls, but this effect was no longer present by abstinence day 7 (F(2,36) = 6.684, p = 0.0034; two-way repeated-measures ANOVA with Bonferroni post-test) (Fig. 1H). The sustained mechanical hypersensitivity during withdrawal days 1–4 coincides with a previous report [33].
ACC L5 PyN-intrinsic excitability is enhanced during spontaneous opioid withdrawal
Next, we investigated the effects of spontaneous withdrawal on ACC L5 thick-tufted, putative dopamine D2-receptor expressing (Supplementary Fig. 1) PyN excitability. To assess neuronal excitability, we measured the number of action potentials generated during depolarizing current injections 24 h after the last morphine injection (Fig. 2A). This approach is often used to measure the intrinsic membrane excitability, which sets the action potential threshold and determines the firing frequency [34,35,36,37,38]. We observed that ACC L5 thick-tufted PyNs in morphine-exposed mice had a significant increase in the number of action potentials compared with saline controls (F(8,232) = 8.110, p < 0.0001; two-way repeated-measure ANOVA with Bonferroni post-test) (Fig. 2B).

A (Top) Experimental timeline. All recordings occurred on withdrawal day 1, starting ~2 h following withdrawal monitoring. (Right) Representative low (5×)- and high (40×)-magnification image of the ACC recording area and patched L5 PyN, respectively. (Left) Locations of recorded cells within L5 of the ACC (saline=black, morphine=magenta). B Summary data showing that 24 h following escalating morphine injections, the number of action potentials fired in L5 PyNs is significantly increased at current injections of 150 through 400 pA. Scale bars: 20 mV, 250 ms. C Representative traces (left) and a summary graph showing that escalating morphine administration decreases the minimal amount of current required to fire an action potential (rheobase). Scale bars: full trace = 20 mV, 250 ms; action potential inset = 40 mV, 2.50 ms. D Summary graph showing that the slope of membrane depolarization during the ramp current injection was significantly higher in morphine-treated mice. E Graph showing a significant negative correlation between the rheobase slope and rheobase. Scale bar: 40 mV, 500 ms. F Summary graph showing that morphine-treated mice had a significant increase in membrane resistance on L5 PyNs in the ACC. G Summary graph showing that morphine-treated mice had a significantly larger voltage deflection in response to hyperpolarizing current injections. (Inset) Example trace showing that the voltage deflection was calculated as the change in mV between the baseline and steady-state potential during a hyperpolarizing current injection. Scale bars: 4 mV, 250 ms. H An action potential phase-plot analysis averaged from ACC L5 PyNs of saline (black)- or morphine (magenta)-treated mice. (Inset) Individual phase-plot analyses from ACC L5 PyNs of saline (black)- or morphine (magenta)-treated mice. Scale bar: 300 mV/ms, 50 mV. For all assays, saline: 15/3; Morphine: 16/3. Data are presented as individual cells with ±SEM error bars. *p < 0.05, **p < 0.01, ***p < 0.001.
We next measured the minimum current required to elicit an action potential (i.e., rheobase), with decreases and increases in the rheobase associated with enhanced and reduced neuronal excitability, respectively [39, 40]. We observed that during spontaneous opioid withdrawal, morphine-exposed mice had a significant decrease in the rheobase, measured as the injected current required to elicit an action potential (t(29) = 3.118, p = 0.0041; Student’s unpaired t-test) (Fig. 2C). This morphine-induced decrease in the rheobase was due to a significant increase in the rheobase slope (t(29) = 2.448, p = 0.021; Student’s unpaired t-test) (Fig. 2D, E), which is associated with a greater voltage deflection in response to a given injected current.
The observed morphine-induced increase in the IME and increase in the slope of the rheobase suggest that a depolarizing current produces a greater voltage change at the soma and a higher likelihood of an action potential discharge. This result would be expected if the membrane resistance was increased during morphine-induced spontaneous withdrawal. To investigate this, we measured the membrane resistance of ACC L5 thick-tufted PyNs during spontaneous withdrawal and found that morphine-treated mice had a statistically significant increase in the membrane resistance (t(29) = 2.830, p = 0.0084; Student’s unpaired t-test) (Fig. 2F). This result coincides with the morphine-induced increase in the voltage deflection measured in response to hyperpolarizing current injections (F(3,87) = 9.245, p < 0.0001; two-way repeated-measures ANOVA with Bonferroni post-test) (Fig. 2G).
Given that increases in membrane resistance are caused, in part, by the closure of membrane-bound voltage-gated channels [41], we assessed action-potential properties of ACC L5 thick-tufted PyNs during spontaneous withdrawal. We saw no significant changes in action-potential characteristics, including action-potential threshold (t(29) = 0.2926, p = 0.7719; Student’s unpaired t-test), peak (t(29) = 0.4039, p = 0.6893; Student’s unpaired t-test), duration (t(29) = 0.0330, p = 0.9739; Student’s unpaired t-test), after hyper polarization amplitude (t(29) = 0.9218, p = 0.3642; Student’s unpaired t-test), and after hyperpolarization time (t(29) = 1.053, p = 0.3011; Student’s unpaired t-test). These results are consistent with the lack of action potential changes observed after phase-plot analyses (Fig. 2H). These results suggest that voltage-gated Na+, K+, and Ca2+ channels, that are involved in action potential generation, do not contribute to the observed increases in membrane resistance during spontaneous withdrawal.
ACC L5 PyN HCN channel-mediated current is decreased during spontaneous opioid withdrawal
Increases in IME and membrane resistance can be caused by reductions in the number of open channels at resting membrane potentials, thereby allowing smaller currents to have larger influences on membrane voltage. The HCN channel is expressed on ACC L5 thick-tufted PyNs [12, 42,43,44], and blocking HCN-channel-mediated currents increases neuronal IME and membrane resistance [45, 46]. Furthermore, a decrease in HCN-mediated current is observed in other hyper-excitable ACC states, such as chronic pain [43, 47]. Because of this, we next investigated whether HCN-channel-mediated current (IH) is altered on ACC L5 thick-tufted PyNs 24 h following escalating morphine exposure. To isolate IH currents, we injected hyperpolarizing voltage steps in the presence of TTX, tetraethylammonium, barium, and 4-aminopyridine. This evoked a slowly activating inward current indicative of IH (Fig. 3B). We measured the IH amplitude by subtracting the instantaneous inward-current peak amplitude (mediated by activated HCN channels during the initial hyperpolarization step) from the steady-state amplitude (mediated by constant HCN-channel activation and deactivation overtime during the hyperpolarizing step) at each voltage step and found a significant decrease in total IH in morphine- vs. saline-treated mice (F(6,138) = 7.518, p < 0.0001; two-way repeated-measure ANOVA with Bonferroni post-test) (Fig. 3B).

A Locations of recorded cells within L5 of the ACC for IH analysis. B (Left) Voltage steps and the resulting representative traces from PyN recordings taken from saline- or morphine-treated mice. (Right) Escalating morphine administration significantly decreased the maximal IH amplitude at voltage steps of −110, −120, and −130 mV. Scale bars = 1000 pA, 500 ms. C Summary graph showing that the IH current density on ACC L5 PyNs was significantly decreased at voltage steps of −110, −120, and −130 mV in morphine-treated mice. D (Left) Representative traces showing that tail currents are observed following hyperpolarizing voltage steps. (Right) Steady-state deactivation curves derived from IH tail currents showed no difference in IH voltage dependence or the V50 values (inset) between saline- and morphine-treated animals. Scale bars = 1000 pA, 500 ms, and 50 pA, 50 ms (inset). E (Left) Representative traces from ACC L5 PyN recordings taken from saline-treated mice after bath perfusion of 1 μM or 10 μM ZD7288. (Right) Summary graph showing that 10 μM ZD7288 (n = 4/2) was abolished, while 1 μM ZD7288 (n = 5/2) partially blocked HCN-mediated currents. Representative traces and summary graphs of saline and morphine groups are the same as shown in Fig. 3B. Scale bars: saline, morphine = 500 pA, 500 ms; 1 and 10 μM ZD7288 = 250 pA, 500 ms. F (Left) Representative traces showing the firing frequency of ACC L5 PyNs following a 400 pA current injection. Scale bars: 40 mV, 200 ms. (Right) Summary graph showing that bath perfusion of ZD7288 (1 μM) (n = 10/2) evoked increases in the number of action potentials fired in ACC L5 PyNs, which was not significantly different from morphine-treated mice. The summary of saline and morphine groups are the same as shown in Fig. 2B. Asterisk denotes a significant difference between saline and all other groups. For assays (A–D), saline: n = 13/3; morphine: n = 12/3. Data are presented as mean with ± SEM error bars. *p < 0.05, **p < 0.01. n/m = cells/animal.
To determine whether the observed changes in IH were mediated by changes in the neuronal surface area [48, 49], we measured HCN-channel current density (i.e., HCN-channel-mediated current divided by the neuronal capacitance, which is proportional to the neuronal surface area). We found that IH current density was significantly decreased in neurons from morphine-exposed mice compared with saline controls at hyperpolarizing voltage steps of −110 to −130 mV (−110 mV: t(23) = 2.233, p = 0.0355; Student’s unpaired t-test; −120 mV: t(23) = 2.186, p = 0.0393; Student’s unpaired t-test; −130 mV: t(23) = 2.605, p = 0.0158; Student’s unpaired t-test) (Fig. 3C). These results suggest that the neuronal surface area did not significantly influence morphine-induced reductions in IH, which is supported by a non-significant change in neuronal capacitance between groups measured using whole-cell electrophysiology (saline: 174.8 ± 5.894 pF; morphine: 171.9 ± 4.794 pF; t(29) = 0.3872, p = 0.7014; Student’s unpaired t-test).
Because the observed decrease in maximal IH amplitude may be caused by either a decrease in HCN-channel numbers or a leftward (hyperpolarized) shift in IH activation, we measured whether spontaneous opioid withdrawal influenced the voltage-dependency of HCN channels. To do this, we measured tail currents upon returning to −70 mV following hyperpolarizing steps. We measured the tail-current peak amplitude, which was plotted as a function of the preceding voltage step. We then fit a Boltzmann function to obtain the IH steady-state deactivation curve. We observed no difference in the deactivation curves between saline and morphine groups (saline V50: −98.37 ± 1.999, morphine V50: −97.80 ± 1.914, t(23) = 0.2047, p = 0.8396; Student’s unpaired t-test) (Fig. 3D).
Last, we investigated whether pharmacological block of HCN-channel-mediated currents could mimic the increases in IME observed in mice undergoing spontaneous opioid withdrawal. To do this, we recorded from ACC L5 thick-tufted PyNs while bath-applying ZD7288, the HCN-channel blocker, from brain slices derived from saline-treated mice. Bath perfusion of 10 μM ZD7288 resulted in a complete block of HCN channel-mediated currents, while 1 μM ZD7288 resulted in a partial block of HCN-mediated currents. This partial block was not significantly different from morphine-treated mice but was significantly different from saline-treated mice, thus mimicking the effect observed during spontaneous opioid withdrawal (F(18,180) = 14.33, p < 0.0001; two-way repeated-measure ANOVA with Bonferroni post-test) (Fig. 3E). Therefore, we bath perfused 1 μM ZD7288 and found that a partial pharmacological block of HCN channels resulted in increases in IME that were not significantly different from morphine-treated mice, but were significantly different from saline-treated mice (F(16,304) = 5.242, p < 0.0001; two-way repeated-measure ANOVA with Bonferroni post-test) (Fig. 3F).
Overall, these results suggest that during spontaneous opioid withdrawal, there is a decrease in maximal IH, but no change in the voltage-dependent deactivation of HCN channels in ACC L5 thick-tufted PyNs, perhaps due to a decrease in total HCN-channel expression or a decrease in available membrane-bound channels.
Spontaneous opioid withdrawal does not alter excitatory or inhibitory synaptic transmission on PyNs in the ACC
To determine whether ACC alterations during spontaneous withdrawal were isolated to membrane properties and not due to changes occurring at the synapse, we next measured both sEPSCs and sIPSCs. To isolate sPSCs, we dialyzed neurons with a Cs-based internal solution, which contains blockers of voltage-gated channels. Therefore, the currents recorded are expected to be synaptically mediated without being influenced by active-channel properties. We found no significant difference in sEPSC amplitude (t(29) = 1.254, p = 0.2200; Student’s unpaired t-test) or frequency (t(29) = 0.6144, p = 0.5437; Student’s unpaired t-test) on thick-tufted PyNs in the ACC from saline- or morphine-treated mice at 1 d abstinence (Fig. 4A–C). Likewise, we found no significant changes in either sIPSC amplitude (t(20) = 0.3336, p = 0.7422; Student’s unpaired t-test) or frequency (t(20) = 0.4385, p = 0.6667; Student’s unpaired t-test) (Fig. 4D–F).

A Representative traces showing spontaneous excitatory postsynaptic currents (sEPSC) recorded from ACC L5 PyNs in saline-(black) or morphine (magenta)-treated mice (saline: n = 14/4; morphine: n = 17/3). Scale bars: top = 25 pA, 100 ms; bottom = 50 pA, 10 ms. Cumulative probability and summary graph (inset) showing the frequency (B) and amplitude (C) of sEPSCs during spontaneous withdrawal. D Representative traces showing spontaneous inhibitory postsynaptic currents (sIPSC) recorded from ACC L5 PyNs in saline-(black) or morphine (magenta)-treated mice (saline: n = 10/3; morphine: n = 12/3). Scale bars: top = 25 pA, 100 ms; bottom = 50 pA, 10 ms. Cumulative probability and summary graph (inset) showing the frequency (E) and amplitude (F) of sIPSCs during spontaneous withdrawal. Data are presented as individual cells with ± SEM error bars.
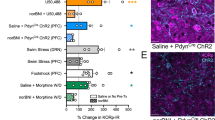
ACC directly regulates opioid-induced spontaneous withdrawal and withdrawal-induced mechanical hypersensitivity
Our results suggest that opioid-induced spontaneous withdrawal and the associated withdrawal-induced mechanical hypersensitivity increase ACC L5 thick-tufted PyN excitability through alterations in intrinsic properties rather than through signals derived from other brain regions (i.e., synaptic alterations). In addition, we found that this effect was specific to thick-tufted PyNs. In Drd1a-tdTomato mice, which are known to express spontaneous opioid withdrawal [29], dopamine D1-expressing, thin-tufted PyNs in ACC L5 had no significant changes in their intrinsic properties or synaptically-mediated currents 24 h following escalating morphine injections (Supplementary Fig. 2). Based on this, we investigated whether activity in the ACC is directly linked to opioid-induced spontaneous withdrawal-like behaviors by locally silencing ACC neurons. To test this, we injected a virus encoding the inhibitory Gi-coupled DREADD into the ACC (Fig. 5A), which, when expressed, would inhibit neuronal activity upon activation with the actuator CNO. We found that following systemic injection of CNO (2 mg/kg) 30 min before withdrawal tests, the global withdrawal score was significantly decreased in morphine-treated mice expressing hM4Di vs. sham morphine-treated mice (F(1,33) = 13.21, p = 0.0009; two-way ANOVA with Bonferroni post-test) (Fig. 5C). However, the locomotor depression observed in morphine-treated sham mice was still observed in morphine-treated mice expressing hM4Di following CNO administration, suggesting that ACC inhibition had no effect on withdrawal-induced locomotor depression (F(1,19) = 0.1046, p = 0.750; two-way ANOVA with Bonferroni post-test) (Fig. 5D, E). In addition, we found that inhibition of the ACC upon CNO injection in morphine-treated mice expressing hM4Di prevented withdrawal-induced mechanical hypersensitivity on withdrawal day 1 (F(6,66) = 7.749, p < 0.0001; two-way repeated-measure ANOVA; Bonferroni multiple comparisons test, morphine sham vs. morphine DREADD, p = 0.0102) (Fig. 5F), which was reversed on withdrawal day 4 when CNO was not administered (Bonferroni multiple-comparison test, morphine sham vs. morphine DREADD, p = 0.0239) (Fig. 5F). However, by withdrawal day 7, in the absence of CNO, withdrawal-induced mechanical hypersensitivity was no longer observed (Fig. 5F). These results suggest an overlapping role of the ACC in mediating both opioid-induced withdrawal and withdrawal-induced mechanical hypersensitivity.

A (Top) Experimental timeline of viral transduction of the ACC prior to behavioral assessments. (Bottom) The extent of viral expression within the ACC. B Representative traces showing bath application of CNO decreases the excitability of transduced ACC L5 thick-tufted PyNs. Scale bars: top = 40 mV, 200 ms; bottom = 40 mV, 250 ms. C Summary graph showing that CNO treatment 30 min prior to withdrawal monitoring blocked morphine-induced withdrawal in morphine-treated mice expressing hM4Di (sham saline: n = 8 mice; sham morphine: n = 9; hM4Di saline: n = 10; hM4Di morphine: n = 10). D Representative traces showing mouse locomotor behavior over the course of the 30 min withdrawal. E Summary graph showing that CNO treatment does not significantly affect locomotor behavior during withdrawal monitoring. F Summary showing that CNO treatment 30 min prior to von Frey testing blocked withdrawal-induced mechanical hypersensitivity in morphine-treated mice expressing hM4Di. In the absence of CNO (withdrawal days 4 and 7), morphine-treated mice displayed withdrawal-induced mechanical hypersensitivity on withdrawal day 4, which was no longer expressed by withdrawal day 7. *p < 0.05, ***p < 0.001.
Discussion
Our results demonstrate that cell-type-specific alterations are evoked in L5 of the ACC in a preclinical model of spontaneous opioid withdrawal. We have shown that ACC L5 thick-tufted PyNs, putative D2-receptor-expressing PyNs, have increased IME, decreases in HCN-channel-mediated currents, and no changes in spontaneous excitatory or inhibitory currents. In contrast, we have shown that thin-tufted, dopamine D1receptor-expressing ACC L5 PyNs do not display any changes in intrinsic or synaptic properties during spontaneous opioid withdrawal. Last, we have shown that inhibition of the ACC is necessary to prevent spontaneous opioid withdrawal and withdrawal-induced mechanical hypersensitivity.
It is established that the locus coeruleus (LC) and the norepinephrine (NE)-expressing neurons located within the LC are critically involved in mediating opioid withdrawal [7, 50]. This seminal research has led to current clinical therapeutic treatments for opioid withdrawal that target the adrenergic system. These treatments include α2-receptor agonists (e.g., clonidine, lofexidine), which act on presynaptic terminals to inhibit NE release, thus blocking adrenergic-mediated symptoms (e.g., yawning, diaphoresis, and mydriasis). Our results expand upon the neurocircuit-mediating opioid withdrawal and show that the ACC is a critical locus for the expression of opioid withdrawal and withdrawal-induced mechanical hypersensitivity.
It is known from both clinical and preclinical research that the ACC is activated upon opioid withdrawal. Brain imaging studies, in male human subjects, have shown increases in ACC activity during naloxone-precipitated opioid withdrawal [24]. Likewise, in rats (sex unspecified), functional magnetic resonance imaging has shown increases in signal intensity in the ACC during naloxone-precipitated opioid withdrawal [23]. Additional studies using c-fos labeling have identified significant increases in c-fos expression, a marker of neuronal activity, in the ACC following naloxone-precipitated withdrawal [51]. Although our model assesses ACC activity following spontaneous opioid withdrawal rather than naloxone-precipitated withdrawal, our results are in line with these previous studies. We found that 24 h after the last injection of morphine, thick-tufted PyNs in layer V of the ACC display characteristics indicative of a hyper-excitable state and that this is correlated with decreases in HCN-mediated IH.
HCN channels consist of 4 subunits (HCN1–4) that can assemble in various combinations with HCN1, 2, and 4 commonly found in layer V of the ACC (Allen Brain Atlas). They are constitutively active at resting-membrane potentials (~−70 mV) with permeability to Na+ and K+ [52]. When the neuron is hyperpolarized, HCN channels open, thus depolarizing the neuron and maintaining the membrane potential close to the firing threshold. Alternatively, when the neuron is depolarized, HCN channels are deactivated resulting in neuronal hyperpolarization [53,54,55]. Therefore, when expressed at the soma, HCN channels serve as high-pass filters that shape the voltage response to rhythmic oscillations [53]. When expressed in the dendritic compartments, HCN channels reduce the input resistance and accelerate the decay phase of incoming synaptic signals reducing the temporal summation of postsynaptic potentials at the soma [53]. Therefore, decreases in HCN-channel function and/or expression are likely to result in increases in membrane resistance and greater shifts in electrical potentials upon synaptic input. Our observed reduction in HCN-channel-mediated currents during spontaneous opioid withdrawal coincides with the observed increases in membrane resistance on L5 thick-tufted PyNs in the ACC. The increases in membrane resistance led to enhanced voltage deflection measured in response to hyperpolarizing current injections, which would explain why we did not observe any changes in SAG currents (data not shown) but did observe significant changes in IH when IH was specifically measured. Furthermore, HCN channel steady-state deactivation was not significantly different between saline- and morphine-treated mice. Changes in HCN-channel deactivation would suggest changes in channel kinetics, which would be associated with changes in subunit composition and/or regulatory factors [56,57,58,59]. Given that HCN-channel deactivation on ACC L5 thick-tufted PyNs was not different in saline- vs. morphine-treated mice, we interpreted the result to mean that HCN-channel kinetics remained unaltered. Therefore, we concluded that the opioid-induced reduction of HCN-channel-mediated currents was likely caused by a reduction in HCN channel expression, which was further supported by our experiments showing that the HCN-channel blocker ZD7288 mimicked the morphine-induced effects on ACC L5 thick-tufted PyN-intrinsic properties. Our observed decreases in HCN-channel-mediated currents, with concomitant increases in membrane excitability, are a phenomenon that has been observed previously in the cortex during the chronic action-potential blockade and in the hippocampus, in a rat model of traumatic brain injury [45, 46]. Therefore, our observed increases in membrane excitability on L5 thick-tufted PyNs during spontaneous withdrawal are potentially due to increases in membrane resistance caused by reduced HCN channel expression.
Although we did not investigate the mechanisms mediating the withdrawal-induced changes in IH, it is plausible to speculate that reduced IH is mediated by opioid-receptor activation on PyNs in the ACC, which in turn, results in decreases in cAMP, an HCN-channel modulator [60]. During withdrawal, it is common for cAMP to overshoot, which causes increases in cAMP activity [61]. However, the cAMP overshoot appears to be brain region-specific as this effect has not been observed in the prefrontal cortical region [62, 63]. Therefore, prolonged reductions in cAMP caused by continued opioid-receptor activation may lead to decreases in HCN-channel function, which future studies can address. Alternatively, withdrawal-induced decreases in HCN-channel function on PyNs may be mediated by NE release in the ACC from overactive LC terminals. Evidence suggests that NE reduces HCN-channel activity [64]. Given the increases in NE release from overactive LC neurons during withdrawal, it is plausible that NE contributes to the increases in PyN intrinsic excitability through HCN-channel modulation. Therefore, future investigations of the LC→ACC L5 PyN neurocircuit may help identify neurocircuit mechanisms mediating withdrawal-induced increases in ACC L5 PyN excitability.
Overall, our observed changes in intrinsic properties would be expected to sensitize ACC L5 thick-tufted PyNs to incoming glutamatergic and GABAergic signals. In addition, evidence from previous studies suggests a direct effect on the excitatory glutamatergic synaptic transmission during opioid withdrawal. A preclinical fMRI study in rats found an increase in the fMRI BOLD response in the ACC during withdrawal [23]. Evidence suggests that the fMRI signal is mediated by synaptic activity as the local field potentials are predictors of the BOLD response [65, 66]. Therefore, based on the results from the fMRI BOLD signals, it would be expected that opioid withdrawal would evoke increases in glutamatergic transmission in the ACC. In agreement with this, a study found that naloxone-precipitated withdrawal in morphine-dependent rats caused an increase in glutamate release in the ACC [25]. Based on these findings, we expected to observe increases in sEPSC amplitude and/or frequency on L5 thick-tufted PyNs during spontaneous withdrawal. Instead, we found no significant changes in spontaneously-evoked currents on ACC L5 thick-tufted PyNs. This lack of synaptic effect suggests that pre and/or postsynaptic modifications are not required for spontaneous withdrawal and that the neurocircuit communication between afferents and ACC neurons is influenced more so by the coordinated timing of neuronal activity, which is regulated by intrinsic properties [67]. However, it is possible that our assessments were not sensitive enough to detect synaptic alterations. For example, opioid-induced synaptic alterations that contribute to withdrawal may be mediated by specific inputs or within specific layers of the ACC. Given that spontaneous excitatory and inhibitory currents are characterized by small amplitudes (~25 and ~40 pA, respectively), they are likely generated closer to the recording site (i.e., soma). Potentially then, glutamatergic and/or GABAergic ACC afferents, which target layers I and/or II/III may undergo synaptic plasticity. Future studies should look to address these potential synaptic changes by investigating layer- and pathway-specific plasticity within the ACC in a model of opioid withdrawal.
In addition to future investigations looking at pathway-specific alterations, our data suggest that cell-type-specific investigations in the ACC are also warranted. Here, we observed a cell-type-specific alteration on the intrinsic properties of thick-tufted, putative dopamine D2-receptor-expressing L5 PyNs, rather than thin-tufted, dopamine D1-receptor-expressing L5 PyNs during spontaneous opioid withdrawal. The reasons for these cell-type-specific effects are unclear but may be related to the differences in projection targets of thick- and thin-tufted PyNs. Data suggest that D1R-expressing thin-tufted PyNs project to cortical regions, while thick-tufted PyNs, with larger HCN-channel-mediated current and D2R expression, the project to subcortical regions, including the brainstem and thalamus, which are implicated in pain processing and opioid withdrawal-like symptoms [68,69,70,71].
Interestingly, we found that inhibition of ACC activity using chemogenetic techniques blocked both spontaneous opioid withdrawal and withdrawal-induced mechanical hypersensitivity. The ability of the ACC to regulate both withdrawal behaviors and withdrawal-induced mechanical hypersensitivity suggests overlapping mechanisms between two seemingly distinguishable behaviors. This commonality potentially suggests that the ACC is a locus for multiple symptoms elicited by opioid withdrawal. Taken separately, first, our observed reduction in spontaneous opioid withdrawal during ACC inhibition demonstrates a causal association between ACC activity and symptoms related to opioid withdrawal. In rodents, symptoms of opioid withdrawal include tremors and shakes (wet dog shakes), jumps, and paw flutters, which were all characteristics that we observed in mice undergoing spontaneous withdrawal 24 h after the last morphine injection. These opioid withdrawal symptoms are controlled by specific brain regions, including those that control jumps (amygdala, medial thalamus, globus pallidus, locus coeruleus, and periaqueductal gray), “wet dog shakes” (amygdala, medial thalamus, and globus pallidus), and paw tremors (amygdala, globus pallidus, and caudate putamen) [72,73,74]. The ACC is directly linked to these structures, receiving inputs from the thalamus, amygdala, and locus coeruleus, while projecting to the amygdala, periaqueductal gray, thalamus, and spinal cord [75, 76]. The ACC’s neuroanatomical position would suggest that it is well-positioned to regulate opioid-induced withdrawal symptoms, which our results support. However, our results also suggest that inhibiting ACC activity does not influence withdrawal-induced reductions in locomotor activity, potentially due to the underlying neurobiological mechanisms. It is known that opioids evoke locomotor sensitization following non-contingent administration [38, 77,78,79], due, in part, to increases in dopamine release, as increases in dopamine are strongly correlated with locomotor activity in rodents [80,81,82]. During opioid withdrawal, dopamine levels are reduced [83,84,85,86,87], which may contribute to withdrawal-induced reductions in locomotor activity. Importantly, the ACC sends glutamatergic projections to brain regions involved in dopamine release, including the substantia nigra and ventral tegmental area [76]. Therefore, it would be expected that ACC inhibition would be unable to rescue withdrawal-induced dopamine depletion, which may explain why locomotor activity remained decreased during chemogenetic inhibition of the ACC in morphine-treated mice. Another potential reason we did not see a rescue of locomotor activity during ACC inhibition during opioid withdrawal may be due to off-target effects within the motor cortex. However, this is unlikely as our histology shows that DREADD expression was restricted to the ACC. In addition, morphine-treated mice injected with hM4Di did not show a bimodal distribution in the distance traveled, which suggests that off-target effects of DREADD expression were not involved.
Second, our observed reduction in withdrawal-induced mechanical hypersensitivity during ACC chemogenetic inhibition coincides with other pain models, including chronic inflammatory pain. For example, one study found that ACC inhibition reduced inflammatory-mediated hypersensitivity [88]. An additional study found that optogenetic activation of PyNs in the ACC lowers hyperalgesia thresholds [89]. However, ACC regulation of pain sensitivity is not consistent across all pain models as it has been shown that in a neuropathic pain model, ACC inhibition had no effect on pain-induced hypersensitivity [90]. Therefore, it is plausible that our observed withdrawal-induced mechanical hypersensitivity may involve similar pain circuits as those involved in inflammatory pain. In addition, on withdrawal day 4, we observed a significant increase in mechanical hypersensitivity in DREADD(Gi)-expressing morphine-treated mice when CNO was not administered. This “rebound hyperalgesia” that we observed in DREADD(Gi)-expressing morphine-treated mice relates to similar rebound hyperalgesia that occurs in postoperative rats after buprenorphine has worn off [91]. Similarly, a common clinical problem has been documented whereby postoperative patients experience enhanced pain following intra-operative opioid administration [92]. It is plausible that the ACC may contribute to this post-opioid hypersensitive pain state, but further investigations are warranted.
Limitations and future directions
We acknowledge that there are limitations to our findings. For example, given that there are no pharmacological agents available to enhance IH, we were limited in our ability to demonstrate a cause-and-effect relationship between decreases in IH and increases in PyN excitability. Therefore, our results provide more of a correlative relationship rather than causative. Future experiments will look at developing viral-mediated tools aimed at overexpression of HCN channels in thick-tufted PyNs. In addition, our synaptic assessments did not isolate pathway-specific inputs in layers I and II/III that may differentially modulate ACC L5 thick-tufted PyNs. Although we did perform paired-pulse ratios using electrical stimulation of either L1 or LII/III and found no significant changes between experimental and control groups (data not shown), future experiments are required to address pathway-specific inputs as well as potential postsynaptic alterations that may contribute to ACC-regulated spontaneous opioid withdrawal. Another limitation to our study is that we did not use a viral or vehicle control for our DREADD experiments, thus controlling for an effect produced by the viral vector or fluorophore. However, since the sham-surgery groups received CNO, this limitation is unlikely to alter our observations. We also acknowledge that our approach did not include female mice, thereby limiting comparisons between sexes. Assessments of sex-specific ACC function during spontaneous opioid withdrawal are required as it has been shown that male rats demonstrate increases in metabolism in the ACC compared with female rats during acute opioid withdrawal [93]. Additional work has shown sex-specific behavioral responses during protracted opioid withdrawal [94]. Last, we acknowledge that our chemogenetic approach did not specifically target ACC L5 thick-tufted PyNs, which future experiments will address. However, despite these limitations, our results have the potential to contribute to next-generation therapeutic options localized to the ACC, including deep brain stimulation strategies, capable of broadly targeting opioid-withdrawal symptoms, while avoiding the necessity of multiple therapeutics to target the underlying pathophysiological mechanisms of the syndrome.
References
Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacol: Off Publ Am Coll Neuropsychopharmacol. 2010;35:217–38.
Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 2016;3:760–73.
Welsch L, Bailly J, Darcq E, Kieffer BL. The negative affect of protracted opioid abstinence: progress and perspectives from rodent models. Biol Psychiatry. 2020;87:54–63.
Burma NE, Kwok CH, Trang T. Therapies and mechanisms of opioid withdrawal. Pain Manag. 2017;7:455–59.
Bagley EE, Chieng BC, Christie MJ, Connor M. Opioid tolerance in periaqueductal gray neurons isolated from mice chronically treated with morphine. Br J Pharmacol. 2005;146:68–76.
Drdla R, Gassner M, Gingl E, Sandkühler J. Induction of synaptic long-term potentiation after opioid withdrawal. Sci (N. Y, NY). 2009;325:207–10.
Christie MJ, Williams JT, Osborne PB, Bellchambers CE. Where is the locus in opioid withdrawal? Trends Pharmacol Sci. 1997;18:134–40.
Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Sci (N. Y, NY). 1997;278:58–63.
Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoact drugs. 2003;35:253–9.
Vernon MK, Reinders S, Mannix S, Gullo K, Gorodetzky CW, Clinch T. Psychometric evaluation of the 10-item Short Opiate Withdrawal Scale-Gossop (SOWS-Gossop) in patients undergoing opioid detoxification. Addictive Behav. 2016;60:109–16.
Kosten TR, Baxter LE. Review article: effective management of opioid withdrawal symptoms: a gateway to opioid dependence treatment. Am J Addictions. 2019;28:55–62.
Gao S-H, Shen L-L, Wen H-Z, Zhao Y-D, Chen P-H, Ruan H-Z. The projections from the anterior cingulate cortex to the nucleus accumbens and ventral tegmental area contribute to neuropathic pain-evoked aversion in rats. Neurobiol Dis. 2020;140:104862.
Carter CS, Braver TS, Barch DM, Botvinick MM, Noll D, Cohen JD. Anterior cingulate cortex, error detection, and the online monitoring of performance. Sci (NY). 1998;280:747–9.
Botvinick M, Nystrom LE, Fissell K, Carter CS, Cohen JD. Conflict monitoring versus selection-for-action in anterior cingulate cortex. Nature. 1999;402:179–81.
Paus T. Primate anterior cingulate cortex: where motor control, drive and cognition interface. Nat Rev Neurosci. 2001;2:417–24.
Kerns JG, Cohen JD, MacDonald AW 3rd, Cho RY, Stenger VA, Carter CS. Anterior cingulate conflict monitoring and adjustments in control. Sci (NY). 2004;303:1023–6.
Bryden DW, Johnson EE, Tobia SC, Kashtelyan V, Roesch MR. Attention for learning signals in anterior cingulate cortex. J Neurosci: Off J Soc Neurosci. 2011;31:18266–74.
Hayden BY, Heilbronner SR, Pearson JM, Platt ML. Surprise signals in anterior cingulate cortex: neuronal encoding of unsigned reward prediction errors driving adjustment in behavior. J Neurosci: Off J Soc Neurosci. 2011;31:4178–87.
Vogt BA. Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci. 2005;6:533–44.
Devinsky O, Morrell MJ, Vogt BA. Contributions of anterior cingulate cortex to behaviour. Brain: J Neurol. 1995;118:279–306.
Elston TW, Bilkey DK. Anterior cingulate cortex modulation of the ventral tegmental area in an effort task. Cell Rep. 2017;19:2220–30.
van Heukelum S, Mars RB, Guthrie M, Buitelaar JK, Beckmann CF, Tiesinga PHE, et al. Where is cingulate cortex? a cross-species view. Trends Neurosci. 2020;43:285–99.
Lowe AS, Williams SC, Symms MR, Stolerman IP, Shoaib M. Functional magnetic resonance neuroimaging of drug dependence: naloxone-precipitated morphine withdrawal. NeuroImage. 2002;17:902–10.
Chu LF, Lin JC, Clemenson A, Encisco E, Sun J, Hoang D, et al. Acute opioid withdrawal is associated with increased neural activity in reward-processing centers in healthy men: a functional magnetic resonance imaging study. Drug Alcohol Depend. 2015;153:314–22.
Hao Y, Yang JY, Guo M, Wu CF, Wu MF. Morphine decreases extracellular levels of glutamate in the anterior cingulate cortex: an in vivo microdialysis study in freely moving rats. Brain Res. 2005;1040:191–6.
Hermann D, Frischknecht U, Heinrich M, Hoerst M, Vollmert C, Vollstadt-Klein S, et al. MR spectroscopy in opiate maintenance therapy: association of glutamate with the number of previous withdrawals in the anterior cingulate cortex. Addict Biol. 2012;17:659–67.
Streit F, Treutlein J, Frischknecht U, Hermann D, Mann K, Kiefer F, et al. Glutamate concentration in the anterior cingulate cortex in alcohol dependence: association with alcohol withdrawal and exploration of contribution from glutamatergic candidate genes. Psychiatr Genet. 2018;28:94–95.
Zubieta JK, Ketter TA, Bueller JA, Xu Y, Kilbourn MR, Young EA, et al. Regulation of human affective responses by anterior cingulate and limbic µ-Opioid neurotransmission. Arch Gen Psychiatry. 2003;60:1145–53.
Madayag AC, Gomez D, Anderson EM, Ingebretson AE, Thomas MJ, Hearing MC. Cell-type and region-specific nucleus accumbens AMPAR plasticity associated with morphine reward, reinstatement, and spontaneous withdrawal. Brain Struct Funct. 2019;224:2311–24.
Quock RM, Brewer AL. Establishing a time course for spontaneous opioid withdrawal in male and female outbred mice using an abbreviated dependence paradigm. FASEB J. 2019;33:663.6–63.6.
Hassan R, Pike See C, Sreenivasan S, Mansor SM, Müller CP, Hassan Z. Mitragynine attenuates morphine withdrawal effects in rats—a comparison with methadone and buprenorphine. Front Psych. 2020;11:411.
Papaleo F, Contarino A. Gender- and morphine dose-linked expression of spontaneous somatic opiate withdrawal in mice. Behav Brain Res. 2006;170:110–8.
Zissen MH, Zhang G, McKelvy A, Propst JT, Kendig JJ, Sweitzer SM. Tolerance, opioid-induced allodynia and withdrawal associated allodynia in infant and young rats. Neuroscience. 2007;144:247–62.
Daoudal G, Debanne D. Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learn Mem. 2003;10:456–65.
Huang YH, Schluter OM, Dong Y. Cocaine-induced homeostatic regulation and dysregulation of nucleus accumbens neurons. Behav Brain Res. 2011;216:9–18.
Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–20.
Ishikawa M, Mu P, Moyer JT, Wolf JA, Quock RM, Davies NM, et al. Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J Neurosci: Off J Soc Neurosci. 2009;29:5820–31.
McDevitt DS, Graziane NM. Timing of morphine administration differentially alters paraventricular thalamic neuron activity. eNeuro. 2019;6:ENEURO.0377-19.2019:1-14.
Kiernan MC, Krishnan AV, Lin CS, Burke D, Berkovic SF. Mutation in the Na+ channel subunit SCN1B produces paradoxical changes in peripheral nerve excitability. Brain: J Neurol. 2005;128:1841–6.
Ibeakanma C, Vanner S. TNFalpha is a key mediator of the pronociceptive effects of mucosal supernatant from human ulcerative colitis on colonic DRG neurons. Gut. 2010;59:612–21.
Purves D, Fitzpatrick D, Katz LC, Lamantia AS, McNamara JO, Williams SM, et al. Neuroscience. 2nd edition. Sinauer Associates; 2001. Voltage-Gated Ion Channels. Available from: https://www.ncbi.nlm.nih.gov/books/NBK10883/.
Cordeiro Matos S, Zhang Z, Seguela P. Peripheral neuropathy induces HCN channel dysfunction in pyramidal neurons of the medial prefrontal cortex. J Neurosci. 2015;35:13244–56.
Gao S-H, Wen H-Z, Shen L-L, Zhao Y-D, Ruan H-Z. Activation of mGluR1 contributes to neuronal hyperexcitability in the rat anterior cingulate cortex via inhibition of HCN channels. Neuropharmacology 2016;105:361–77.
van Aerde KI, Feldmeyer D. Morphological and physiological characterization of pyramidal neuron subtypes in rat medial prefrontal cortex. Cereb Cortex. 2015;25:788–805.
Karimi SA, Hosseinmardi N, Sayyah M, Hajisoltani R, Janahmadi M. Enhancement of intrinsic neuronal excitability-mediated by a reduction in hyperpolarization-activated cation current (Ih) in hippocampal CA1 neurons in a rat model of traumatic brain injury. Hippocampus. 2021;31:156–69.
Gibson JR, Bartley AF, Huber KM. Role for the subthreshold currents ILeak and IH in the homeostatic control of excitability in neocortical somatostatin-positive inhibitory neurons. J Neurophysiol. 2006;96:420–32.
Santello M, Nevian T. Dysfunction of cortical dendritic integration in neuropathic pain reversed by serotoninergic neuromodulation. Neuron. 2015;86:233–46.
Chu N-N, Zuo Y-F, Meng L, Lee DY-W, Han J-S, Cui C-L. Peripheral electrical stimulation reversed the cell size reduction and increased BDNF level in the ventral tegmental area in chronic morphine-treated rats. Brain Res. 2007;1182:90–98.
Sklair-Tavron L, Shi WX, Lane SB, Harris HW, Bunney BS, Nestler EJ. Chronic morphine induces visible changes in the morphology of mesolimbic dopamine neurons. Proc Natl Acad Sci. 1996;93:11202–07.
Rasmussen K, Beitner-Johnson D, Krystal J, Aghajanian G, Nestler E. Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci. 1990;10:2308–17.
Erdtmann-Vourliotis M, Mayer P, Riechert U, Grecksch G, Höllt V. Identification of brain regions that are markedly activated by morphine in tolerant but not in naive rats. Brain Res Mol Brain Res. 1998;61:51–61.
Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol life Sci. 2009;66:470–94.
Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–85.
Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327.
Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–80.
Lewis AS, Estep CM, Chetkovich DM. The fast and slow ups and downs of HCN channel regulation. Channels (Austin). 2010;4:215–31.
Arinsburg SS, Cohen IS, Yu HG. Constitutively active Src tyrosine kinase changes gating of HCN4 channels through direct binding to the channel proteins. J Cardiovasc Pharm. 2006;47:578–86.
Zong X, Eckert C, Yuan H, Wahl-Schott C, Abicht H, Fang L, et al. A novel mechanism of modulation of hyperpolarization-activated cyclic nucleotide-gated channels by Src kinase. J Biol Chem. 2005;280:34224–32.
Gravante B, Barbuti A, Milanesi R, Zappi I, Viscomi C, DiFrancesco D. Interaction of the pacemaker channel HCN1 with filamin A. J Biol Chem. 2004;279:43847–53.
DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–7.
Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–10.
Ferrer-Alcón M, García-Fuster MJ, La Harpe R, García-Sevilla JA. Long-term regulation of signalling components of adenylyl cyclase and mitogen-activated protein kinase in the pre-frontal cortex of human opiate addicts. J neurochemistry. 2004;90:220–30.
Duman RS, Tallman JF, Nestler EJ. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Therapeutics. 1988;246:1033–9.
Zhang Z, Cordeiro Matos S, Jego S, Adamantidis A, Séguéla P. Norepinephrine drives persistent activity in prefrontal cortex via synergistic α1 and α2 adrenoceptors. PloS One. 2013;8:e66122–e22.
Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A. Neurophysiological investigation of the basis of the fMRI signal. Nature 2001;412:150–57.
Logothetis NK, Wandell BA. Interpreting the BOLD signal. Annu Rev Physiol. 2004;66:735–69.
Wang X-J. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev. 2010;90:1195–268.
Seong HJ, Carter AG. D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J Neurosci: Off J Soc Neurosci. 2012;32:10516–21.
Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS. Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci: Off J Soc Neurosci. 2012;32:4959–71.
Dembrow N, Johnston D. Subcircuit-specific neuromodulation in the prefrontal cortex. Front Neural Circ. 2014;8:54.
Radnikow G, Feldmeyer D. Layer- and cell type-specific modulation of excitatory neuronal activity in the neocortex. Front Neuroanat. 2018;12:1.
Maldonado R, Stinus L, Gold LH, Koob GF. Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Therapeutics. 1992;261:669–77.
Tremblay EC, Charton G. Anatomical correlates of morphine-withdrawal syndrome: differential participation of structures located within the limbic system and striatum. Neurosci Lett. 1981;23:137–42.
Wei E, Loh HH, Way EL. Neuroanatomical correlates of morphine dependence. Sci (NY). 1972;177:616–7.
Fillinger C, Yalcin I, Barrot M, Veinante P. Afferents to anterior cingulate areas 24a and 24b and midcingulate areas 24a’ and 24b’ in the mouse. Brain Struct Funct. 2017;222:1509–32.
Fillinger C, Yalcin I, Barrot M, Veinante P. Efferents of anterior cingulate areas 24a and 24b and midcingulate areas 24a’ and 24b’ in the mouse. Brain Struct Funct. 2018;223:1747–78.
McKendrick G, Garrett H, Jones HE, McDevitt DS, Sharma S, Silberman Y, et al. Ketamine blocks morphine-induced conditioned place preference and anxiety-like behaviors in mice. Front Behav Neurosci. 2020;14:75.
McKendrick G, Garrett H, Tanniru S, Ballard S, Sun D, Silberman Y, et al. A novel method to study reward-context associations and drug-seeking behaviors. J Neurosci Methods. 2020;343:108857.
McKendrick G, Sharma S, Sun D, Randall PA, Graziane NM. Acute and chronic bupropion treatment does not prevent morphine-induced conditioned place preference in mice. Eur J Pharm. 2020;889:173638.
Freed CR, Yamamoto BK. Regional brain dopamine metabolism: a marker for the speed, direction, and posture of moving animals. Sci (NY). 1985;229:62–65.
Pijnenburg AJJ, Honig WMM, Van Der Heyden JAM, Van Rossum JM. Effects of chemical stimulation of the mesolimbic dopamine system upon locomotor activity. Eur J Pharmacol. 1976;35:45–58.
Isaacson RL, Yongue B, McClearn D. Dopamine agonists: Their effect on locomotion and exploration. Behav Biol. 1978;23:163–79.
Rossetti ZL, Hmaidan Y, Gessa GL. Marked inhibition of mesolimbic dopamine release: a common feature of ethanol, morphine, cocaine and amphetamine abstinence in rats. Eur J Pharm. 1992;221:227–34.
Acquas E, Carboni E, Di Chiara G. Profound depression of mesolimbic dopamine release after morphine withdrawal in dependent rats. Eur J Pharm. 1991;193:133–4.
Acquas E, Di Chiara G. Depression of mesolimbic dopamine transmission and sensitization to morphine during opiate abstinence. J neurochemistry. 1992;58:1620–5.
Crippens D, Robinson TE. Withdrawal from morphine or amphetamine: different effects on dopamine in the ventral-medial striatum studied with microdialysis. Brain Res. 1994;650:56–62.
Pothos E, Rada P, Mark GP, Hoebel BG. Dopamine microdialysis in the nucleus accumbens during acute and chronic morphine, naloxone-precipitated withdrawal and clonidine treatment. Brain Res. 1991;566:348–50.
Koga K, Yamada A, Song Q, Li XH, Chen QY, Liu RH, et al. Ascending noradrenergic excitation from the locus coeruleus to the anterior cingulate cortex. Mol Brain. 2020;13:49.
Kang SJ, Kwak C, Lee J, Sim SE, Shim J, Choi T, et al. Bidirectional modulation of hyperalgesia via the specific control of excitatory and inhibitory neuronal activity in the ACC. Mol Brain. 2015;8:81.
Donahue RR, LaGraize SC, Fuchs PN. Electrolytic lesion of the anterior cingulate cortex decreases inflammatory, but not neuropathic nociceptive behavior in rats. Brain Res. 2001;897:131–8.
St ASL, Martin WJ. Evaluation of postoperative analgesia in a rat model of incisional pain. Contemp Top Lab Anim Sci. 2003;42:28–34.
Guignard B, Bossard Anne E, Coste C, Sessler Daniel I, Lebrault C, Alfonsi P, et al. Acute opioid tolerance: intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology 2000;93:409–17.
Santoro GC, Carrion J, Dewey SL. Imaging sex differences in regional brain metabolism during acute opioid withdrawal. J Alcohol Drug Depend. 2017;5:262.
Bravo IM, Luster BR, Flanigan ME, Perez PJ, Cogan ES, Schmidt KT, et al. Divergent behavioral responses in protracted opioid withdrawal in male and female C57BL/6J mice. Eur J Neurosci. 2020;51:742–54.
Acknowledgements
We thank Dr. Diane McCloskey for edits and comments on the paper and Dr. Kirsteen Browning for her thoughtful input on our electrophysiology data.
Funding
This project is supported by the NARSAD Young Investigator Award (27364; NG) and by the Pennsylvania Department of Health using Tobacco CURE Funds. The authors have no financial or non-financial competing interests to declare.
Author information
Authors and Affiliations
Contributions
DSM, GEM, and NMG designed the experiments, performed the analyses, and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
McDevitt, D.S., McKendrick, G. & Graziane, N.M. Anterior cingulate cortex is necessary for spontaneous opioid withdrawal and withdrawal-induced hyperalgesia in male mice. Neuropsychopharmacol. 46, 1990–1999 (2021). https://doi.org/10.1038/s41386-021-01118-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01118-y
This article is cited by
-
HCN-Channel-Dependent Hyperexcitability of the Layer V Pyramidal Neurons in IL-mPFC Contributes to Fentanyl-Induced Hyperalgesia in Male Rats
Molecular Neurobiology (2023)
-
Opioid withdrawal: role in addiction and neural mechanisms
Psychopharmacology (2023)
