
Abstract
Anger is a common and debilitating symptom of post-traumatic stress disorder (PTSD). Although studies have identified brain circuits underlying anger experience and expression in healthy individuals, how these circuits interact with trauma remains unclear. Here, we performed the first study examining the neural correlates of anger in patients with PTSD. Using a data-driven approach with resting-state fMRI, we identified two prefrontal regions whose overall functional connectivity was inversely associated with anger: the left anterior middle frontal gyrus (aMFG) and the right orbitofrontal cortex (OFC). We then used concurrent TMS-EEG to target the left aMFG parcel previously identified through fMRI, measuring its cortical excitability and causal connectivity to downstream areas. We found that low-anger PTSD patients exhibited enhanced excitability in the left aMFG and enhanced causal connectivity between this region and visual areas. Together, our results suggest that left aMFG activity may confer protection against the development of anger, and therefore may be an intriguing target for circuit-based interventions for anger in PTSD.
Similar content being viewed by others
Introduction
Post-traumatic stress disorder (PTSD) is a common and disabling disorder worldwide [1, 2]. Although the illness has a diverse array of associated symptoms, perhaps the largest single predictor of overall severity is anger [3]. People with PTSD report more anger than the general population [4], an association that holds true even when controlling for pre-trauma anger levels [5]. Perhaps due to the type of trauma, the relationship between anger and PTSD is particularly strong in military populations [6, 7], with almost half of veterans with PTSD reporting that they engaged in physical aggression after returning from deployment [8].
Besides the immediate emotional and interpersonal consequences of anger [9], PTSD-related anger is associated with poor physical health [10] and poor treatment response [11]. Indeed, PTSD patients entering treatment often report anger as their greatest clinical concern [12] and the symptom that most limits their daily functioning [13]. Despite strong evidence for the critical importance of anger in PTSD, it is understudied compared to other aspects of the disorder [14] and little is known about its cognitive, psychological, or neurobiological underpinnings.
To our knowledge, no studies have yet examined the neural correlates of anger in patients with PTSD. When patients with PTSD are presented with negative stimuli such as angry faces, they generally show hyperactivation in the amygdala, insula, and the anterior cingulate, and hypoactivation in medial prefrontal regions [15,16,17]. However, these evoked responses have not been linked to participants’ anger levels or aggressive behavior. Thus, it is unclear whether the patterns of brain activity in response to negative stimuli predispose to or protect from anger, or indeed have any association with the patients’ symptoms.
Although the neurobiology of anger in PTSD remains unknown, multiple studies have measured neural correlates of anger in healthy participants or patients with other disorders. Some of the first clues came from lesion studies, which revealed that people with damage to the orbital and lateral prefrontal cortex (PFC) were more likely to experience anger outbursts [18]. Similarly, epileptic patients with reduced prefrontal gray matter were noted to exhibit more impulsive aggression [19]. In contrast, people with greater ventromedial PFC activity in response to angry faces [20] or unfair offers in an ultimatum game [21] reported lower levels of anger and aggression. Consistent with these findings, enhancing either ventromedial [22] or dorsolateral [23, 24] PFC activity with transcranial direct current stimulation may reduce provoked anger. Taken together, it appears that at least in healthy individuals, activity in specific prefrontal areas may mitigate anger and aggression.
Conversely, anger and aggression appear to be enhanced by the activity of certain subcortical regions, particularly the amygdala [25, 26] and ventral striatum [27]. One recent paper, for example, showed that patients with intermittent explosive disorder had higher amygdala responses to angry faces than healthy controls and that the amygdala response correlated with the number of prior aggressive acts [28]. Furthermore, a series of studies demonstrated that amygdala responses to an anger-inducing game [25] or film [29] correlated with later subthreshold traumatic stress symptoms after a year of combat training. Combining these results with the PFC findings above, it has been hypothesized that reduced prefrontal control of subcortical regions may predispose individuals to angry and aggressive behavior [21, 30]. Indeed, multiple resting-state fMRI connectivity analyses have revealed that people with high trait anger or aggression exhibit low connectivity between the amygdala and either ventral [31] or dorsolateral [32] PFC, although at least one paper found the opposite direction of effect [25]. Similarly, people with reduced functional connectivity between the nucleus accumbens and the PFC tended to be more aggressive [27].
Here, we examine how patterns of brain connectivity and excitability relate to anger levels in patients with PTSD. Why do so many patients with PTSD develop anger problems, and how do some patients avoid this fate? To tackle this question, we took advantage of multimodal neuroimaging data in veterans with and without PTSD, exploring which neural signatures distinguish between PTSD participants with high anger levels and those with low anger levels, and how these signatures compare with healthy, trauma-exposed controls. We first took a data-driven, hypothesis-generating approach, searching the entire brain for fMRI connectivity correlates of anger in patients with PTSD. We then used these findings as the basis for subsequent analysis of TMS-EEG data, which allowed us to assess both cortical excitability and causal connectivity from the areas of stimulation. We discovered two prefrontal parcels whose connectivity to the rest of the brain was inversely correlated with anger and verified that one of these—located within the left anterior middle frontal gyrus (aMFG)—exhibited both higher excitability and stronger causal connectivity to downstream sensory areas in patients with PTSD who reported low anger, compared to their high-anger peers. Thus, targeting this region may hold promise for the treatment of anger in PTSD.
Materials and methods
Participants
The study included 162 Iraq/Afghanistan-era combat veterans (97 trauma-exposed healthy participants and 65 with PTSD). PTSD diagnoses were based on DSM-5 criteria using the Clinician-Administered PTSD Scale (CAPS) [33]. Exclusion criteria included a history of psychotic disorders, bipolar disorder, or substance use disorder within 3 months for patients and lifetime for controls; a history of a neurological disorder; greater than mild traumatic brain injury (i.e., >30 min loss of consciousness or >24 h post-trauma amnesia); and claustrophobia. Controls were required to have experienced a military service-related criterion A trauma, but not meet lifetime criteria for any psychiatric disorder, including PTSD. Participants were allowed to continue their current medications as long as the dose was stable for at least 2 months.
Participants were recruited at either New York University or Stanford University after signing an informed consent approved by the relevant University’s institutional review board, in accordance with the ethical principles in the Declaration of Helsinki. Data from some of the same participants have been reported previously [34], although not any data related to anger. The study was designed for the discovery and validation of PTSD-related biomarkers; there were no pre-specified hypotheses or primary analyses.
All 162 participants underwent resting-state fMRI scans, and 86 of them (49 controls, 37 with PTSD) also underwent TMS-EEG recordings. See participant characteristics in Supplementary Tables 1–2 and 4.
Clinical and behavioral assessments
Participants completed multiple clinical and behavioral assessments and self-report scales (for more details, see ref. [34]). In addition to the CAPS for PTSD symptoms and the Beck Depression Inventory (BDI-II) [35] for depressive symptoms, participants reported on their quality of life using the World Health Organization’s WHOQOL-BREF survey [36]. Intelligence quotient (IQ) was estimated with the Wechsler Abbreviated Scale of Intelligence (WASI) [37]. Participants also completed a computerized neurocognitive test battery [38].
Our anger measure, the State and Trait Anger Expression Inventory (STAXI-2) [39], is a self-report instrument that assesses the experience, expression, and control of anger. For this study, we used the 10-item trait anger scale score, which assesses perceptions of anger responses experienced over time, as well as the frequency that angry feelings are experienced in situations involving frustration and negative evaluation. All items are measured on a 4-point Likert scale, for a total range of 10–40.
fMRI
Participants underwent an 8-min resting-state scan at either New York University (Siemens Skyra 3T scanner; N = 114) or Stanford University (General Electric 750 3T scanner; N = 48). To probe functional connectivity, the mean time series was extracted for 100 cortical and 33 subcortical brain parcels, identical to what we performed in a recent publication [40]. For interpretation purposes, parcels were mapped to seven previously-identified functional brain networks [41] based on the spatial overlap between each parcel and each network. Specifically, each voxel in a parcel was assigned to one of the seven networks, and the parcel was mapped to whichever network covered the most voxels. Notably, this mapping of parcel to network is identical to that provided by Schaefer et al. [42]. Connectivity values between each pair of parcels were estimated using Pearson’s correlation coefficient. Fisher-transformed absolute connectivity values were then averaged for each parcel to obtain a single measure of that parcel’s global connectivity to all other parcels. We chose to measure global connectivity because this approach is often used to reduce whole-brain data dimensionality while maintaining most of the signal and avoiding redundancy in the data [25, 43]. More details about fMRI acquisition, preprocessing, brain parcellation, and time-series extraction can be found in the Supplementary Information.
TMS-EEG
In a subsequent session at both the NYU and Stanford sites, a subset of participants underwent EEG recordings concurrent with single-pulse TMS stimulation of two parcels in the left dorsolateral prefrontal cortex (DLPFC): the aMFG (in the ventral attention/salience network) and the posterior middle frontal gyrus (pMFG, in the frontoparietal control network). To determine the target coordinates, we first identified these two resting-state networks using an independent component analysis of fMRI data from a separate group of individuals [44], then extracted the MNI coordinates of the prefrontal component of those networks, and finally transformed these coordinates back to individual subject native space using non-linear spatial normalization with FSL (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki). The targets were placed on each participant’s T1-weighted anatomical MRI for neuronavigation using the Visor2 LT 3D neuronavigation system (ANT Neuro, The Netherlands). For further details about the TMS-EEG data acquisition and preprocessing, please see the Supplementary Information.
Statistics
All statistical analyses were conducted in SPSS (IBM Corporation). Participants with PTSD were divided on the basis of their trait anger scores on the STAXI-2. Cutoffs were established from the STAXI-2 manual [39], which defines high anger as >75th percentile (trait anger score > = 21) and low anger as <25th percentile (trait anger score < = 14). This allowed us to divide our sample into three groups: healthy trauma-exposed participants (n = 97), low-anger PTSD (n = 19), and high-anger PTSD participants (n = 27). The approach of splitting participants into high- and low-anger groups is similar to that used in other papers examining anger [45,46,47] and was useful since the distribution of anger scores was positively skewed, with multiple subjects scoring the lowest possible score, making it difficult to discriminate among participants with low scores. When group differences were detected, we examined the relationship between functional connectivity of these brain parcels and STAXI-2 scores across all members of the PTSD group, ensuring that data from all participants—not just those at the extremes of self-reported anger—were incorporated into the study.
For our fMRI analyses, we constructed a generalized linear model (GLM) for each brain parcel, using that parcel’s global connectivity as the dependent variable; group (healthy, low-anger PTSD, high-anger PTSD) as a categorical predictor; and age, gender, education level, IQ, CAPS total score, BDI total score, and site of fMRI acquisition as covariates. The P values for the set of 133 GLMs were corrected for multiple comparisons using the false discovery rate (FDR) procedure. Any surviving GLM results were then subjected to post hoc pairwise tests of significant effects using the Sidak correction for multiple comparisons. Similarly, for our TMS-EEG analyses, we constructed a GLM for each of the four time windows (P30, P60, N100, and P200), FDR-corrected the resulting P values, and then ran post hoc tests with Sidak correction. This approach resembles what we previously used to study fMRI and TMS-EEG data from PTSD patients split by performance on a memory task [34]. We used a nonparametric test (Kendall’s tau) to examine correlations between brain measures and STAXI, due to the non-normal distribution of the STAXI data. For similar reasons, group mean differences in anger were estimated with the nonparametric Wilcoxon rank-sum test. Fisher’s exact tests were used to compare proportions of controls or PTSD participants with low or high anger.
Results
Subjects and demographics
A total of 162 trauma-exposed combat veterans, 97 without any current or prior psychiatric diagnosis and 65 with current PTSD, filled out the STAXI-2 trait anger questionnaire, along with other clinical surveys and behavioral paradigms. Compared to participants with PTSD, trauma-exposed veterans who did not meet criteria for any psychiatric disorder reported fewer PTSD and depressive symptoms, higher quality of life, and less medication use (Supplementary Table 1).
We next examined how patients and controls rated their trait anger on the STAXI-2. While patients with PTSD tiled the entire range of scores, healthy controls clustered in the lower end (Supplementary Fig. 1: controls, median 13; patients, median 19; Wilcoxon rank-sum, Z = −5.7, P < 1.5E-8). To examine clinical, behavioral, and neural markers of anger, we used established cutoffs for high anger (>=75th percentile) and low anger (<=25th percentile) on the STAXI-2 [39]. Using these cutoffs, only 3/97 controls reported high anger, compared to 27/65 patients with PTSD (Fisher’s exact test, P < 0.001) Conversely, 65/97 controls reported low anger, versus 19/65 patients with PTSD (Fisher’s exact test, P < 0.001). Comparing PTSD patients with high versus low anger revealed that the high-anger group had higher depression scores and lower quality of life, but no substantial differences in other demographic or cognitive measures (Supplementary Table 2).
Group differences in resting-state functional connectivity
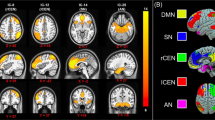
As this is the first study to measure neural correlates of anger in patients with PTSD, we began with a hypothesis-generating, data-driven approach, searching the entire brain for parcels whose overall (i.e., global) connectivity differed significantly between healthy controls, low-anger PTSD patients, and high-anger PTSD patients. After extracting BOLD signal from 100 previously identified cortical parcels [42] and 33 subcortical parcels [48,49,50,51], we calculated the global connectivity of each parcel (i.e., the average pairwise connectivity from that parcel to every other parcel in the brain) for each subject. After correcting for multiple comparisons, two parcels showed significant differences between the groups (Fig. 1): the left aMFG, which is part of the DLPFC and mapped onto the salience network (MNI coordinates: x = −30, y = 44, z = 30; Wald χ2 = 16.8, P < 0.0002, Pfdr < 0.03); and the right orbitofrontal cortex (OFC), mapped onto the limbic network (MNI coordinates: x = 13, y = 37, z = −19; Wald χ2 = 13.2, P < 0.001, Pfdr < 0.05).

Resting-state fMRI global connectivity values were calculated for 133 brain regions in each participant and then entered into generalized linear models to discover regions that could distinguish healthy controls, low-anger PTSD patients, and high-anger PTSD patients. Two regions survived FDR correction: the left aMFG (A) and the right OFC (C). In both cases, the low-anger group had stronger connectivity than either high-anger patients or healthy controls. In a post hoc, exploratory analysis, global connectivity levels from both the left aMFG (B) and the right OFC (D) inversely correlated with trait anger. *P < 0.05, ***P < 0.001, Sidak correction for multiple comparisons.
For the left aMFG (Fig. 1A), post hoc comparisons between the groups revealed that the low-anger group had stronger global connectivity than both the high-anger group (P < 0.0001) and the healthy controls (P < 0.02). Furthermore, across all PTSD patients, there was a significant negative correlation between the global connectivity of this parcel and trait anger scores (Fig. 1B, Kendall’s τ = −0.28, P = 0.0033). For the right OFC (Fig. 1C), post hoc pairwise comparisons showed that again, the low-anger group exhibited stronger global connectivity than both the high-anger group (P < 0.001) and the healthy controls (P < 0.013). Across all patients, we again found a significant negative correlation between the global connectivity of this parcel and trait anger scores (Fig. 1D, Kendall’s τ = −0.21, P < 0.029).
To explore what might be driving the global connectivity group differences, we examined the pairwise connectivity strengths between each of the two parcels above and all 132 other parcels. This exploratory analysis revealed group differences in a variety of functional connections, especially involving sensory areas (e.g., visual and somatosensory cortex) and subcortical parcels (Fig. 2). In total, we identified 18 connections that significantly differed between the groups after correction for multiple comparisons (Supplementary Table 3). Five of these involved the left aMFG and its connectivity to parcels in the visual cortex, cerebellum, and striatum (Fig. 3A and Supplementary Fig. 2). The remaining 13 connections involved the right OFC and its connectivity to multiple parcels, including the striatum and cortical parcels involved in somatosensory processing, executive control, and the default mode network (Fig. 3B and Supplementary Fig. 3). All but one of the functional connections showed significant correlations between STAXI-2 scores and connectivity in the full PTSD group (all P < 0.03, Supplementary Figs. 4–5).

In a post hoc, exploratory analysis, functional connectivity strengths were calculated from the left aMFG (top) and the right OFC (bottom) to all other parcels in the brain. The corresponding significance levels are presented on the right using −log(P value) plots, in which the vertical line indicates the −log(FDR adjusted P value threshold), which are 6.91 for left aMFG and 5.51 for right OFC. Numerous group differences are apparent. All regions surviving FDR correction are displayed in Fig. 3.

Positive (yellow) and negative (blue) connections are shown from the left aMFG (A) and the right OFC (B).
Group differences in TMS response
Our exploratory fMRI analysis suggested that left aMFG and right OFC may play a role in tempering anger expression in patients with PTSD, since higher connectivity from these regions was associated with lower anger levels. To test the consistency of these findings across modalities, we recorded EEG from a subset of the participants (49 healthy controls and 37 PTSD patients, demographics in Supplementary Table 4) while they received single pulses of TMS to the left aMFG, one of the regions identified with fMRI. This hypothesis-driven approach allowed us to measure both cortical excitability (i.e., the neural response to TMS directly underneath the coil) and causal connectivity (i.e., the neural response to TMS in downstream areas) [52, 53], complementing the measures from fMRI. Given the fMRI findings, we predicted that the left aMFG would be more excitable and more strongly connected to specific downstream regions in patients with low anger.
We first examined cortical excitability [52] by quantifying the amplitudes of four time-locked evoked potentials (P30, P60, N100, and P200) that were source-localized to the aMFG, the brain parcel being stimulated. We entered the peaks of these TMS-evoked potentials into a GLM with the same covariates as above and found a significant effect of group for both the P30 and the P60 potentials (Fig. 4). In both cases, this was driven by high-anger PTSD patients, who exhibited lower-amplitude potentials than the other groups (P30, Sidak post hoc comparisons: high-anger vs controls, P = 0.001; high-anger vs low-anger, P = 0.039; P60: high-anger vs controls, P < 0.001).

EEG signals were recorded while single pulses of TMS were applied to the region of the dorsolateral prefrontal cortex (DLPFC) associated with anger (left aMFG, white sphere, which maps onto the salience network, shown in red). Early but not late TMS-evoked potentials were significantly associated with anger. P30: Wald χ2 = 16, P = 3.35e-4, Pfdr < 6.7e-4. P60: Wald χ2 = 28.8, P = 5.55e-7, Pfdr < 2.2e-6. *P < 0.05, ***P < 0.001, Sidak correction for multiple comparisons. For the color figure, please refer to the online version.
To ensure the spatial specificity of these findings, we performed two control analyses. First, we examined the TMS-evoked potentials in the left aMFG during stimulation of the left posterior middle frontal gyrus (pMFG), a neighboring part of the DLPFC that maps onto the frontoparietal control network rather than the salience network. This parcel was not found to be related to anger in our fMRI analysis, so we predicted that the TMS-EEG results would be similarly unrevealing. Indeed, the GLM results were insignificant (Pfdr > 0.05 for all four potentials, Supplementary Fig. 6), implying that the results above were not solely due to baseline differences in left aMFG excitability. Second, we examined EEG responses in left pMFG to stimulation of left pMFG. Again, the GLM was insignificant (Pfdr > 0.05 for all potentials, Supplementary Fig. 6), implying that the results were not solely due to differences in TMS-evoked activity under the coil, regardless of the stimulation site. Instead, our results suggest that in patients with PTSD, anger-related changes in excitability and connectivity are specific to aMFG. Note, however, that the negative results in these control analyses may be due to limited sample size, as direct comparisons between the aMFG and pMFG cortical excitability values did not reach statistical significance (frontal parcel x group interaction, P = 0.06 for the P30 and P60 potentials).
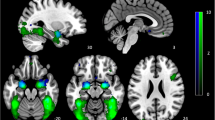
Finally, we used TMS-EEG to examine causal connectivity between the left aMFG and the two cortical parcels identified by our fMRI analysis, both mapped to the visual network. Specifically, we stimulated the left aMFG with single-pulse TMS, and then measured the evoked EEG potentials in the two visual parcels revealed by fMRI. Given our fMRI findings, our hypothesis was that PTSD patients with low anger would have stronger causal connectivity with these visual parcels than PTSD patients with high anger. As EEG is limited to cortical regions, we could not test connectivity to any of the subcortical regions identified by fMRI. For both visual areas, consistent with our hypothesis, we found a significant effect of group for the P30, P60, and N100 potentials, with low-anger PTSD patients showing the strongest TMS-evoked potentials (Fig. 5). As a control, we repeated the analysis for stimulation of left pMFG, and there was no significant effect of group (Pfdr > 0.05 for all four potentials in both visual areas, Supplementary Fig. 7). Again, however, direct comparisons of the pMFG and aMFG findings did not reach significance (frontal parcel x group interaction, P < 0.1 for the P30, P60, and N100 parcels).

EEG signals in two areas of the visual cortex were recorded while single pulses of TMS were applied to the region of the dorsolateral prefrontal cortex (DLPFC) associated with anger (left aMFG). For both visual areas, there was a significant effect of group for the P30 (area 52, Wald χ2 = 13.5, P = 0.001, Pfdr = 0.004; area 53, Wald χ2 = 16.7, P = 2.3e-4, Pfdr < 9e-4), P60 (area 52, Wald χ2 = 10.1, P = 0.006, Pfdr = 0.012; area 53, Wald χ2 = 12.1, P = 0.002, Pfdr = 0.004), and N100 TMS-evoked potentials (area 52, Wald χ2 = 6.8, P = 0.034, Pfdr = 0.045; area 53, Wald χ2 = 10.7, P = 0.005, Pfdr = 0.007). *P < 0.05, **P < 0.01, Sidak correction for multiple comparisons.
Discussion
We performed a two-site, cross-sectional, exploratory study of combat veterans with and without PTSD, using self-report scales, fMRI, and concurrent TMS-EEG to discover neural correlates of anger in this patient population. We found that compared to trauma-exposed healthy controls, PTSD participants endorsed substantially higher trait anger levels, with 42% reporting anger that exceeded the clinical cutoff, versus just 3% of the controls. Taking a data-driven, whole-brain approach to fMRI connectivity data, we found that anger was associated with global connectivity in two parcels: the left aMFG, a part of the DLPFC associated with the salience network; and the right OFC, in the limbic network. Both parcels had stronger global connectivity in the low-anger group than in the high-anger group or controls. Finally, we used concurrent TMS-EEG to complement our fMRI findings with an approach that allows interrogation of the causal influence of the stimulated region on downstream regions. The results suggested a role for the left aMFG in anger expression, as PTSD patients with low-anger scores showed higher cortical excitability than did patients with high-anger scores, as well as higher connectivity to visual areas. Together, our findings raise the hypothesis that stronger connectivity and excitability in specific frontal parcels may protect against anger in PTSD.
To our knowledge, this is the first study to examine the neural substrates of anger in patients with PTSD. Our results, however, fit well with prior experiments in other populations, which have reported the involvement of cortical and limbic areas in the experience and expression of anger [54]. Specifically, our OFC findings are consistent with prior work showing that patients with OFC lesions have higher aggression and violence scores than patients with lesions in other regions [55] and that healthy people with higher levels of aggression have reduced OFC reactivity [20]. Likewise, our aMFG findings dovetail with meta-analyses showing impairments of DLPFC in antisocial behavior [56], although it is important to note that some areas of the frontal cortex may in fact be more reactive in healthy people with high anger levels [46, 57]. The laterality of our findings may also fit prior literature showing left DLPFC and right OFC deficits in antisocial behavior [56].
In addition to global connectivity, we also explored specific pairwise connections from these two cortical parcels. Our most common finding was in the ventral and dorsomedial striatum, consistent with prior work associating impulsive aggression with increased responsiveness in these striatal regions, which may increase sensitivity to frustration or rejection [27, 58, 59]. Specifically, we found that the OFC (part of the cortico-limbic system) showed increased positive connectivity with striatal regions in high-anger patients, while the aMFG (part of the salience network) showed increased positive connectivity with striatal regions in the low-anger group. Thus, activation of specific striatal regions by the limbic network, and not the salience network, may predispose patients to higher levels of aggression. Future work using causal tools in humans or animal models will be crucial to understand how these networks compete to determine behavior.
Our data also suggested an association between anger and frontal connections with primary sensory areas. In particular, both the fMRI and TMS-EEG results revealed stronger (negative) connectivity between the aMFG and the visual cortex in the low-anger group than in the high-anger group. Although our study could not test the functional role of these connections, one possibility is that top-down control of primary visual areas helps to minimize distorted imagery, which has been theorized to contribute to anger in PTSD [14]. According to this theory, anger in PTSD—unlike anger in other settings—also involves impairments at the level of perception. Such impairments could make it more likely for patients to perceive or experience threat when it does not exist, exacerbating the so-called “survivor mode” in which veterans carry home the threat-focused mindset they honed on the battlefield [60]. Hypothetically, stronger frontal regulation of visual areas may allow veterans to reverse this mindset and set a higher threshold before perceiving environmental cues as threatening and worthy of aggression.
The idea that anger in PTSD may differ from other types of anger is also supported by the absence of the amygdala from our findings. Trauma is thought to affect amygdala functioning [61, 62], but perhaps in a sample in which everyone is trauma-exposed, amygdala activity plays a smaller role than sensory or regulatory activity in the development of anger. Future studies with non-trauma-exposed controls would be crucial to test this hypothesis.
One intriguing finding was that the trauma-exposed controls, who had low levels of anger, showed fMRI connectivity patterns similar to those seen in PTSD patients with high anger. One possible explanation may be that these controls, who are by definition protected from developing PTSD despite trauma exposure, have no need to upregulate cortical regions to prevent anger. In other words, other brain circuits (likely involving the amygdala [25, 29]) may determine a person’s predisposition to developing PTSD. Once PTSD is established, aMFG and OFC may then be important in determining the presence or extent of anger. Such a two-step process explanation is consistent with neural models of PTSD that posit a predisposing, pre-trauma vulnerability followed by acquired factors that determine particular symptoms [63].
Although the trauma-exposed controls sometimes resembled low-anger patients and sometimes resembled high-anger patients, the difference between the low- and high-anger PTSD patients was consistent: the low-anger group exhibiting stronger functional connectivity and cortical excitability than the high-anger group. This raises the possibility that among patients with PTSD, enhanced capacity for specific cortical areas to engage in top-down control may be protective, endowing this group with resilience against anger. If this hypothesis is true, our results point to potential anger-reduction strategies in PTSD. There are no FDA-approved medications to treat anger or aggression in PTSD, and evidence for the efficacy of psychotherapy remains limited [64]. There are hints that neurostimulation may be a new avenue for treatment, with at least two small studies showing that upregulating DLPFC through transcranial direct current stimulation (tDCS) can reduce aggressive statements or behavior [23, 24], while downregulating left DLPFC through TMS can increase aggression [65]. Compared to the tDCS studies, here we had the benefit of higher spatial resolution, and our results imply that stimulation of the aMFG (but not pMFG) may be a worthwhile target for future investigation.
This exploratory, hypothesis-generating study had several limitations. First, self-report of trait anger may not correspond well with actual day-to-day experience or expression of anger [66]. Future work should supplement self-report scales with objective measures of aggression as well as collateral from others in the participants’ lives. Second, we did not attempt to induce anger; thus, our results speak only to more stable, dispositional characteristics. Others have previously examined provoked aggression in healthy participants, showing activation of a large portion of the frontal cortex [67, 68]. It would be interesting to examine the neural basis of acute anger in PTSD. Third, this was a cross-sectional study, and thus we could not examine causal relationships. For example, it is unclear if the PTSD group had higher anger because PTSD caused the anger or because people with high trait anger were more likely to develop PTSD, as previously reported in a prospective study of police officers [5]. Additional prospective studies with pre-specified hypotheses will be vital to track the development of anger after trauma exposure. Fourth, some of the FDR-corrected P values were close to the significance cutoff, and thus should be interpreted with caution, especially given the inclusion of multiple covariates and the potential confound of a two-site design. Fifth, the sample was entirely trauma-exposed veterans, and primarily male, so our results may not generalize to other populations with PTSD. Indeed, the fact that our controls were trauma-exposed implies that we cannot make any conclusions about the specific effect of trauma on anger; rather, we can only speak to the development of anger in patients who have PTSD, compared to those who did not develop PTSD despite trauma exposure. Furthermore, in most of our results, the trauma-exposed controls resembled the high-anger PTSD group, but when it came to cortical excitability, they resembled the low-anger PTSD group. Future studies that include a healthy, non-trauma-exposed group with varying levels of anger would better enable an analysis of the effect of trauma on the neural correlates of anger.
The uniquely high prevalence and morbidity of anger in PTSD imply an urgent need to understand the neural mechanisms that predispose to or protect from this symptom. Here, we uncover specific cortical parcels and cortical-subcortical connections that may be areas of vulnerability amenable to rTMS and other neuromodulatory treatments.
Funding and disclosure
This work was funded by R01 MH091860 from the National Institute of Mental Health (to AE), a grant from the Steven A. and Alexandra M. Cohen Foundation to NYU School of Medicine (to CRM), and funds from Cohen Veterans Bioscience and Ann and Peter Tarlton (to AE). AE was also funded by the Sierra-Pacific Mental Illness Research, Education, and Clinical Center of the Palo Alto Veterans Affairs Health Care System. NE was supported by NIMH grants T32 MH019908 and K08 MH123791. WW was funded by National Natural Science Foundation of China (Nos. 61876063 and 61836003). AE has consulted for and owns equity in Akili Interactive and Mindstrong Health, and has consulted for Brainsway, Cervel, Takaeda, Posit, Acadia, Otsuka, Lundbeck, and Janssen. AE received a grant from Brain Resource, received salary from Alto Neuroscience, and was a paid editor at Neuropsychopharmacology. AE and CRM have a pending patent (WO2017172487A1) entitled “Detecting or treating post-traumatic stress syndrome.” NE, AMK, WW, and DAA have nothing to declare.
References
Alonso J, Petukhova M, Vilagut G, Chatterji S, Heeringa S, Üstün TB, et al. Days out of role due to common physical and mental conditions: results from the WHO World Mental Health surveys. Mol Psychiatry. 2011;16:1234–46.
Koenen KC, Ratanatharathorn A, Ng L, McLaughlin KA, Bromet EJ, Stein DJ, et al. Posttraumatic stress disorder in the World Mental Health Surveys. Psychol Med. 2017;47:2260–74.
Novaco RW, Chemtob CM. Anger and combat-related posttraumatic stress disorder. J Trauma Stress. 2002. https://pubmed.ncbi.nlm.nih.gov/12013063/?dopt=Abstract. Accessed 23 November 2019.
Van Voorhees EE, Dennis P, Elbogen E, Fuemmeler B, Neal L, Calhoun P, et al. Characterizing anger-related affect in individuals with posttraumatic stress disorder using ecological momentary assessment. Psychiatry Res. 2018. https://pubmed.ncbi.nlm.nih.gov/29329048/?from_term=van+voorhees+beckham+2018&from_pos=2. Accessed 23 November 2019.
Meffert SM, Metzler TJ, Henn-Haase C, McCaslin S, Inslicht S, Chemtob C, et al. A prospective study of trait anger and PTSD symptoms in police. J Trauma Stress. 2008;21:410–6.
Orth U, Wieland E. Anger, hostility, and posttraumatic stress disorder in trauma-exposed adults: a meta-analysis. J Consult Clin Psychol. 2006;74:698–706.
Miles SR, Sharp C, Tharp AT, Stanford MS, Stanley M, Thompson KE, et al. Emotion dysregulation as an underlying mechanism of impulsive aggression: Reviewing empirical data to inform treatments for veterans who perpetrate violence. Aggress Violent Behav. 2017;34:147–53.
Elbogen E, Johnson S, Wagner H, Sullivan C, Taft C, Beckham J. Violent behaviour and post-traumatic stress disorder in US Iraq and Afghanistan veterans. Br J Psychiatry J Ment Sci. 2014. https://pubmed.ncbi.nlm.nih.gov/24578444/?from_term=elbogen+2014+aggression&from_pos=1. Accessed 23 November 2019.
Taft CT, Creech SK, Kachadourian L. Assessment and treatment of posttraumatic anger and aggression: a review. J Rehabil Res Dev. 2012;49:777–88.
Ouimette P, Cronkite R, Prins A, Moos RH. Posttraumatic stress disorder, anger and hostility, and physical health status. J Nerv Ment Dis. 2004;192:563–6.
Forbes D, Creamer M, Hawthorne G, Allen N, McHugh T. Comorbidity as a predictor of symptom change after treatment in combat-related posttraumatic stress disorder. J Nerv Ment Dis. 2003. https://pubmed.ncbi.nlm.nih.gov/12586962/?from_term=forbes+mchugh+2003&from_pos=2. Accessed 24 November 2019.
Rosen C, Adler E, Tiet Q. Presenting concerns of veterans entering treatment for posttraumatic stress disorder. J Trauma Stress. 2013. https://pubmed.ncbi.nlm.nih.gov/24123262/?from_term=rosen+tiet+2013&from_pos=2. Accessed 24 November 2019.
Biddle D, Elliott P, Creamer M, Forbes D, Devilly GJ. Self-reported problems: a comparison between PTSD-diagnosed veterans, their spouses, and clinicians. Behav Res Ther. 2002;40:853–65.
McHugh T, Forbes D, Bates G, Hopwood M, Creamer M. Anger in PTSD: is there a need for a concept of PTSD-related posttraumatic anger? Clin Psychol Rev. 2012;32:93–104.
Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–88.
Patel R, Spreng RN, Shin LM, Girard TA. Neurocircuitry models of posttraumatic stress disorder and beyond: a meta-analysis of functional neuroimaging studies. Neurosci Biobehav Rev. 2012;36:2130–42.
Stark EA, Parsons CE, Van Hartevelt TJ, Charquero-Ballester M, McManners H, Ehlers A, et al. Post-traumatic stress influences the brain even in the absence of symptoms: a systematic, quantitative meta-analysis of neuroimaging studies. Neurosci Biobehav Rev. 2015;56:207–21.
Grafman J, Schwab K, Warden D, Pridgen A, Brown HR, Salazar AM. Frontal lobe injuries, violence, and aggression: a report of the Vietnam Head Injury Study. Neurology. 1996;46:1231–8.
Woermann FG, van Elst LT, Koepp MJ, Free SL, Thompson PJ, Trimble MR, et al. Reduction of frontal neocortical grey matter associated with affective aggression in patients with temporal lobe epilepsy: an objective voxel by voxel analysis of automatically segmented MRI. J Neurol Neurosurg Psychiatry. 2000;68:162–9.
Beyer F, Münte TF, Göttlich M, Krämer UM. Orbitofrontal cortex reactivity to angry facial expression in a social interaction correlates with aggressive behavior. Cereb Cortex. 2015;25:3057–63.
Gilam G, Lin T, Raz G, Azrielant S, Fruchter E, Ariely D, et al. Neural substrates underlying the tendency to accept anger-infused ultimatum offers during dynamic social interactions. NeuroImage. 2015;120:400–11.
Gilam G, Abend R, Gurevitch G, Erdman A, Baker H, Ben-Zion Z, et al. Attenuating anger and aggression with neuromodulation of the vmPFC: a simultaneous tDCS-fMRI study. Cortex J Devoted Study Nerv Syst Behav. 2018;109:156–70.
Choy O, Raine A, Hamilton RH. Stimulation of the prefrontal cortex reduces intentions to commit aggression: a randomized, double-blind, placebo-controlled, stratified, parallel-group trial. J Neurosci J Soc Neurosci. 2018;38:6505–12.
Dambacher F, Schuhmann T, Lobbestael J, Arntz A, Brugman S, Sack AT. Reducing proactive aggression through non-invasive brain stimulation. Soc Cogn Affect Neurosci. 2015;10:1303–9.
Gilam G, Maron-Katz A, Kliper E, Lin T, Fruchter E, Shamir R, et al. Tracing the Neural carryover effects of interpersonal anger on resting-state fMRI in men and their relation to traumatic stress symptoms in a subsample of soldiers. Front Behav Neurosci. 2017;11:252.
Fanning JR, Keedy S, Berman ME, Lee R, Coccaro EF. Neural correlates of aggressive behavior in real time: a review of fMRI studies of laboratory reactive aggression. Curr Behav Neurosci Rep. 2017;4:138–50.
Chester DS, DeWall CN. The pleasure of revenge: retaliatory aggression arises from a neural imbalance toward reward. Soc Cogn Affect Neurosci. 2016;11:1173–82.
McCloskey MS, Phan KL, Angstadt M, Fettich KC, Keedy S, Coccaro EF. Amygdala hyperactivation to angry faces in intermittent explosive disorder. J Psychiatr Res. 2016;79:34–41.
Lin T, Gilam G, Raz G, Or-Borichev A, Bar-Haim Y, Fruchter E, et al. Accessible neurobehavioral anger-related markers for vulnerability to post-traumatic stress symptoms in a population of male soldiers. Front Behav Neurosci. 2017;11:38.
Peper JS, de Reus MA, van den Heuvel MP, Schutter DJLG. Short fused? associations between white matter connections, sex steroids, and aggression across adolescence. Hum Brain Mapp. 2015;36:1043–52.
Fulwiler CE, King JA, Zhang N. Amygdala-orbitofrontal resting-state functional connectivity is associated with trait anger. Neuroreport. 2012;23:606–10.
Varkevisser T, Gladwin TE, Heesink L, van Honk J, Geuze E. Resting-state functional connectivity in combat veterans suffering from impulsive aggression. Soc Cogn Affect Neurosci. 2017;12:1881–9.
Weathers FW, Bovin MJ, Lee DJ, Sloan DM, Schnurr PP, Kaloupek DG, et al. The Clinician-Administered PTSD Scale for DSM–5 (CAPS-5): development and initial psychometric evaluation in military veterans. Psychol Assess. 2018;30:383.
Etkin A, Maron-Katz A, Wu W, Fonzo GA, Huemer J, Vértes PE, et al. Using fMRI connectivity to define a treatment-resistant form of post-traumatic stress disorder. Sci Transl Med. 2019;11.
Beck AT, Steer RA, Brown GK. Manual for the beck depression inventory-II. San Antonio TX Psychol Corp. 1996;1:82.
Skevington SM, Lotfy M, O’Connell K. The World Health Organization’s WHOQOL-BREF quality of life assessment: psychometric properties and results of the international field trial. A report from the WHOQOL group. Qual Life Res. 2004;13:299–310.
Wechsler D. Manual for the Wechsler abbreviated intelligence scale (WASI). Psychol Corp: San Antonio, TX; 1999.
Silverstein SM, Berten S, Olson P, Paul R, Williams LM, Cooper N, et al. Development and validation of a World-Wide-Web-based neurocognitive assessment battery: WebNeuro. Behav Res Methods. 2007;39:940–9.
Spielberger CD, Sydeman SJ, Owen AE, Marsh BJ. Measuring anxiety and anger with the State-Trait Anxiety Inventory (STAI) and the State-Trait Anger Expression Inventory (STAXI). In: Maruish ME (editor). Use Psychol. Test. Treat. Plan. Outcomes Assess. 2nd ed. Mahwah, NJ, USA: Lawrence Erlbaum Associates Publishers: 1999. p. 993–1021.
Maron-Katz A, Zhang Y, Narayan M, Wu W, Toll RT, Naparstek S, et al. Individual patterns of abnormality in resting-state functional connectivity reveal two data-driven PTSD subgroups. Am J Psychiatry. 2020;177:244–53.
Thomas Yeo BT, Krienen FM, Sepulcre J, Sabuncu MR, Lashkari D, Hollinshead M, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106:1125–65.
Schaefer A, Kong R, Gordon EM, Laumann TO, Zuo X-N, Holmes AJ, et al. Local-global parcellation of the human cerebral cortex from intrinsic functional connectivity MRI. Cereb Cortex. 2017;28:3095–114.
Maron-Katz A, Vaisvaser S, Lin T, Hendler T, Shamir R. A large-scale perspective on stress-induced alterations in resting-state networks. Sci Rep. 2016;6:21503.
Chen AC, Oathes DJ, Chang C, Bradley T, Zhou Z-W, Williams LM, et al. Causal interactions between fronto-parietal central executive and default-mode networks in humans. Proc Natl Acad Sci. 2013;110:19944–9.
Jakupcak M, Conybeare D, Phelps L, Hunt S, Holmes HA, Felker B, et al. Anger, hostility, and aggression among Iraq and Afghanistan war veterans reporting PTSD and subthreshold PTSD. J Trauma Stress. 2007;20:945–54.
Alia-Klein N, Preston-Campbell RN, Moeller SJ, Parvaz MA, Bachi K, Gan G, et al. Trait anger modulates neural activity in the fronto-parietal attention network. PLoS ONE. 2018;13:e0194444.
Forbes D, Alkemade N, Hopcraft D, Hawthorne G, O’Halloran P, Elhai JD, et al. Evaluation of the dimensions of anger reactions-5 (DAR-5) scale in combat veterans with posttraumatic stress disorder. J Anxiety Disord. 2014;28:830–5.
Buckner RL, Krienen FM, Castellanos A, Diaz JC, Yeo BTT. The organization of the human cerebellum estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106:2322–45.
Choi EY, Yeo BTT, Buckner RL. The organization of the human striatum estimated by intrinsic functional connectivity. J Neurophysiol. 2012;108:2242–63.
Chen AC, Etkin A. Hippocampal network connectivity and activation differentiates post-traumatic stress disorder from generalized anxiety disorder. Neuropsychopharmacology. 2013;38:1889–98.
Behrens TEJ, Johansen-Berg H, Woolrich MW, Smith SM, Wheeler-Kingshott CAM, Boulby PA, et al. Non-invasive mapping of connections between human thalamus and cortex using diffusion imaging. Nat Neurosci. 2003;6:750–7.
Esser SK, Huber R, Massimini M, Peterson MJ, Ferrarelli F, Tononi G. A direct demonstration of cortical LTP in humans: a combined TMS/EEG study. Brain Res Bull. 2006;69:86–94.
Rogasch NC, Fitzgerald PB. Assessing cortical network properties using TMS-EEG. Hum Brain Mapp. 2013;34:1652–69.
Coccaro EF, Sripada CS, Yanowitch RN, Phan KL. Corticolimbic function in impulsive aggressive behavior. Biol Psychiatry. 2011;69:1153–9.
Lane SD, Kjome KL, Moeller FG. Neuropsychiatry of aggression. Neurol Clin. 2011;29:49–64, vii.
Yang Y, Raine A. Prefrontal structural and functional brain imaging findings in antisocial, violent, and psychopathic individuals: a meta-analysis. Psychiatry Res. 2009;174:81–88.
Heesink L, Gladwin TE, Vink M, van Honk J, Kleber R, Geuze E. Neural activity during the viewing of emotional pictures in veterans with pathological anger and aggression. Eur Psychiatry J Assoc Eur Psychiatr. 2018;47:1–8.
Blair RJR. Considering anger from a cognitive neuroscience perspective. Wiley Interdiscip Rev Cogn Sci. 2012;3:65–74.
Rosell DR, Siever LJ. The neurobiology of aggression and violence. CNS Spectr. 2015;20:254–79.
Chemtob CM, Novaco RW, Hamada RS, Gross DM, Smith G. Anger regulation deficits in combat-related posttraumatic stress disorder. J Trauma Stress. 1997;10:17–36.
Disner SG, Marquardt CA, Mueller BA, Burton PC, Sponheim SR. Spontaneous neural activity differences in posttraumatic stress disorder: A quantitative resting-state meta-analysis and fMRI validation. Hum Brain Mapp. 2018;39:837–50.
Diamond DM, Zoladz PR. Dysfunctional or hyperfunctional? The amygdala in posttraumatic stress disorder is the bull in the evolutionary China shop. J Neurosci Res. 2016;94:437–44.
Admon R, Milad MR, Hendler T. A causal model of post-traumatic stress disorder: disentangling predisposed from acquired neural abnormalities. Trends Cogn Sci. 2013;17:337–47.
Taft CT, Creech SK, Murphy CM. Anger and aggression in PTSD. Curr Opin Psychol. 2017;14:67–71.
Perach-Barzilay N, Tauber A, Klein E, Chistyakov A, Ne’eman R, Shamay-Tsoory SG. Asymmetry in the dorsolateral prefrontal cortex and aggressive behavior: a continuous theta-burst magnetic stimulation study. Soc Neurosci. 2013;8:178–88.
Veenstra L, Bushman BJ, Koole SL. The facts on the furious: a brief review of the psychology of trait anger. Curr Opin Psychol. 2018;19:98–103.
Repple J, Pawliczek CM, Voss B, Siegel S, Schneider F, Kohn N, et al. From provocation to aggression: the neural network. BMC Neurosci. 2017;18:73.
Denson TF, Pedersen WC, Ronquillo J, Nandy AS. The Angry brain: neural correlates of anger, angry rumination, and aggressive personality. J Cogn Neurosci. 2008;21:734–44.
Acknowledgements
We are enormously indebted to the participants in this study.
Author information
Authors and Affiliations
Contributions
NE conceived the study, analyzed the data, and wrote the paper. AMK analyzed the fMRI data and edited the paper. WW analyzed the TMS-EEG data and edited the paper. DAA helped collect the data. CRM and AE conceived and funded the study and edited the paper.
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Eshel, N., Maron-Katz, A., Wu, W. et al. Neural correlates of anger expression in patients with PTSD. Neuropsychopharmacol. 46, 1635–1642 (2021). https://doi.org/10.1038/s41386-020-00942-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-00942-y
This article is cited by
-
A systematic review of neural, cognitive, and clinical studies of anger and aggression
Current Psychology (2023)
