
Abstract
Preclinical and clinical evidence suggests that exogenous administration of oxytocin (OT) may hold promise as a therapeutic strategy for reducing heavy alcohol drinking. However, it remains unknown whether these effects are mediated by stimulation of endogenous sources of OT and signaling at oxytocin receptors (OTR) in brain or in the periphery. To address this question, we employed a targeted chemogenetic approach to examine whether selective activation of OT-containing neurons in the paraventricular nucleus of the hypothalamus (PVN) alters alcohol consumption in a binge-like drinking (“Drinking-in-the-Dark”; DID) model. Adult male Oxt-IRES-Cre mice received bilateral infusion of a Cre-dependent virus containing an excitatory DREADD (AAV8-hSyn-DIO-hM3Dq-mCherry) or control virus (AAV8-hSyn-DIO-mCherry) into the PVN. Chemogenetic activation of PVNOT+ neurons following clozapine-N-oxide injection reduced binge-like alcohol drinking in a similar manner as systemic administration of the neuropeptide. Pretreatment with a brain-penetrant OTR antagonist (L-368,899) reversed this effect while systemic administration of a peripherally restricted OTR antagonist (Atosiban) did not alter reduced alcohol drinking following chemogenetic activation of PVNOT+ neurons. Altogether, these data are the first to demonstrate that targeted activation of hypothalamic (endogenous) OT reduces alcohol consumption, providing further evidence that this neuropeptide plays a role in regulation of alcohol self-administration behavior. Further, results indicate that the ability OT to reduce alcohol drinking is mediated by signaling at OTR in the brain.
Similar content being viewed by others


Introduction
Binge alcohol drinking is the most common form of excessive alcohol consumption and contributes to a host of long-term negative health consequences, including increased risk for developing alcohol use disorder [1,2,3]. Repeated bouts of heavy drinking and intoxication are known to produce aberrant plasticity changes in multiple brain regions (e.g., cortical, limbic, basal ganglia structures) that are intimately associated with reward processing and stress responsivity [4, 5]. Animal models involving binge-like drinking have been valuable in elucidating neuroadaptations in brain reward and stress pathways that play a role in enhanced motivation to engage in such risky pattens of alcohol consumption [6, 7]. Understanding mechanisms that promote and mediate binge-like drinking is key to developing more effective treatment strategies for impeding progression to more prolonged and excessive alcohol consumption.
The neuropeptide oxytocin (OT) has been implicated in several neuropsychiatric disorders, including alcohol and drug addiction [8,9,10,11]. Clinical studies have indicated that intranasal OT treatment reduces alcohol withdrawal symptoms [12], craving [13], and brain activation to alcohol-related cues [14]. A growing number of preclinical studies have demonstrated that systemic administration of OT reduces alcohol consumption in mice [15, 16], rats [17,18,19], and prairie voles [20, 21]. While there is evidence indicating that these effects are mediated by central actions of OT [14, 18, 19, 22], the mechanism by which peripherally delivered exogenous OT reduces the motivational effects of alcohol is not fully understood.
Historically, peripherally administered OT was not thought to penetrate the blood–brain barrier in quantities sufficient to influence behavior [23]. Rather, it has been hypothesized that OT impacts behavior via a feed-forward mechanism whereby exogenous OT stimulates hypothalamic release of endogenous OT [24,25,26]. Although studies have demonstrated elevated central OT levels following peripheral administration of OT in rodents [27, 28], nonhuman primates [29, 30], and humans [31], more recent studies have indicated that the measured OT appears to originate solely from the exogenous source [32, 33]. While most of these studies examined central OT levels through measurement in cerebrospinal fluid, intranasal administration of OT was also shown to bypass the blood–brain barrier and reach several brain regions in measurable concentrations in rhesus macaques [34]. Nevertheless, the question remains as to whether stimulation of endogenous OT release can produce similar effects as exogenous OT administration in reducing alcohol consumption.
OT is predominantly synthesized and released from neurons in the paraventricular nucleus (PVN) and supraoptic nucleus (SON) of the hypothalamus, with axons extending to the neurohypophysis for release into general circulation, as well as projections to a number of forebrain and hindbrain structures [35, 36]. OT signals through a single receptor, and these OT receptors (OTR) are expressed throughout the periphery to mediate classic hormonal regulation of uterine contraction during parturition, milk-let down during lactation, and sexual reflexes [37]. OTRs are also expressed in numerous brain regions where signaling mediates various social and affective behavioral effects of the neuropeptide. This includes regions sensitive to chronic alcohol effects, including prefrontal cortical areas and extended amygdala structures [38,39,40,41,42,43]. OTR signaling in the periphery may also play a role in influencing centrally mediated behavioral effects of OT via afferent vagal nerve inputs. However, studies investigating the relative contributions of peripheral versus central OT signaling have yielded mixed results. While there is evidence to suggest that peripheral OT directly interacts with vagal afferents to regulate feeding behavior in mice [44], and methamphetamine self-administration in rats [45], Tunstall et al. [19] demonstrated that the peripherally restricted OTR antagonist L-371,257 did not alter the effects of intranasal OT in reducing alcohol consumption in dependent rats. Thus, the central versus peripheral contribution to OT’s effect on alcohol consumption also remains an important question.
Here, we utilize targeted chemogenetic and pharmacological approaches to selectively activate OT-containing neurons within the hypothalamus (PVN) to examine the effect of endogenous OT on binge-like alcohol consumption. Additionally, using brain penetrant and peripherally restricted OTR antagonists (L-368,899 and Atosiban, respectively), we test the hypothesis that activation of PVNOT+ neurons reduce alcohol consumption via central, not peripheral, actions. Together, these studies indicate that OT signaling in the brain plays a role in regulation of binge-like alcohol consumption.
Materials and methods
Subjects
Adult male Oxt-IRES-Cre mice at least 10 weeks old were used in all experiments. The breeding colony was established by crossing homozygote Oxt-IRES-Cre male mice (B6;129S-Oxttm1.1(cre)Dolsn/J; stock #024234) with C57BL/6J female mice, both obtained from Jackson Laboratories (Bar Harbor, ME). All offspring were positive for expression of Cre recombinase under the control of the oxytocin promoter as determined by standard (PCR) genotyping protocols. Mice were individually housed under a 12-h reverse light/dark cycle in an AAALAC-accredited facility. All testing was conducted during the dark phase of the circadian cycle. Mice were provided free access to food and water throughout the duration of the experiments. All experimental protocols were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee and consistent with guidelines of the NIH Guide for the Care and Use of Laboratory Animals.
Alcohol binge-like drinking procedure
Alcohol consumption was assessed using a standard 4-day “Drinking-In-The-Dark” (DID) protocol [46, 47]. Briefly, mice were presented in the home cage with a single bottle of 20% (v/v) alcohol 3-h into the dark cycle for 2-h on 3 consecutive days. On Day-4 access to alcohol was extended to 4-hr, and this 4-day procedure was repeated for 3 weeks (with 3 days off between the weekly cycles). Mice received no treatment during the first 4-day cycle. Chemogenetic and pharmacological manipulations were conducted prior to Day-4 drinking sessions during the second and third DID cycles. Alcohol intake (±0.1 ml; expressed as g/kg) was recorded for the first 2-h and the last 2-h of the 4-h test session. Vehicle injections were given 30-min prior to the 2-h drinking sessions during the first 3 days to acclimate mice to handling and the injection procedure.
Drugs and administration
Clozapine-N-oxide (CNO; Tocris Bioscience, Minneapolis, MN), synthetic human oxytocin (CellSciences, Canton, MA), and the oxytocin receptor antagonists L-368,899 hydrochloride (Tocris) and Atosiban (Tocris), were dissolved in 0.9% saline, which served as the vehicle. Injections were administered intraperitoneally (i.p.) at a volume of 0.01 ml/g body weight in all experiments. Alcohol (95% ethanol) was obtained from AAPER (Shelbyville, KY) and diluted with tap water to the appropriate concentration. Doses of the oxytocin receptor antagonists were based on published studies [48, 49] and preliminary work in our lab.
Surgical procedures
Male Oxt-IRES-Cre mice were anesthetized with isoflurane and received bilateral infusion of a Cre-dependent virus containing an excitatory DREADD (AAV8-hSyn-DIO-hM3Dq-mCherry) or control virus (AAV8-hSyn-DIO-mCherry) into the PVN (AP: −0.78 mm, ML: ± 0.2 mm, DV: −5.2 mm) [50]. Viral constructs were obtained from Addgene (Cambridge, MA) and procedural details for infusions are provided in Supplemental Materials.
Chemogenetic activation of hypothalamic (PVN) oxytocin-containing neurons
To examine the effect of chemogenetic activation of hypothalamic OT-containing neurons on alcohol consumption, adult male Oxt-IRES-Cre mice were infused with active (hM3Dq DREADD) virus (N = 12) or control virus (N = 11) targeting the PVN. Subjects were left undisturbed for three weeks to allow for viral infection prior to the start of weekly DID cycles. At 30-min prior to the 4-h test sessions during two consecutive DID cycles, mice were injected with vehicle (saline) or CNO (3 mg/kg). A within-subjects crossover design was used, with order of drug treatment (saline vs. CNO) counter-balanced across the two test sessions.
Systemic oxytocin administration
To examine whether systemic administration of exogenous OT produced similar effects on alcohol drinking in this transgenic mouse line, a separate group of male Oxt-IRES-Cre mice (N = 9) that did not receive viral infusions was injected with vehicle (saline) or OT (1 mg/kg) 30-min prior to the 4-h test sessions during two consecutive weekly DID cycles. Testing was conducted in the same way as described above. This dose of OT (1 mg/kg) was previously shown to reduce alcohol consumption in C57BL/6J mice under similar testing conditions without affecting total fluid intake, sucrose self-administration, or general locomotor activity [51].
Pharmacological antagonism of central oxytocin receptors
A brain-penetrant OTR antagonist was used to examine whether chemogenetic activation of PVNOT+ neurons reduce alcohol consumption via actions at central OTRs. Prior to testing alcohol drinking in the DID procedure, adult male Oxt-IRES-Cre mice received intra-PVN infusion of active virus (AAV8-hSyn-DIO-hM3Dq-mCherry) (N = 13) or control virus (AAV8-hSyn-DIO-mCherry) (N = 11). Mice were then pretreated with the selective nonpeptide OTR antagonist L-368,899 (10 mg/kg) or vehicle 15-min prior to injection of CNO (3 mg/kg) or vehicle (saline), which was given 30-min before the start of the 4-h test sessions. Testing was conducted over two DID cycles, with order of antagonist drug treatment counter-balanced over the two test sessions.
Pharmacological antagonism of peripheral oxytocin receptors
In this study, a peripherally restricted OTR antagonist was used to investigate whether the reduction in alcohol drinking produced by targeted chemogenetic activation of PVNOT+ neurons was mediated, at least in part, by signaling at peripheral oxytocin receptors. Adult male Oxt-IRES-Cre mice first received intra-PVN infusion of either active virus (AAV8-hSyn-DIO-hM3Dq-mCherry) (N = 12) or control virus (AAV8-hSyn-DIO-mCherry) (N = 10). Mice were pretreated with the peripherally restricted OTR antagonist Atosiban (1 mg/kg) or saline 15-min prior to CNO (3 mg/kg) or vehicle (saline) administration (i.p.), which was given 30-min before the start of the 4-h test drinking sessions. Similar to the previous study, testing was conducted over two DID cycles, with order of antagonist drug treatment counter-balanced over the two test sessions.
Histology
At the conclusion of all studies, mice were euthanized, and brains were histologically examined for viral expression. Behavioral data was only analyzed from subjects in which viral expression was anatomically verified. Details of immunohistochemical and confocal microscopy procedures are provided in Supplemental Materials.
Statistical analysis
Alcohol intake (g/kg) over the entire 4-h test sessions was analyzed by separate ANOVAs for each of the experimental (virus) groups. Also, intake during the first 2-h and last 2-h of the test sessions were analyzed by ANOVA, with Drug (CNO vs. saline) and Time (2-h vs. 4-h) as repeated measures. A similar strategy was used to analyze effects of systemic administration of OT on alcohol consumption. The effects of pretreatment with OTR antagonists on alcohol intake were analyzed separately in mice that received infusions of active (excitatory DREADD) virus or control virus by ANOVA, with Treatment (saline, CNO, saline + antagonist, CNO + antagonist) and Time as repeated factors. Statistical analyses were carried out using SPSS software package (IBM SPSS Statistics, Version 25). Post hoc comparisons were performed when appropriate (Newman–Keuls) and significance level for all analyses was set at p < 0.05.
Results
Chemogenetic activation of hypothalamic oxytocin-containing neurons reduces binge-like alcohol consumption
To determine whether activating endogenous OT alters binge-like alcohol consumption, Oxt-IRES-Cre transgenic mice were used to enable selective chemogenetic targeting of OT-containing neurons in the hypothalamic PVN. Bilateral infusion of active virus containing an excitatory DREADD (AAV8-hSyn-DIO-hM3Dq-mCherry) (N = 12) or control virus (AAV8-hSyn-DIO-mCherry) (N = 11) showed targeted (Cre-dependent) expression in the PVN (Fig. 1) similar to that reported for this mouse model [52]. Three weeks after virus injection, mice were tested in the DID alcohol binge-drinking model. Data from one mouse infused with active virus was excluded from analysis after histological evaluation revealed little evidence of viral expression, suggesting the infusion was misplaced during surgery. Alcohol intake collapsed across the two 4-day DID cycles displayed the typical pattern of consumption for both active and control virus groups (Supplemental Fig. S1A, S1C). Mice that received intra-PVN infusion of the excitatory (hM3Dq) DREADD virus and injected (ip.) with CNO (3 mg/kg) consumed significantly less alcohol compared to the same mice that were injected with saline over the 4-h test sessions (Fig. 2A). This was supported by ANOVA, which revealed a significant main effect of Drug [F(1,20) = 14.4622, p = 0.0011] (Fig. 2B). This reduction in alcohol intake was most robust during the first half of the test session (Supplemental Fig. S1B). In contrast, CNO injection did not significantly alter alcohol intake in mice expressing the control virus (Drug: [F(1,20) = 0.3728, p = 0.5484]) (Fig. 2C, D), and this was true for both the first and last half of the 4-h drinking session (Supplemental Fig. S1D). These latter effects indicate that CNO administration did not produce “off-target” effects, as it only reduced alcohol consumption when activating the excitatory DREADD selectively expressed in PVNOT+ neurons.

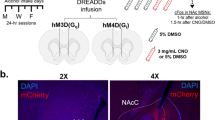
A Illustration showing location of bilateral placement of the virus in the PVN. B Confocal imaging showing bilateral expression of the mCherry tag around the third ventricle (3V) at 10X magnification. C Confocal imaging at 20X magnification of the mCherry fluorescent tag on one side of the 3V. D Cre expression in the same area shown in C. E merge of mCherry and Cre fluorescence shown in C and D.

A Individual and B group data showing that chemogenetic activation of PVNOT+ neurons reduced alcohol consumption in Oxt-IRES-Cre mice treated with active (AAV8-hSyn-DIO-hM3Dq-mCherry) virus. C, D Systemic CNO (3 mg/kg) administration did not alter alcohol drinking in Oxt-IRES-Cre mice harboring control (AAV8-hSyn-DIO-mCherry) virus. E, F Systemic administration of OT (1 mg/kg) decreased alcohol consumption compared to vehicle (saline) in Oxt-IRES-Cre mice. Values (panels B, D, F) are mean ± s.e.m. (N = 9–12/group); *p < 0.05 vs. saline.
In a follow up study in a separate cohort of mice (N = 9), we next examined whether systemic administration of OT produced comparable effects in Oxt-IRES-Cre mice that did not receive viral infusion. Alcohol consumption over the 4-day DID procedure produced the typical pattern of intake (Supplemental Fig. S1E). Analysis of alcohol intake following injection of OT (1 mg/kg) or vehicle (saline) revealed a significant main effect of Drug [F(1,16) = 7.4370, p = 0.0149], indicating that cumulative intake across the 4-h test session was decreased in mice treated with exogenous OT compared to those treated with saline (Fig. 2D, E). Although the reduction in alcohol drinking following injection of OT was clearly most robust during the first half of the test session, the Drug x Time interaction was only marginally significant (Supplemental Fig. S1F).
Activation of endogenous oxytocin reduces binge-like alcohol consumption via signaling at central oxytocin receptors
To determine whether chemogenetic activation of PVNOT+ neurons reduced binge-like alcohol intake via action at central OTRs, mice were treated with an OTR antagonist that penetrates the blood–brain barrier. Specifically, Oxt-IRES-Cre mice that received intra-PVN infusion of active (excitatory DREADD) (N = 13) or control (N = 11) virus were injected (i.p.) with the brain-penetrant OTR antagonist L-368,889 (10 mg/kg) or vehicle before CNO or vehicle (saline) administration (N = 5–7/group). As previously observed, CNO (3 mg/kg) injection significantly reduced alcohol consumption in animals harboring the excitatory DREADD virus, and this effect was blocked in animals that were pretreated with the OTR antagonist (Fig. 3A). This was supported by a significant effect of Treatment [F(3,20) = 6.9616, p = 0.0022], and post hoc analysis indicated that the ability of chemogenetic activation of endogenous OT to reduce alcohol binge-like drinking was completely reversed by pretreatment with the brain-penetrant OTR antagonist L-368,899. This effect was most evident during the first 2-h period of the test session (Supplemental Fig. S2A). Importantly, alcohol intake was not altered in mice treated with the control virus following injection of CNO or pretreatment with L-68,899 (Treatment: [F(3,18) = 0.9595, p = 0.4332]) (Fig. 3B). This was true for alcohol consumed during the first and last half of the drinking session (Supplemental Fig. S2B). In all cases, L-368,899 treatment alone did not alter alcohol intake.

A In mice expressing the excitatory DREADD, CNO-induced activation of PVNOT+ neurons decreased alcohol consumption compared to vehicle and pretreatment with L-368,899 (10 mg/kg) reversed this effect. B No effect was observed in animals treated with control virus. Values are mean ± s.e.m. (N = 5–7/group); *p < 0.05 vs. saline.
Peripheral oxytocin receptor antagonism does not alter endogenous oxytocin-induced decrease in binge-like alcohol consumption
Next, we examined whether the ability of chemogenetic activation of endogenous OT to reduce binge-like alcohol drinking may be mediated via actions at OTRs outside of the brain. A similar experimental strategy was employed as described above, except that separate groups of Oxt-IRES-Cre mice that received active (hM3Dq DREADD) (N = 12) or control (N = 10) virus in the PVN were pretreated with the peripherally restricted OTR antagonist Atosiban (1 mg/kg) or vehicle prior to CNO or saline injection (N = 5–6/group). Data from one mouse infused with active virus and two mice infused with control virus were excluded from the analyses after histological evaluation indicated misplaced viral expression. Replicating our earlier finding, CNO treatment reduced alcohol consumption in mice expressing the excitatory (hM3Dq) DREADD in PVNOT+ neurons, and this effect was observed whether the mice were pretreated with vehicle or with Atosiban (Fig. 4A). This was supported by a main effect of Treatment [F(3,18) = 10.5303, p = 0.0003], and the CNO-induced effect was evident throughout the testing period (Supplemental Fig. S3A). In contrast, CNO administration did not alter alcohol intake in mice treated with the control virus (Treatment: [F(3,12) = 0.2489, p = 0.8606] (Fig. 4B and Supplemental Fig. S3B). Atisoban treatment alone did not alter alcohol drinking at any time during the test drinking session in groups of mice that received active or control virus.

A In mice harboring the excitatory DREADD, CNO-induced activation of PVNOT+ neurons decreased alcohol consumption compared to vehicle. Pretreatment with Atosiban (1 mg/kg) did not affect alcohol consumption in vehicle (Saline + Atosiban) or CNO (CNO + Atisoban) groups. B Neither CNO nor Atisoban pretreatment altered alcohol drinking in animals treated with control virus. Values are mean ± s.e.m. (N = 5–6/group); *p < 0.05 vs. saline.
Discussion
While preclinical and clinical evidence suggests that OT treatment may be promising as a potential therapeutic for AUD, the mechanism and site of action by which exogenous OT reduces alcohol consumption is not fully understood. Here, we demonstrate that direct activation of OT-containing neurons in the hypothalamus (PVN) via targeted expression of an excitatory DREADD decreases alcohol consumption in a binge-like drinking model. Further, this chemogenetic effect was specific in that the activating effects of CNO were only observed in mice harboring the hM3Dq (excitatory) DREADD in PVNOT+ neurons. A similar targeted approach has been employed to show OT-mediated regulation of social behavior [53, 54]. This is the first demonstration that stimulating hypothalamic release of endogenous sources of OT is effective in reducing alcohol consumption.
Additionally, combining chemogenetic and pharmacological approaches, we demonstrate that the effects of activating PVNOT+ neurons on alcohol consumption can be reversed with a blood–brain barrier penetrant OTR antagonist, while peripheral administration of an OTR antagonist that does not cross the blood–brain barrier did not reverse this effect. Taken together, these data suggest a central mechanism for OT’s effect on alcohol drinking and implicate OTR signaling in brain as a mechanism of action.
Hypothalamic OT-containing neurons release the neuropeptide into the bloodstream via axon terminals that interface with fenestrated capillaries in the neurohypophysis, as well as target extrahypothalamic brain regions via axon collaterals [35, 36, 40]. Therefore, chemogenetic-induced activation of these neurons could lead to secretion of OT into the general circulation and produce effects through OTR signaling in the periphery. The present study provides evidence that peripheral receptors do not significantly contribute to the effect of activating PVNOT+ neurons on alcohol consumption. Specifically, whereas the peripherally restricted OTR antagonist Atosiban had no effect on the ability of chemogenetically induced release of endogenous OT to reduce alcohol consumption, pretreatment with a brain-penetrant OTR antagonist, L-368,899, reversed this effect. These results are consistent with a recent report by Tunstall et al. demonstrating intracerebroventricular (i.c.v.) administration of a large molecule OTR agonist, PF-06655075, that does not cross the BBB, significantly reduced alcohol drinking in dependent rats. However, the same compound administered systemically (not expected to enter the CNS) did not have an effect. The authors also showed that systemic administration of an OTR antagonist that does not enter the brain (L-371,257) did not reverse the ability of intranasal OT to reduce alcohol drinking in dependent rats [19]. Taken together, while chemogenetic activation of PVNOT+ neurons may also release other neurotransmitters (glutamate) [40], results from the present study involving OTR antagonists suggest that OTR signaling in the brain reduces alcohol consumption independent of OTR binding in the periphery.
The central mechanisms by which OT reduces alcohol drinking are not fully understood. OTRs are widely distributed in the brain, with overlapping expression in several alcohol-sensitive regions, including frontal cortex, basal ganglia, and limbic areas [38,39,40,41,42,43]. Post mortem analyses in humans and rodent models indicate that chronic alcohol exposure results in reduced hypothalamic OT expression but elevated OTR expression in many of these target regions [14, 55,56,57]. OT is known to influence mesolimbic reward circuitry, altering GABA and glutamate modulation of ventral tegmental area neuronal activity [58], as well as dopamine transmission in target regions such as the nucleus accumbens [18]. OTR signaling in the nucleus accumbens has been shown to contribute to the rewarding effects of alcohol [22]. Others have reported that OT activity in prefrontal cortex [41, 59] and limbic regions such as the amygdala [19, 40, 60] play an important role in reward, fear, and anxiety related behaviors. Thus, OTR signaling in many of these regions would appear relevant to the motivational effects of alcohol. Additionally, OT is known to exert anti-neuroinflammatory effects via OTR signaling in microglia [61]. To the extent that microglia and neuroimmune factors play a role in regulation of alcohol consumption [62], OT may also contribute to these effects. Future work will need to determine which brain cells, regions and circuits mediate the effects of activated endogenous OT in reducing alcohol consumption.
Given its similar structure to another hypophyseal neuropeptide (arginine vasopressin; AVP), it is possible that OT reduces alcohol drinking via interaction with AVP receptors. The AVP system has been implicated in a number of alcohol effects, but studies examining the influence of AVP receptors on alcohol drinking have produced mixed results. For example, genetic deletion of either V1a or V1b receptors did not alter alcohol preference or intake [63], although knockdown of V1a receptors was reported to increase alcohol intake [64]. In contrast, pharmacological blockade of V1b receptors was shown to reduce alcohol consumption in mice [65] and rats selectively bred for high alcohol preference [66] and those rendered dependent following chronic alcohol exposure [67]. Further, a recent placebo-controlled clinical trial showed the V1b receptor antagonist ABT-436 to have a modest effect in reducing alcohol drinking in dependent individuals [68]. Nevertheless, it is unlikely that activating endogenous OT in the present study reduced alcohol consumption via AVP release and/or signaling through AVP receptors. First, Cre expression is driven by the OT promoter in Oxt-IRES-Cre mice used in this study and these mice have been shown to express Cre exclusively in PVN neurons with OT immunoreactivity, but not AVP neurons in the PVN [52]. Thus, Cre-dependent expression of the excitatory DREADD effectively targeted OT + neurons in the present study. Additionally, the fact that the brain-penetrant OTR antagonist L-368,899 reversed the effects of chemogenetic activation in the present study provides strong evidence for OTR signaling in mediating the resultant reduction in alcohol intake. Of note, the peripherally restricted OTR antagonist Atosiban is also known to block AVP V1a receptors [69] but since it did not alter alcohol drinking when administered alone, it is unlikely that these receptors play a significant role in regulating alcohol consumption in the DID model. Finally, blockade (not activation) of AVP (V1b) receptors was shown to reduce alcohol drinking, making it unlikely that OT signaling at these receptors would produce the same effect.
A limitation of the present study is that only male subjects were tested. Our previous work suggested a trend for female mice to exhibit greater sensitivity to OT to decrease stress-induced alcohol relapse-like responding [16]. Another study reported that genetic deletion of OTR increased alcohol drinking in female, but not male mice [70]. In a recent study, repeated systemic administration of OT was shown to significantly reduce alcohol intake to the same extent in male and female mice [15]. There is evidence for sexually dimorphic expression of OT and OTRs in brain, and fluctuations in sex hormones in females may influence OT sensitivity [71]. A recent report indicates that while male alcohol-dependent rats and male human postmortem brain analyses revealed reduced OT-immunoreactivity in PVN and upregulated OTR-binding sites in striatum, no such changes were observed in analyses of female alcohol-dependent rats or postmortem striatal tissue from female alcohol-dependent patients [72]. Thus, possible sex-related differences in the ability of endogenous (and exogenous) OT to reduce alcohol self-administration remains to be determined.
Another limitation relates to the fact that OT is known to reduce food intake [73] and, in the present study, the effects of chemogenetic activation of PVNOT+ neurons on consumption of another caloric solution (e.g., sucrose) was not examined. However, we previously reported that systemic administration of OT reduced alcohol self-administration at doses that did not alter operant self-administration of sucrose or general fluid intake [51]. It remains to be determined whether activation of endogenous OT release reduces alcohol consumption under conditions in which there are no effects on general food or fluid intake.
In summary, results from this study show that targeted chemogenetic activation of OT neurons in the PVN significantly reduced alcohol consumption in a binge-like drinking model. Further, decreased alcohol intake produced by activation of endogenous OT was reversed by pretreatment with an OTR antagonist that is known to penetrate the blood–brain barrier. In contrast, this effect was not altered when mice were pretreated with a peripherally restricted OTR antagonist. Taken together, results from these studies demonstrate a role for hypothalamic (endogenous) OT in regulation of alcohol consumption and implicate signaling at central OTRs in mediating this effect.
Funding and disclosure
Supported by grants from the National Institute on Alcohol Abuse and Alcoholism (R01 AA026536, U01 AA014095, U24 AA020929, P50 AA0107061, F31 AA026483) and VA Medical Research (I01BX000813). All authors declare that they do not have any conflicts of interest to report in connection with this manuscript.
References
Addolorato G, Vassallo GA, Antonelli G, Antonelli M, Tarli C, Mirijello A, et al. Binge drinking among adolescents is related to the development of alcohol use disorders: results from a cross-sectional study. Sci Rep. 2018;8:12624.
Jennison KM. The short-term effects and unintended long-term consequences of binge drinking in college: a 10-year follow-up study. Am J Drug Alcohol Abus. 2004;30:659–84.
McCarty CA, Ebel BE, Garrison MM, DiGiuseppe DL, Christakis DA, Rivara FP. Continuity of binge and harmful drinking from late adolescence to early adulthood. Pediatrics. 2004;114:714–9.
Becker HC. Influence of stress associated with chronic alcohol exposure on drinking. Neuropharmacology. 2017;122:115–26.
Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3:760–73.
Becker HC. Animal models of excessive alcohol consumption in rodents. Curr Top Behav Neurosci. 2013;13:355–77.
Sprow GM, Thiele TE. The neurobiology of binge-like ethanol drinking: evidence from rodent models. Physiol Behav. 2012;106:325–31.
Bowen MT, Neumann ID. The multidimensional therapeutic potential of targeting the brain oxytocin system for the treatment of substance use disorders. Curr Top Behav Neurosci. 2018;35:269–87.
King CE, Gano A, Becker HC. The role of oxytocin in alcohol and drug abuse. Brain Res. 2020;1736:146761.
Lee MR, Weerts EM. Oxytocin for the treatment of drug and alcohol use disorders. Behav Pharm. 2016;27:640–8.
McGregor IS, Bowen MT. Breaking the loop: oxytocin as a potential treatment for drug addiction. Horm Behav. 2012;61:331–9.
Pedersen CA, Smedley KL, Leserman J, Jarskog LF, Rau SW, Kampov-Polevoi A, et al. Intranasal oxytocin blocks alcohol withdrawal in human subjects. Alcohol Clin Exp Res. 2013;37:484–9.
Mitchell JM, Arcuni PA, Weinstein D, Woolley JD. Intranasal oxytocin selectively modulates social perception, craving, and approach behavior in subjects with alcohol use disorder. J Addict Med. 2016;10:182–9.
Hansson AC, Koopmann A, Uhrig S, Buhler S, Domi E, Kiessling E, et al. Oxytocin reduces alcohol cue-reactivity in alcohol-dependent rats and humans. Neuropsychopharmacology. 2018;43:1235–46.
Caruso MA, Robins MT, Fulenwider HD, Ryabinin AE. Temporal analysis of individual ethanol consumption in socially housed mice and the effects of oxytocin. Psychopharmacology (Berl). 2021. https://doi.org/10.1007/s00213-020-05741-3
King CE, Becker HC. Oxytocin attenuates stress-induced reinstatement of alcohol seeking behavior in male and female mice. Psychopharmacol (Berl). 2019;236:2613–22.
MacFadyen K, Loveless R, DeLucca B, Wardley K, Deogan S, Thomas C, et al. Peripheral oxytocin administration reduces ethanol consumption in rats. Pharm Biochem Behav. 2016;140:27–32.
Peters ST, Bowen MT, Bohrer K, McGregor IS, Neumann ID. Oxytocin inhibits ethanol consumption and ethanol-induced dopamine release in the nucleus accumbens. Addict Biol. 2017;22:702–11.
Tunstall BJ, Kirson D, Zallar LJ, McConnell SA, Vendruscolo JCM, Ho CP, et al. Oxytocin blocks enhanced motivation for alcohol in alcohol dependence and blocks alcohol effects on GABAergic transmission in the central amygdala. PLoS Biol. 2019;17:e2006421.
Stevenson JR, Wenner SM, Freestone DM, Romaine CC, Parian MC, Christian SM, et al. Oxytocin reduces alcohol consumption in prairie voles. Physiol Behav. 2017;179:411–21.
Walcott AT, Ryabinin AE. Assessing effects of oxytocin on alcohol consumption in socially housed prairie voles using radio frequency tracking. Addict Biol. 2021;26:e12893.
Bahi A, Al Mansouri S, Al Maamari E. Nucleus accumbens lentiviral-mediated gain of function of the oxytocin receptor regulates anxiety- and ethanol-related behaviors in adult mice. Physiol Behav. 2016;164:249–58.
Opacka-Juffry J, Mohiyeddini C. Experience of stress in childhood negatively correlates with plasma oxytocin concentration in adult men. Stress. 2012;15:1–10.
Carson DS, Hunt GE, Guastella AJ, Barber L, Cornish JL, Arnold JC, et al. Systemically administered oxytocin decreases methamphetamine activation of the subthalamic nucleus and accumbens core and stimulates oxytocinergic neurons in the hypothalamus. Addict Biol. 2010;15:448–63.
Ermisch A, Barth T, Ruhle HJ, Skopkova J, Hrbas P, Landgraf R. On the blood-brain barrier to peptides: accumulation of labelled vasopressin, DesGlyNH2-vasopressin and oxytocin by brain regions. Endocrinol Exp. 1985;19:29–37.
Ludwig M, Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci. 2006;7:126–36.
Neumann ID, Maloumby R, Beiderbeck DI, Lukas M, Landgraf R. Increased brain and plasma oxytocin after nasal and peripheral administration in rats and mice. Psychoneuroendocrinology. 2013;38:1985–93.
Tanaka A, Furubayashi T, Arai M, Inoue D, Kimura S, Kiriyama A, et al. Delivery of oxytocin to the brain for the treatment of autism spectrum disorder by nasal application. Mol Pharm. 2018;15:1105–11.
Dal Monte O, Noble PL, Turchi J, Cummins A, Averbeck BB. CSF and blood oxytocin concentration changes following intranasal delivery in macaque. PLoS ONE 2014;9:e103677.
Freeman SM, Samineni S, Allen PC, Stockinger D, Bales KL, Hwa GG, et al. Plasma and CSF oxytocin levels after intranasal and intravenous oxytocin in awake macaques. Psychoneuroendocrinology 2016;66:185–94.
Striepens N, Kendrick KM, Hanking V, Landgraf R, Wullner U, Maier W, et al. Elevated cerebrospinal fluid and blood concentrations of oxytocin following its intranasal administration in humans. Sci Rep. 2013;3:3440.
Lee MR, Scheidweiler KB, Diao XX, Akhlaghi F, Cummins A, Huestis MA, et al. Oxytocin by intranasal and intravenous routes reaches the cerebrospinal fluid in rhesus macaques: determination using a novel oxytocin assay. Mol Psychiatry. 2018;23:115–22.
Smith AS, Korgan AC, Young WS. Oxytocin delivered nasally or intraperitoneally reaches the brain and plasma of normal and oxytocin knockout mice. Pharm Res. 2019;146:104324.
Lee MR, Shnitko TA, Blue SW, Kaucher AV, Winchell AJ, Erikson DW, et al. Labeled oxytocin administered via the intranasal route reaches the brain in rhesus macaques. Nat Commun. 2020;11:2783.
Althammer F, Grinevich V. Diversity of oxytocin neurons: beyond magno- and parvocellular cell types? J Neuroendocrinol. 2017. https://doi.org/10.1111/jne.12549
Grinevich V, Knobloch-Bollmann HS, Eliava M, Busnelli M, Chini B. Assembling the puzzle: pathways of oxytocin signaling in the brain. Biol Psychiatry. 2016;79:155–64.
Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81:629–83.
Boccia ML, Petrusz P, Suzuki K, Marson L, Pedersen CA. Immunohistochemical localization of oxytocin receptors in human brain. Neuroscience. 2013;253:155–64.
Duque-Wilckens N, Steinman MQ, Busnelli M, Chini B, Yokoyama S, Pham M, et al. Oxytocin receptors in the anteromedial bed nucleus of the stria terminalis promote stress-induced social avoidance in female california mice. Biol Psychiatry. 2018;83:203–13.
Knobloch HS, Charlet A, Hoffmann LC, Eliava M, Khrulev S, Cetin AH, et al. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron. 2012;73:553–66.
Li K, Nakajima M, Ibanez-Tallon I, Heintz N. A cortical circuit for sexually dimorphic oxytocin-dependent anxiety behaviors. Cell. 2016;167:60–72.
Martinon D, Lis P, Roman AN, Tornesi P, Applebey SV, Buechner G, et al. Oxytocin receptors in the dorsolateral bed nucleus of the stria terminalis (BNST) bias fear learning toward temporally predictable cued fear. Transl Psychiatry. 2019;9:140.
Tan Y, Singhal SM, Harden SW, Cahill KM, Nguyen DM, Colon-Perez LM, et al. Oxytocin receptors are expressed by glutamatergic prefrontal cortical neurons that selectively modulate social recognition. J Neurosci. 2019;39:3249–63.
Iwasaki Y, Kumari P, Wang L, Hidema S, Nishimori K, Yada T. Relay of peripheral oxytocin to central oxytocin neurons via vagal afferents for regulating feeding. Biochem Biophys Res Commun. 2019;519:553–8.
Everett NA, Turner AJ, Costa PA, Baracz SJ, Cornish JL. The vagus nerve mediates the suppressing effects of peripherally administered oxytocin on methamphetamine self-administration and seeking in rats. Neuropsychopharmacology. 2021;46:297–304.
Anderson RI, Lopez MF, Griffin WC, Haun HL, Bloodgood DW, Pati D, et al. Dynorphin-kappa opioid receptor activity in the central amygdala modulates binge-like alcohol drinking in mice. Neuropsychopharmacology. 2019;44:1084–92.
Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63.
Broadbear JH, Kabel D, Tracy L, Mak P. Oxytocinergic regulation of endogenous as well as drug-induced mood. Pharm Biochem Behav. 2014;119:61–71.
Tan O, Musullulu H, Raymond JS, Wilson B, Langguth M, Bowen MT. Oxytocin and vasopressin inhibit hyper-aggressive behaviour in socially isolated mice. Neuropharmacology. 2019;156:107573.
Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates, 3rd ed. San Diego: Academic Press; 2008.
King CE, Griffin WC, Luderman LN, Kates MM, McGinty JF, Becker HC. Oxytocin reduces ethanol self-administration in mice. Alcohol Clin Exp Res. 2017;41:955–64.
Wu Z, Xu Y, Zhu Y, Sutton AK, Zhao R, Lowell BB, et al. An obligate role of oxytocin neurons in diet induced energy expenditure. PLoS ONE. 2012;7:e45167.
Penagarikano O, Lazaro MT, Lu XH, Gordon A, Dong H, Lam HA, et al. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci Transl Med. 2015;7:271ra8.
Wei D, Lee D, Cox CD, Karsten CA, Penagarikano O, Geschwind DH, et al. Endocannabinoid signaling mediates oxytocin-driven social reward. Proc Natl Acad Sci USA. 2015;112:14084–9.
Silva SM, Madeira MD, Ruela C, Paula-Barbosa MM. Prolonged alcohol intake leads to irreversible loss of vasopressin and oxytocin neurons in the paraventricular nucleus of the hypothalamus. Brain Res. 2002;925:76–88.
Stevenson JR, Young KA, Bohidar AE, Francomacaro LM, Fasold TR, Buirkle JM, et al. Alcohol consumption decreases oxytocin neurons in the anterior paraventricular nucleus of the hypothalamus in prairie voles. Alcohol Clin Exp Res. 2017;41:1444–51.
Zhou Y, Liang Y, Low MJ, Kreek MJ. Nuclear transcriptional changes in hypothalamus of Pomc enhancer knockout mice after excessive alcohol drinking. Genes Brain Behav. 2019;18:e12600.
Peris J, MacFadyen K, Smith JA, de Kloet AD, Wang L, Krause EG. Oxytocin receptors are expressed on dopamine and glutamate neurons in the mouse ventral tegmental area that project to nucleus accumbens and other mesolimbic targets. J Comp Neurol. 2017;525:1094–108.
Sabihi S, Dong SM, Maurer SD, Post C, Leuner B. Oxytocin in the medial prefrontal cortex attenuates anxiety: Anatomical and receptor specificity and mechanism of action. Neuropharmacology. 2017;125:1–12.
Viviani D, Charlet A, van den Burg E, Robinet C, Hurni N, Abatis M, et al. Oxytocin selectively gates fear responses through distinct outputs from the central amygdala. Science. 2011;333:104–7.
Yuan L, Liu S, Bai X, Gao Y, Liu G, Wang X, et al. Oxytocin inhibits lipopolysaccharide-induced inflammation in microglial cells and attenuates microglial activation in lipopolysaccharide-treated mice. J Neuroinflammation. 2016;13:77.
Warden AS, Wolfe SA, Khom S, Varodayan FP, Patel RR, Steinman MQ, et al. Microglia control escalation of drinking in alcohol-dependent mice: genomic and synaptic drivers. Biol Psychiatry 2020;88:910–21.
Caldwell HK, Stewart J, Wiedholz LM, Millstein RA, Iacangelo A, Holmes A, et al. The acute intoxicating effects of ethanol are not dependent on the vasopressin 1a or 1b receptors. Neuropeptides. 2006;40:325–37.
Sanbe A, Takagi N, Fujiwara Y, Yamauchi J, Endo T, Mizutani R, et al. Alcohol preference in mice lacking the Avpr1a vasopressin receptor. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1482–90.
Zhou Y, Rubinstein M, Low MJ, Kreek MJ. V1b receptor antagonist SSR149415 and naltrexone synergistically decrease excessive alcohol drinking in male and female mice. Alcohol Clin Exp Res. 2018;42:195–205.
Zhou Y, Colombo G, Carai MA, Ho A, Gessa GL, Kreek MJ. Involvement of arginine vasopressin and V1b receptor in alcohol drinking in Sardinian alcohol-preferring rats. Alcohol Clin Exp Res. 2011;35:1876–83.
Edwards S, Guerrero M, Ghoneim OM, Roberts E, Koob GF. Evidence that vasopressin V1b receptors mediate the transition to excessive drinking in ethanol-dependent rats. Addict Biol. 2012;17:76–85.
Ryan ML, Falk DE, Fertig JB, Rendenbach-Mueller B, Katz DA, Tracy KA, et al. A phase 2, double-blind, placebo-controlled randomized trial assessing the efficacy of ABT-436, a novel V1b receptor antagonist, for alcohol dependence. Neuropsychopharmacology. 2017;42:1012–23.
Vrachnis N, Malamas FM, Sifakis S, Deligeoroglou E, Iliodromiti Z. The oxytocin-oxytocin receptor system and its antagonists as tocolytic agents. Int J Endocrinol. 2011;2011:350546.
Rodriguez KM, Smith BL, Caldwell HK. Voluntary alcohol consumption is increased in female, but not male, oxytocin receptor knockout mice. Brain Behav. 2020;10:e01749.
Dumais KM, Veenema AH. Vasopressin and oxytocin receptor systems in the brain: sex differences and sex-specific regulation of social behavior. Front Neuroendocrinol. 2016;40:1–23.
Hansson AC, Spanagel R. No changes in the oxytocin system in alcohol-dependent female rodents and humans: Towards a sex-specific psychopharmacology in alcoholism. Addict Biol. 2021;26:e12945.
Wald HS, Chandra A, Kalluri A, Ong ZY, Hayes MR, Grill HJ. NTS and VTA oxytocin reduces food motivation and food seeking. Am J Physiol Regul Integr Comp Physiol. 2020;319:R673–83.
Author information
Authors and Affiliations
Contributions
CEK conducted all surgical and behavioral work, assisted in histological and data analyses, and wrote main portions of the manuscript. WCG conducted immunohistochemical and confocal microscopy procedures and assisted in data interpretation and editing manuscript drafts. MFL assisted in experimental design and statistical analyses, along with data interpretation and editing manuscript drafts. HCB conceived of the project, acquired funding, guided all aspects of experimental work, and edited final draft of manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
King, C.E., Griffin, W.C., Lopez, M.F. et al. Activation of hypothalamic oxytocin neurons reduces binge-like alcohol drinking through signaling at central oxytocin receptors. Neuropsychopharmacol. 46, 1950–1957 (2021). https://doi.org/10.1038/s41386-021-01046-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01046-x
