
Abstract
Depression is a heterogeneous and etiologically complex psychiatric syndrome, not a unitary disease entity, encompassing a broad spectrum of psychopathology arising from distinct pathophysiological mechanisms. Motivated by a need to advance our understanding of these mechanisms and develop new treatment strategies, there is a renewed interest in investigating the neurobiological basis of heterogeneity in depression and rethinking our approach to diagnosis for research purposes. Large-scale genome-wide association studies have now identified multiple genetic risk variants implicating excitatory neurotransmission and synapse function and underscoring a highly polygenic inheritance pattern that may be another important contributor to heterogeneity in depression. Here, we review various sources of phenotypic heterogeneity and approaches to defining and studying depression subtypes, including symptom-based subtypes and biology-based approaches to decomposing the depression syndrome. We review “dimensional,” “categorical,” and “hybrid” approaches to parsing phenotypic heterogeneity in depression and defining subtypes using functional neuroimaging. Next, we review recent progress in neuroimaging genetics (correlating neuroimaging patterns of brain function with genetic data) and its potential utility for generating testable hypotheses concerning molecular and circuit-level mechanisms. We discuss how genetic variants and transcriptomic profiles may confer risk for depression by modulating brain structure and function. We conclude by highlighting several promising areas for future research into the neurobiological underpinnings of heterogeneity, including efforts to understand sexually dimorphic mechanisms, the longitudinal dynamics of depressive episodes, and strategies for developing personalized treatments and facilitating clinical decision-making.
Similar content being viewed by others
Introduction
The modern Diagnostic and Statistical Manual of Mental Disorders recognizes at least 256 unique symptom presentations that meet criteria for Major Depressive Disorder (MDD) [1]. For most of recorded history, scholars have recognized that depression is a disabling syndrome that comes in many forms [2,3,4]. Multiple factors contribute to diagnostic heterogeneity. Depression frequently presents with anxiety disorders, substance use disorders, ADHD, and other psychiatric co-morbidities, and exhibits strong sexual dimorphism in its prevalence and molecular correlates [5,6,7]. Individuals also present with markedly varying temporal dynamics in the frequency and duration of their depressive episodes and the stability of remission [8].
Lacking better alternatives, most studies to date have tended to treat depression as a unitary disease entity. More recently, investigators have recognized that this approach may be an obstacle to progress in research. This is especially true for efforts to identify the neurobiological mechanisms that give rise to depression, which may not be the same for all individuals, and for optimizing existing antidepressants and developing fundamentally new ones, since distinct pathophysiological mechanisms could warrant different types of treatment [9,10,11]. Indeed, diagnostic heterogeneity may be an important obstacle to achieving better outcomes in a clinical setting. On average, 11 years elapse between the onset of mental illness and the initiation of treatment [12, 13]. The majority of patients do not achieve full remission after an initial treatment and a substantial portion are treatment-resistant [14,15,16]. Untreated depression, in turn, has substantial personal and societal costs, affecting 17.7 million people in the U.S. in 2018 [17] and costing the global economy 1 trillion USD in lost productivity annually [18]. Depression is a leading cause of disability worldwide [19], and is associated with a 40% higher lifetime risk of developing cardiovascular and metabolic diseases [20] and worse outcomes for comorbid medical conditions [21,22,23,24]. In 2018, suicide due to mental illness was the second leading cause of death among 10- to 34-year-olds and the tenth cause of death overall in the United States [25]. Depression is a leading risk factor for suicide [26, 27], and over 2/3 of suicides in the United States occur in patients with depression [28].
Thus, there is a pressing need for studies aimed at understanding the neurobiological basis of heterogeneity in depression, delineating subtype-specific mechanisms, and developing new treatment strategies. Historically, most efforts to define depression subtypes have searched for clusters of symptoms that tend to co-occur in subgroups of patients and tested for neurophysiological correlates. More recently, investigators have attempted a converse approach, subgrouping or dimensionally stratifying patients on the basis of neuroimaging and other biological measures and validating the resulting models based on their ability to predict clinical symptoms, treatment outcomes, or other clinical variables. With the advent of extremely large genome-wide association studies in depression, geneticists have been able to identify multiple genetic variants, each conferring a small increase in depression risk (with many odds ratios <1.2) [29,30,31,32,33]. This highly polygenic inheritance pattern could be another important contributor to heterogeneity.
The goals of this work are to highlight some of the major causes of heterogeneity in depression; to consider how heterogeneity could impede progress in elucidating the neurobiological mechanisms of depression; to review progress on biologically informed subtyping approaches focused primarily on functional neuroimaging; and to discuss efforts from related fields to integrate neuroimaging analyses with genomic and transcriptomic data. We begin by reviewing clinical approaches to subtyping depression and consider how subtyping based on quantitative neuroimaging measures could compliment these approaches and generate testable mechanistic hypotheses. We provide a brief overview of dimensional, categorical, and hybrid approaches to parsing diagnostic heterogeneity in depression based on functional neuroimaging data, and refer readers to recent reviews from our group [34] and others providing more detailed accounts of this topic [35, 36]. Next, we review several approaches to integrating data from neuroimaging and genetics in order to advance our understanding of how genetic risk variants and molecular signaling pathways contribute to pathophysiology and heterogeneity in depression. Many of these methods have only recently been applied in efforts to parse depression heterogeneity and are fraught with potential technical obstacles. Therefore, we have attempted to highlight important technical challenges, along with emerging views on how to overcome them. We conclude by considering especially promising areas for future research, including the neurobiological basis of sex differences and episodic temporal dynamics and the potential for reverse translational approaches to test subtype-specific mechanisms generated from human neuroimaging studies.
Symptom-based subtypes of depression
Historical Perspective
For as long as scholars have been studying and writing about depression, they have recognized that it comes in different forms and may have many distinct causes. Hippocrates identified a “melancholic temperament” associated with symptoms of moroseness, despondency, and fear. It was thought to be caused by an excess of black bile, one of the four humors (blood, yellow bile, black bile, phlegm) that were thought to underlie most medical illnesses at the time [37]. Robert Burton’s The Anatomy of Melancholy, another seminal historical work, underscored distinct psychosocial causes of melancholy and depression [4]. Beginning in the 19th century, psychiatrists began to identify different forms of depression, including Kahlbaum differentiating dysthymia from the fluctuating mood of cyclothymia and Kraepelin differentiating depression from manic-depressive psychosis and dementia praecox [38, 39]. One of the first modern attempts to standardize depression nosology was with the introduction of “neurotic depression” in the DSM-II (1968), and subsequently the recognition in the DSM-III (1980) that episodic depression was not a personality disorder and was distinct from dysthymia [40, 41]. At the same time, motivated in part by an effort to devise diagnostic categories with increased reproducibility and inter-rater reliability, the DSM-III and -IV defined broad categories of depression—including the modern definition for MDD that requires a patient to present with five or more of nine possible symptoms. This definition yields 256 unique symptom combinations (126 five-symptom combinations, 84 six-symptom combinations, 36 seven-symptom combinations, etc.), and comprises a very large and highly heterogeneous diagnostic category that encompasses >17 million people annually in the United States alone [17]. With increasing interest in psychopharmacological treatments, investigators in the 1980s began to systematically evaluate the causes of heterogeneity in depression and their neurobiological substrates.
Sources of heterogeneity
Investigators now recognize multiple sources of depression heterogeneity. First, as noted above, there is symptom heterogeneity: there are at least 256 unique symptom combinations that meet the criteria for MDD, and some of those criteria are themselves heterogeneous. For example, MDD patients could be sleeping too little or too much; losing weight or gaining weight; and suffering from psychomotor agitation or retardation—opposing symptoms that suggest distinct mechanisms.
Second, like most psychiatric disorders, depression commonly co-occurs with other conditions. About 75% of MDD patients have at least one comorbid neuropsychiatric illness, most commonly generalized anxiety disorder, post-traumatic stress disorder, various phobias, and substance use disorders, but also including obsessive compulsive disorder, attention-deficit hyperactivity disorder and personality disorders [6, 7, 16]. These high co-morbidity rates suggest that transdiagnostic mechanisms may contribute to the biology underlying heterogeneous symptom presentations not only in depression, but also in other closely related disorders. Importantly, depression and other psychiatric diagnoses are also unstable over time: in one recent study [42] involving 1037 participants tracked from birth to age 45, 85% of participants diagnosed with at least one psychiatric disorder also had multiple comorbidities, and there was substantial flux not only between closely related psychiatric conditions (e.g. MDD and generalized anxiety disorder), but also between seemingly disparate families of disorders (e.g. internalizing and externalizing disorders). Remarkably, among the 1037 participants, this report identified 692 unique psychiatric disorder lifetime trajectories, and 605 (87.4%) were unique to just one person. These findings underscore the need for human neuroimaging studies to recruit samples that include a range of related diagnoses and to follow subjects longitudinally. However, as discussed in more detail below, it is also critical to think carefully about subject recruitment strategies and to quantify current mood state, as the mechanisms that give rise to specific symptoms and behaviors and enable their persistence may fluctuate with mood state.
Third, patients differ widely in variables related to the longitudinal course of their illness, including the age of onset (adolescence vs. young adulthood vs. late-life), the frequency of episodes, and the temporal dynamics of episodic changes in depression and euthymia (dysthymia, ultradian cycling, episode duration, durability of remission, “kindling” and cycle acceleration, etc.)—all of which could have a neurobiological basis [8, 43,44,45,46,47]. For example, the age of onset is associated with depression severity and recurrence rate and may be linked to different biological processes [46, 48,49,50,51,52,53,54]. Fourth, sex is one of the most important risk factors for depression and may contribute to diagnostic heterogeneity [5, 6], but the mechanisms are not yet well defined. Fifth, the mechanisms underlying “spontaneous” depressive episodes may also differ from those that can be linked to exposure to a specific antecedent psychosocial stressor such as a history of adversity, trauma, or abuse. The brain’s response to psychosocial stress reflects the allostatic load (lifetime accumulation of the physiological costs of biological responses to stressors) and the long-term psychobiological effects of stressors [55,56,57]. It follows that the same stressor may elicit heterogeneous neurobiological effects in different individuals as a function of their lifetime history, including exposure to known risk factors such as early childhood neglect or abuse, financial insecurity, discrimination, income inequality, and other socioeconomic disparities [58,59,60,61,62]. Table 1 outlines the relative risks associated with some of the most important risk factors.
Subtyping on clinical symptom profiles
Pioneering early efforts to define depression subtypes and parse diagnostic heterogeneity tended to subgroup patients with distinct co-occurring symptom patterns and then test for neurobiological correlates of these subgroups, which could potentially be used as biomarkers for aiding diagnosis or predicting treatment outcomes [63, 64]. The most clinically significant example is of course the distinction between unipolar and bipolar depressive episodes, which are often clinically indistinguishable but are associated with distinct risk factors, clinical trajectories, and treatment response profiles [65]. Some of the most influential examples of unipolar subtypes include melancholic depression, atypical depression, seasonal depression, and psychotic depression. Melancholic depression is characterized by anhedonia, psychomotor disturbances, and persistent nonreactivity of mood, and has been associated with more severe depressive episodes and a history of childhood abuse [66, 67]. Atypical depression is defined by heightened mood reactivity and rejection sensitivity and the presence of other “atypical” symptoms, including hyperphagia, hypersomnia, and “leaden paralysis” [68]. Seasonal depression, a highly influential concept that has seeped into the public lexicon, is characterized by depressive episodes coinciding with different seasonal transitions, most commonly in winter. It is associated with specific disturbances in sleep, circadian rhythms, and melatonin cycling and may be especially responsive to phototherapy and melatonin-derivatives [69,70,71]. Psychotic depression is a severe form of unipolar depression thought to affect ~5% of patients, characterized by hallucinations, delusions, guilty rumination, and feelings of worthlessness [72, 73] and associated with alterations in circadian glucocorticoid rhythms and a potentially increased responsiveness to glucocorticoid-targeting augmentation strategies [74, 75]. A recent meta-analysis supports the distinction of psychotic depression from nonpsychotic depression, showing that the rate of psychotic episodes was consistently and stably elevated in the former groups across twelve studies between 1981 and 2015 [76].
These pioneering efforts to define subtypes based on clinical symptoms yielded multiple important insights into the neurobiological basis of heterogeneity in depression, and they have also raised several unexpected and important questions. First, some studies suggest that the specific clinical symptoms that a patient experiences in one episode tend to vary over time, and consequently, patients may transition between multiple subtypes over the course of a lifetime and possibly within a single episode [34, 77,78,79,80,81,82]. Some studies report that the melancholic, agitated, and atypical depression subtypes are less stable over time than psychotic and severity-based subtypes [76,77,78,79]. Second, some promising leads notwithstanding [69, 75], it has been challenging to identify neuroimaging, neuroendocrine, or other blood-based biomarkers that can be used for diagnosing these subtypes on an individual level.
Third, despite some promising findings for psychotic and seasonal depression [70, 74], it has been difficult overall to use these subtypes to reliably predict who will respond best to which antidepressants. For example, the iSPOT-D Trial found that there were no differences in treatment response between melancholic, atypical, anxious, and mixed subtypes and found significant overlap between the criteria used to classify individuals into different subtypes [83]. Thus, while these symptom-based subtypes have been the focus of important research and some have been described in the current and previous versions of the DSM, they are not widely used in general clinical practice for informing treatment decisions. This limitation applies equally to the initial results of brain-derived subtype discovery efforts described below: widespread clinical utilization will require robust evidence and prospective replication of the ability to reliably inform clinical decision making.
Of note, recent data-driven approaches to symptom-based clustering and treatment prediction have achieved some promising early results. In one such study involving Quick Inventory of Depressive Symptomatology and Hamilton Depression Rating Scale data from >4700 patients [84], the authors identified three symptom clusters associated with insomnia, core emotional symptoms, and atypical depressive symptoms (n.b. This study focused on symptom clusters, not clusters of patients based on their symptoms). They replicated this clustering solution in a second sample and found that antidepressants were most effective for core emotional symptoms. Interestingly, high-dose duloxetine was superior to escitalopram in treating core emotional symptoms with an effect size of 2.3 HAM-D points. In another large-scale study [85], the authors identified seven treatment response clusters comprising patients with differing trajectories of antidepressant response over time (e.g. fast and complete response vs. slow and incomplete response). These subgroups were predictable based on baseline clinical symptoms, personality traits, mood episode duration, and life events. Data mining of medical records, which leverages breakthroughs in natural language processing and extremely large sample sizes, is another promising symptom-based approach to discovering novel subtypes of depression [86], predicting disease severity [87], and potentially informing treatment selection.
Toward brain-based depression subtypes
Motivated in part by the challenges described above, recent studies have sought to parse heterogeneity in depression with a “brain-first” approach, developing novel dimensional rating and subtyping models based on neurobiological measures and then validating them by testing their ability to predict clinical symptoms, behaviors, and treatment outcomes. One way to achieve this aim, inspired by the NIMH’s Research Domain Criteria Initiative (RDoC), is to search for multivariate combinations of brain measures that explain individual differences in clinical symptoms across a continuous spectrum—a dimensional approach to parsing heterogeneity. A second option is to search for data-driven subgroups of patients that differ with respect to depression-related brain measures—a categorical approach. A third option is a hybrid approach that integrates dimensional modeling of individual differences with data-driven clustering on these dimensions. Here, we provide a brief review of dimensional, categorical, and hybrid approaches to parsing diagnostic heterogeneity (Fig. 1), focusing on functional neuroimaging data. For additional details on these three approaches, see a recent review from our group [34]. For comprehensive reviews of other methods, see [35, 36].

Schematic illustrating two data-driven approaches to parsing heterogeneity in depression described in the text using a “top-down” symptom-based approach (right, blue) or a “bottom-up” brain-based mechanistic approach (left, green). The goals of both approaches are to advance our understanding of neurobiological mechanisms underlying depression-related symptoms and behaviors and to develop new tools for informing diagnosis and treatment decisions.
Dimensional approaches
The NIMH RDoC Initiative was designed to overcome some of the obstacles inherent in studying psychiatric disorders that exhibit high comorbidity rates and have a weak correspondence to their biological substrates by integrating multiple units of analysis including self-reported symptoms, objective behaviors, neurophysiology, circuit function, cellular and molecular neurobiology, and genetics [10, 11]. Instead of searching for neurobiological features that differentiate a heterogeneous group of MDD patients from healthy controls, investigators seek to develop models that predict individual differences in a clinically relevant domain (e.g. “anhedonia”) across multiple diagnostic groups. In accord with this perspective, dimensional approaches to parsing heterogeneity in depression develop models for predicting individual differences in specific clinical symptom domains across a continuous spectrum, based on neuroimaging measures of brain structure and function. In this vein, Xia et al. identified brain-behavior dimensions of psychopathology based on fMRI measures of functional connectivity that predicted specific combinations of psychiatric symptoms and behaviors [88]. The study included a broad sample of 663 participants aged 8–22 years old from the Philadelphia Neurodevelopmental Cohort (PNC). They performed sparse canonical correlation analysis (sCCA) on functional connectivity measures and clinical items from a structured clinical interview (see overview schematic in Fig. 2a). CCA is a multivariate dimensionality reduction technique that identifies linear combinations of functional connectivity features that maximally correlate with linear combinations of clinical features. In sCCA, elastic net regularization is used to prevent overfitting and ensure a more interpretable model based on a sparse set of brain features. Their sCCA analysis identified four brain-behavior dimensions, predicting individual differences in 37 out of the 111 clinical symptoms studied (Fig. 2b–i). Although the analysis was purely data driven (i.e. unbiased by hypothetical priors), the four dimensions mapped onto clinically intuitive combinations of mood, psychosis, fear, and externalizing symptoms. Interestingly, each brain-behavior dimension described coherent clinical features and was correlated with symptoms present in several other clinical diagnostic categories. The results support the hypothesis that shared patterns of atypical functional circuitry underlie multiple neuropsychiatric disorders.

a Schematic of the analytical pipeline depicting the calculation of functional connectivity matrices from the Pearson correlations between the average BOLD fMRI signals for each of 264 spherical regions of interest (ROI) and every other ROI. Sparse canonical correlation (sCCA) was used to define linear combinations of clinical symptoms across a range of psychiatric diagnoses (lower panel) that were maximally correlated with linear combinations of functional connectivity. Psychopathology domains: psychotic and subthreshold symptoms (PSY), depression (DEP), mania (MAN), suicidality (SUI), attention-deficit hyperactivity disorder (ADD), oppositional defiant disorder (ODD), conduct disorder (CON), obsessive-compulsive disorder (OCD), separation anxiety (SEP), generalized anxiety disorder (GAD), specific phobias (PHB), mental health treatment (TRT), panic disorder (PAN), post-traumatic stress disorder (PTSD). ROI communities: somatosensory/motor network (SMT), cingulo-opercular network (COP), auditory network (AUD), default mode network (DMN), visual network (VIS), fronto-parietal network (FPT), salience network (SAL), subcortical network (SBC), ventral attention network (VAT), dorsal attention network (DAT), cerebellar. b–e Four brain-behavior dimensions of psychopathology were identified that captured individual differences in clinical symptoms. Scatter plots depict brain connectivity and clinical dimension scores, which are linear combinations of functional connectivity features and psychiatric symptoms. b Dimension 1 described mood-related symptoms, e.g. feeling sad. c Dimension 2 described psychosis-related symptoms, e.g. auditory hallucinations. d Dimension 3 described fear-related symptoms, e.g. fear of traveling. e Dimension 4 described externalizing behavior and related symptoms, e.g. trouble following instructions. f–i Network module connectivity patterns associated with each brain connectivity-behavior dimension. Heatmaps depict the magnitude and direction of correlation change to each brain-behavior dimension score (positively- or negatively correlated) in the following functional networks: default mode network (DMN), visual network (VIS), fronto-parietal network (FPT), salience network (SAL), ventral attention network (VAT), dorsal attention network (DAT). Abbreviations: a.u., arbitrary units. Figure adapted with permission from ref. [88].
In a second example of this approach, Mihalik et al. [89] investigated how psychopathology dimensions relate to changes in the patterns of functional connectivity during brain development in a mostly healthy sample of adolescents. The study included 306 participants aged 14–24, but most had relatively few clinical symptoms and only 25 were clinically depressed. They used CCA to identify two brain-behavior modes of covariation. The first mode described externalizing/internalizing behaviors and was strongly associated with sex. The second mode described emotional well being vs. distress and was independent of sex. Interestly, both brain-behavior modes were associated with age, but in opposite directions, and each mode was correlated with distinct patterns of functional connectivity that related to adolescent brain development. The first mode was positively correlated with age and attentional and frontoparietal networks, but negatively correlated with limbic and intra-subcortical connections. The second mode was negatively correlated with age and connectivity in frontoparietal control and attention networks, but positively correlated with limbic and subcortical connectivity. One limitation with respect to understanding depression heterogeneity is that only 25 of the 306 participants were significantly depressed, which could bias the results. Still, the results provide insight into the neurobiological mechanisms underlying developing psychopathology in adolescents and young adults, and a qualitative correspondence with some of the findings from the study by Xia et al. [88].
Importantly, these are just two examples of a dimensional approach to understanding diagnostic heterogeneity by modeling neurobiological predictors of individual differences in behavior. This approach is anchored in a much larger body of literature that has sought to identify neurobiological correlates at the group level of specific behaviors and symptoms that transcend conventional diagnostic boundaries. Multiple transdiagnostic meta-analyses are now available. For example, Zhang et al. tested for transdiagnostic correlates of three anhedonia subdomains [90]. They found that in patients with MDD or schizophrenia, consummatory and anticipatory anhedonia were linked to decreased activity in the basal ganglia and abnormalities in fronto-striatal networks including the dorsal anterior cingulate and dorsolateral prefrontal cortex, respectively. Other meta-analyses have identified transdiagnostic substrates of dysfunction in cognitive control [91], cognitive reappraisal [92], emotion regulation [93], and meta-cognition [94].
Categorical approaches
While the studies above parsed diagnostic heterogeneity across a continuous spectrum of psychopathology, an alternative strategy is to cluster subjects into categorical subgroups, which may have some advantages for clinicians, who tend to think in terms of categorical heuristics. Price et al. [95] used task-based functional MRI scans from 68 depressed patients and 24 never-depressed control subjects and clustered patients using a community detection algorithm based on brain activity during a positive mood induction task. Their analysis focused on measures derived from the default mode network, cognitive control network, and ventral affective network. They identified two subgroups distinguished by functional properties of the ventral affective network and other ventral cortical areas and by diagnosis. The subgroup with hyperactivity in ventral regions included 81% of depressed patients, but only 50% of never-depressed controls. In a follow-up study, Price and colleagues used resting state functional MRI scans from the same 68 depressed subjects and an additional 12 depressed patients [96]. Similar to the first study, they performed network analysis of functional connectivity patterns and used community detection analysis to cluster patients into subgroups. They identified two depression subgroups that differed with respect to sex, as well as patterns of atypical functional connectivity in the default mode network. Interestingly, abnormal functional connectivity in the default mode network that was detected in subgroup B was not evident when all depressed subjects were compared to healthy controls, highlighting the utility of clustering patients into subgroups with more homogeneous neurobiological profiles. A separate study by Feder and colleagues [97] clustered on functional connectivity of depressed individuals and healthy controls and also identified two clusters, though there were no strong differences in clinical symptoms between subgroups.
Hybrid approaches
Importantly, dimensional and categorical approaches are not mutually exclusive. In a hybrid approach, Drysdale and colleagues used a large-scale multi-site resting state fMRI dataset (N = 458 depressed patients and N = 730 healthy controls) to identify two brain-behavior dimensions that predicted individual differences in specific depressive symptoms and then clustered patients with treatment-resistant depression along these two dimensions [98]. The first dimension predicted anhedonia symptoms based on frontostriatal and orbitofrontal connectivity, while the second dimension predicted anxiety and insomnia based on amygdala, ventral hippocampus, ventral striatum, and lateral prefrontal cortex connectivity. Hierarchical clustering on these two dimensions identified four patient subgroups that differed in patterns of atypical connectivity (Fig. 3a) and in their clinical symptom profiles (Fig. 3b, c). Subgroup differences in anhedonia, anxiety, and other clinical symptoms were confirmed in an independent replication sample. Subsequent work [99] in a more clinically heterogeneous sample (N = 187 patients with a history of mixed mood or anxiety disorders, 64.2% in remission) showed that this approach tends to overfit, a problem that can be solved by using L2 regularization to improve the robustness and stability of canonical correlations in held-out data and may be sensitive to subjects’ mood state at the time of scanning [100] (see Box 1 for additional details). Another important form of validation came from a subsequent analysis testing for subgroup differences in antidepressant response. This analysis showed that these four clusters predicted treatment response to repetitive transcranial magnetic stimulation (TMS) targeting the dorsomedial prefrontal cortex, and that TMS response was correlated with functional connectivity patterns involving the left amygdala, left dorsolateral prefrontal cortex, and dorsomedial prefrontal cortex (Fig. 3d–f). These results define just one solution to parsing diagnostic heterogeneity in depression that will surely be improved upon in future studies employing larger samples and richer clinical phenotyping. As discussed below, it will also be critical to evaluate whether these treatment predictive algorithms are sufficiently accurate to improve treatment outcomes in a prospective randomized controlled trial.

a Four rsfMRI-based subtypes of depression, identified through hierarchical clustering on latent brain-behavior dimensions (canonical connectivity–symptom components), exhibit distinct patterns of atypical functional connectivity. Heatmaps depict the z score from a Wilcoxon rank sum test for differences between the functional connectivity of depressed subjects in each subtype and of healthy controls. b The four subtypes were associated with distinct clinical symptom profiles as indexed by item-level responses to the Hamilton Depression Rating Scale. c Boxplots depicting subtype differences in depression severity. d–e Patients in Subtypes 1 and 3 were more likely to respond to rTMS targeting the dorsomedial prefrontal cortex, compared to patients in Subtypes 2 and 4. f Distinct functional connectivity patterns prior to treatment in rTMS-responders vs. nonresponders. Heatmap depicts functional connectivity features that were significantly different in responders, including connectivity between the dorsomedial prefrontal target and the left dorsolateral prefrontal cortex and left amygdala. Figure adapted with permission from Ref. [98]. ACC anterior cingulate cortex; amyg amygdala; a.u. arbitrary units; COTC cingulo-opercular task-control network; DAN dorsal attention network; DLPFC dorsolateral prefrontal cortex; DMN default-mode network; DMPFC dorsomedial prefrontal cortex; FPTC frontoparietal task-control network; GP globus pallidus; HAMD Hamilton Depression Rating Scale; HC hippocampus; lat PFC lateral prefrontal cortex; LIMB limbic; M1 primary motor cortex; NAcc nucleus accumbens; OFC orbitofrontal cortex; PCC posterior cingulate cortex; PPC posterior parietal cortex; precun precuneus; rTMS repetitive transcranial magnetic stimulation; rsfMRI resting state functional MRI; SM primary sensorimotor cortex (M1 or S1); SS1 primary somatosensory cortex; SN salience network; subC subcortical; thal, thalamus; VAN ventral attention network; vis visual cortex; VLPFC ventrolateral prefrontal cortex; vStr ventral striatum; n.s. not significant.
In a second study utilizing a hybrid approach, Tokuda et al. [101] used unsupervised Bayesian co-clustering of a high-dimensional and multimodal dataset of 134 subjects (N = 67 depressed patients). The study included measurements of functional connectivity, gene expression data, clinical symptoms, and a number of other biological measures. They identified three subtypes that differed in functional connectivity patterns, especially in the angular gyrus and other default mode network brain regions, as well as trauma history and treatment response to SSRIs. This study is remarkable not only for these interesting subgroups differences but also for the fact that it is among the first to attempt to integrate gene expression data—a promising future direction reviewed in more detail below.
Technical challenges and potential solutions
While these results highlight the potential of neuroimaging data for parsing diagnostic heterogeneity and identifying novel subtypes of depression, it is also important to recognize key technical challenges inherent in these approaches. First, it is challenging to directly compare the studies reviewed above and identify clear points of convergence due to differences in methodology, patient samples, and clustering criteria. This will be a critical goal for future studies as this area of research matures. To aid in the synthesis of convergent findings across studies, Table 2 provides a summary of the results from the studies discussed above and identifies two preliminary areas of convergence in the clinical correlates of data-driven subtypes. Second, functional neuroimaging data are susceptible to various artifacts, particularly those related to head motion, respiratory patterns, and local changes in blood gases [102,103,104,105,106,107]. Third, efforts to study diagnostic heterogeneity and discover novel subtypes invariably benefit from large samples, which often require integrating data from multiple scanners, which can introduce additional artifacts that can be difficult to control [108,109,110]. Fourth, while there are obvious benefits to analyzing a heterogeneous sample and adopting an inclusive recruitment strategy, it is still critical to think carefully about how clinical sample characteristics influence the results of a specific study. Fifth, complex multivariate analyses of high-dimensional datasets are prone to pitfalls including overfitting and false positives, and benefit from stringent controls and other strategies for minimizing these problems. In Box 1, we discuss potential solutions to these four technical challenges. For a more detailed discussion, see ref. [34].
Toward molecular mechanisms: integrating neuroimaging and genomic data
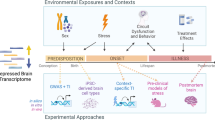
As noted above, genetic factors are another likely contributor to diagnostic heterogeneity in depression, and there is increasing interest in integrating genetic and neuroimaging data to understand how genetic risk variants influence brain structure and function (Fig. 4). These efforts are often motivated by the premise that depression-related genetic variants may manifest in endophenotypes—neurobiological changes that can be measured with neuroimaging and other tools [111]. These intermediate neuroimaging endophenotypes may be more closely linked to genetic risk variants than the highly heterogeneous depression diagnosis and could therefore provide a promising avenue for understanding how genetic factors modulate pathophysiological processes. Family, adoption, and twin studies, which can be used to separate the influence of genetic versus environmental factors, have shown that depression is moderately heritable: ~35–40% of an individual’s risk for unipolar depression is attributable to genetic variation [112, 113], and bipolar depression is ~60–85% heritable [114]. Like most psychiatric disorders, depression exhibits a highly polygenic inheritance pattern, such that an individual’s risk is thought to be determined by complex interactions between a large number of genetic variants and factors like early life adversity, psychosocial stress, and other environmental variables [111, 115,116,117]. Further complicating these efforts, the lifetime prevalence of depression is an order of magnitude higher than psychiatric conditions such as schizophrenia and autism, such that much larger sample sizes are required for identifying robust and replicable effects [30]. Recently, extremely large-scale genome-wide association (GWA) studies have identified multiple genetic variants conferring increased risk with genome-wide significance [31, 33, 118, 119]. A recent meta-analysis involving 135,458 cases and 344,901 controls identified 44 independent loci corresponding to 153 genes associated with excitatory neurotransmission, synapse function, and dendritic spines [31]. Of note, six of these loci were shared with the 108 risk variants identified in schizophrenia GWA studies, supporting transdiagnostic overlap in the genetic etiology of these diseases [31, 120]. An even larger meta-analysis involving 414,055 cases and 892,299 controls, identified 102 risk variants, 87 of which were significant after correcting for multiple comparisons, and the findings supported the transdiagnostic overlap with schizophrenia found previously [33]. While the heritability of depression has been found to be ~37% in twin studies, it is estimated that currently GWAS only capture ~9% of genetic variation [29]. Furthermore, since MDD is more prevalent and has a considerably lower heritability rate than other psychiatric illnesses such as schizophrenia (81% heritability) [121], some studies estimate that much larger samples sizes, on the order of 100,000 to 1 million MDD cases and matched controls, may be needed to have sufficient power to detect the degree of risk that individual genetic variants confer to overall depression risk [29, 30, 122]. Mirroring developments in depression genetics, efforts to understand how genetic risk variants influence brain structure and function focused initially on candidate genes and more recently on confirmed GWA risk variants and transcriptomic data. Below, we review findings from each approach as well as emerging consensus views on statistical challenges and best practices for implementing these analyses.

Schematic of how combining neuroimaging with genetic data can be used to parse heterogeneity in depression and uncover subgroups within the depressed population. Polygenic variation may manifest in intermediate behavior-related brain circuits that can give rise to distinct depression subgroups. The polygenic effects of risk variants on circuit dysfunction, cognition, behavior, and clinical symptoms may interact with each other. Genes indicated are examples of candidate depression risk variants from Table 3 with the known variation and locus in the genome indicated and depicted on the chromosome strand in yellow. 1. Indicates examples of brain circuits known to be dysfunctional in depression. 2. Lists examples of cognitive processes and behaviors that are altered in depression, possibly as a direct or indirect consequence of genetic risk variants. 3. Lists clinical symptoms of depression that may result directly or indirectly from dysfunction in depression brain circuits. The double-sided arrows indicate the bi-directional relationships between 1, 2, and 3 that may modulate the expression of intermediate phenotypes.
Candidate gene studies
Prior to the discovery of confirmed risk variants in large-scale GWA studies, early attempts to integrate genetic and neuroimaging data focused on candidate genes that were hypothesized to confer depression susceptibility. Excellent reviews are available elsewhere [123,124,125,126]. Here, we focus on highlighting findings from the two most frequently studied examples of this approach: common polymorphisms associated with the serotonin transporter (5-HTTLPR) and brain derived neurotrophic factor (BDNF) genes.
The 5-HTTLPR “long” and “short” alleles are degenerate repeat polymorphisms in a regulatory region linked to the serotonin transporter gene promoter. The short allele is associated with reduced expression of the serotonin transporter and reduced serotonin uptake in lymphoblasts [127]. It has also been associated with neuroticism [128] and increased susceptibility to depression in response to stressful life events in some epidemiological cohorts and studies [129, 130] but not in others [131]. In multiple functional neuroimaging studies, this polymorphism has been found to modulate emotion regulation, social cognition, and amygdala reactivity to emotionally salient stimuli, and functional interactions between the anterior cingulate cortex and amygdala [123, 132,133,134]. Structural neuroimaging studies have reported genotype differences in hippocampal volume and white matter microstructure in frontolimbic areas [135, 136].
The BDNF Val66Met polymorphism is a single nucleotide substitution in codon 66 of the BDNF prodomain that disrupts activity-dependent BDNF release [137]. BDNF has been implicated in stress resilience and in ketamine’s antidepressant mechanism of action [138,139,140], and the Val66Met polymorphism is associated with increased anxiety-related behavior that is resistant to fluoxetine treatment in transgenic knock-in mice [141]. In human neuroimaging studies, Met carriers exhibit modest deficits in episodic memory performance and hippocampal activity [137], and reduced hippocampal volume in some [142] but not all studies [143].
Investigators have also recognized several important limitations of the candidate gene approach in neuroimaging genetics [124]. Methodological differences across studies complicate efforts to identify replicated findings. Two recent meta-analyses of structural neuroimaging studies involving the 5-HTTLPR and BDNF Val66Met polymorphisms found that most changes in hippocampal volume did not reach statistical significance across studies [125, 126]. However, reductions in hippocampal volume in BDNF Met allele carriers in bipolar disorder was a consistently replicated finding [125], and exploratory secondary analyses identified consistent increases in activity and gray matter volume in the prefrontal cortex [126]. Likewise, carriers of the 5-HTTLPR “short” allele exhibited consistent alterations in white matter microstructure in the corpus callosum and superior longitudinal fasciculus. The authors also identified multiple factors that could contribute to inconsistent results across studies, including gender distribution, medication status, and perhaps most importantly, relatively small sizes (typically ~35–45 subjects per genotype). Another important limitation is that recent studies indicate that many candidate gene associations may be false positives and do not appear to contribute significantly to depression risk with genome-wide significance in very large cohorts [126, 130, 144].
Polygenic risk
More recently, neuroimaging studies have begun to leverage findings from large-scale depression GWA studies. As noted above, at least 44 genetic variants associated with 153 genes have been identified [31], but none is highly penetrant. Understanding how these common SNPs interact to modulate depression pathophysiology is thus a major challenge for the field. One approach to this problem is to investigate how polygenic risk modulates brain structure and function by calculating a polygenic risk score (PRS), which measures the risk for a phenotype attributable to the accumulation of multiple genetic variants [145,146,147]. (For an excellent review on polygenic risk scores and detailed guidelines on how to calculate PRS quality control measures and other best practices, see ref. [148].) While multiple studies have used this approach for other diseases [149,150,151,152], its application in depression is relatively new. However, the early results are promising. For example, motivated by the fact that depressed patients frequently present with working memory deficits, Yüksel et. al. investigated whether polygenic risk for depression also modulated working memory performance and brain activity in 137 healthy individuals [153]. Brain activation patterns in the right prefrontal cortex and other working-memory related brain areas were modulated by polygenic risk for depression (Fig. 3a–c). This suggests that depression-related genetic variants could modulate depression risk by acting in part on working memory functions—a potential endophenotype. Alternatively, polygenic risk could modulate depression risk directly, and working memory deficits could emerge as one possible consequence (but not necessarily a 100% determined outcome) of depression pathophysiology. Dissociating these two models of causality would require additional data on whether working memory deficits are more or less heritable than depression; whether they precede the onset of depression and persist in a remitted mood state; and whether they occur in first-degree relatives of depressed probands [154].
A second study tested whether polygenic risk for anhedonia modulates brain structure and function using the UK Biobank dataset [155]. They first performed a novel GWAS using the 375,724 UK Biobank participants and identified 11 novel loci for anhedonia. Using the anhedonia GWAS results, they tested for genetic overlap with GWAS results from other psychiatric disorders [156], and found that polygenic risk for anhedonia was correlated with polygenic risk for major depressive disorder, schizophrenia, and bipolar disorder, but not for OCD or Parkinson’s Disease. Next, they tested whether anhedonia PRS scores predicted individual differences in brain structure and functional activity during an fMRI emotional face processing task. They found that increased polygenic risk for anhedonia was not associated with functional activity, but was associated with reduced volume in the orbitofrontal cortex and other brain areas involved in reward-processing, and with alterations in white matter microstructure as indexed by diffusion tractography (Fig. 5a–c). These promising findings notwithstanding, how genetic risk variants modulate depression-related pathophysiology is largely unknown. Table 3 synthesizes findings from recent efforts to link candidate genes and GWA-confirmed risk variants with specific depression-related behaviors and neurophysiological functions. As above, it will be critical to replicate these results in prospective cohorts.

a–c Polygenic risk scores (PRS) for anhedonia were associated with a. regional volumes of cortical and subcortical regions of interest, b tract-specific fractional anisotropy (measure of axonal integrity and myelination), and c tract-specific mean diffusivity (measures of structural integrity in the intra- and extracellular space, neuropil, and global CSF). d Partial least squares (PLS) analysis linear combinations of genes whose spatial expression patterns co-localized with schizotypy-associated myelination patterns as indexed by an MRI magnetization transfer measure. Genes with larger “PLS1 weight” values were more important predictors of the spatial distribution of SRM myelination. e Positively weighted PLS1 genes were associated with genes known to be down-regulated in schizophrenia (“Gandal Down-Reg” and “Fromer Down-Reg”), neuron cell types, and increased SRM myelination. f Negatively weighted PLS1 genes were associated with genes known to be up-regulated in schizophrenia (“Gandal Up-Reg” and “Fromer Up-Reg”), decreased SRM myelination, and astrocyte, microglia, and neuron cell types. FDR false discovery rate; PLS partial least squares; SRM schizotypy-related magnetization; Up/Down-Reg Up/Down-Regulated. Figure adapted with permission from refs. [155] (a–c) and [160] (d–f).
Gene expression and transcriptomics
A third approach that is becoming increasingly common is to investigate how regional differences in gene expression predict disease-related changes in brain structure and function. Although gene expression data are obtained post-mortem and are therefore not directly compatible with human neuroimaging in vivo, public databases are now making gene expression data available to the scientific community. The Allen Human Brain Atlas (AHBA) is one prominent example, including brain-wide microarray samples collected postmortem from 3702 brain regions in six neurotypical adult brains (N = 1 female, N = 5 male). Four contain samples from only one hemisphere while two brains have samples on both hemispheres and also RNA-seq data. Recent work indicates that regional differences in gene expression in the AHBA predict human functional network architecture [157].
To date, no studies have used AHBA gene expression data in combination with functional neuroimaging measures in depressed subjects, but this is a promising approach that has been applied to functional and structural imaging in other psychiatric disorders [158,159,160]. In one elegant example, Romero and colleagues tested whether regional differences in the expression of schizophrenia genetic risk variants predict regional differences in brain structure and schizotypal traits [160]. Their analysis combined gene expression data from the AHBA with an MRI magnetization transfer measure of intracortical myelination in 2135 healthy individuals (age 14–25 years old), who also completed a schizotypy self-report questionnaire. First, they quantified schizotypy-related magnetization by correlating schizotypal personality scores with age-corrected magnetization transfer measurements from 68 brain regions of interest. To compare the schizotypy-related magnetization to the spatial distribution of gene expression, they used a multivariate technique called partial least squares analysis (PLS). PLS was used to define a linear combination of schizotypy-related magnetization measures that maximally covaries with brainwide measurements of gene expression from the AHBA atlas. They identified two significant brain structure/gene expression components, and an enrichment analysis found that genes positively co-located with schizotypy-related magnetization were enriched for genes that are downregulated in schizophrenia and neurons, confirming a significant overlap with genes known to confer schizophrenia risk in this healthy normative sample (Fig. 5d, e). Other examples of this approach have begun to examine whether regional differences in gene expression explain structural abnormalities in psychosis [159], functional changes associated with early life trauma and conversion disorder [158], adolescent brain development [161], functional network organization [162], and structural changes in autism [163]. Together, these studies demonstrate the potential for neuroimaging gene expression analyses to reveal insights into how genetic risk variants influence brain structure and function in depression.
Technical challenges and potential solutions
Efforts to integrate genetic and neuroimaging data are a promising avenue for future research, but they can be logistically complicated, because ideally, they benefit from access to large datasets. Thus, they can be hindered by limited access to datasets that include both a large number of subjects and functional MRI data, behavioral measures, and genotypes. Functional MRI, genomic, and transcriptomic datasets typically include tens of thousands of variables and a comparatively small number of subjects. Functional MRI scans can be composed of over one million 3D voxels over many time volumes, and GWAS datasets often include millions of SNPs. In contrast, until recently, datasets that include both genotype data and fMRI data have typically included only a few dozen subjects. However, the growing availability of collaborative, large-scale, publicly accessible datasets like the UK Biobank will help to mitigate this problem [164, 165]. Likewise, multiple gene expression databases now exist, including the Brainspan database and the Allen Human Brain Atlas of microarray gene expression data [166, 167]. Another important obstacle is that these analyses can be statistically and technically complicated. In Box 2, we highlight important statistical considerations for preprocessing gene expression data and implementing partial least squares (PLS) analyses integrating neuroimaging and gene expression data.
Conclusions and future research directions
Depression is a highly heterogeneous psychiatric syndrome bearing a weak correspondence to its neurobiological and genetic substrates. Efforts to parse diagnostic heterogeneity in terms of continuous brain-behavior dimensions and to discover neurobiologically driven subtypes of depression hold substantial promise. These studies have the potential to uncover clinically useful dimensions and subtypes and to provide a framework for developing novel approaches to diagnosing depression subtypes, predicting clinical outcomes, and selecting treatments.
Above, we reviewed various approaches to integrating neuroimaging and genetic datasets, with a focus on candidate gene studies, polygenic risk scores, and transcriptomic analyses. There are a number of other approaches to combining gene expression data with functional neuroimaging data, including univariate approaches and sparse canonical correlation analysis. With all these approaches, it is important to recognize that these are discovery-oriented exploratory analyses, and thus descriptive, not predictive [168]. Replication studies in independent datasets are critical [124]. Future studies will benefit from datasets involving larger samples and incorporating information about brain-based depression dimensions and subtypes. With the growing availability of public databases, open-source statistical methods, and increasing computational power, neuroimaging genetics will increasingly be able to reveal insights into the molecular machinery that gives rise to depression pathophysiology. These findings, in turn, could be used for formulating mechanistic hypotheses and testing them in animal models [100] or in human subjects using innovative lesion mapping approaches and non-invasive brain stimulation [169,170,171,172]. In the long term, these methods could potentially translate to personalized pharmacological and circuit-based therapeutics. We conclude by highlighting five especially promising priorities for future research.
Defining sexually dimorphic mechanisms
Women are about twice as likely as men to be diagnosed with depression [6, 16, 173], indicating that sex may be an important contributor to diagnostic heterogeneity and that pathophysiological mechanisms may not be identical in men and women [174]. Some existing studies support this hypothesis. For example, a recent study examined the transcriptional correlates of depression in postmortem brain tissue derived from men or women and tested for sex-specific convergence in a chronic stress model in mice [5]. They found that stress susceptibility in mice and depression in humans were associated with largely non-overlapping transcriptional signatures in males vs. females. In both mice and patients, Dusp6—a gene encoding dual specificity phosphatase 6, a regulator of the ERK signaling pathway—was identified as a hub gene among differentially expressed genes in the ventromedial prefrontal cortex in female MDD patients and stressed female mice. Downregulating Dusp6 in the vmPFC was sufficient to increase stress susceptibility in female mice, but not in males. This study suggests that there are sex-specific pathophysiological mechanisms underlying depression in men and women. Future studies will surely benefit from examining how sexually dimorphic processes contribute to depression heterogeneity.
Longitudinal studies of disease course
Depression is a fundamentally episodic mental illness, but most studies to date have been cross-sectional. There is substantial heterogeneity in the temporal dynamics of depressive episodes across individuals [8], but the neurobiological bases of these differences are not well understood. Longitudinal studies, aided by advances in neuroimaging [175,176,177], will be critical for investigating the mechanisms mediating mood state transitions, rapid cycling, and highly recurrent depressions vs. single depressive episodes.
Reverse translation: testing mechanistic hypotheses in animal models
Neuroimaging models of depression heterogeneity and PLS approaches can be used to formulate hypotheses about how distinct neurobiological mechanisms contribute to specific symptoms and behaviors, at both the molecular and neural circuit levels [159, 160, 178]. Optogenetics, chemogenetics, and viral manipulations of specific signaling pathways, in turn, could be used to test subtype-specific mechanisms in animal models [100, 179,180,181].
Validating novel subtyping solutions
With renewed interest in rethinking our approach to diagnosing depression, it will be essential to validate candidate subtyping solutions and identify points of convergence. Initial analyses will be optimized for inference: i.e. identifying genetic, transcriptomic, and neurophysiological features that interact to explain individual differences in depression-related symptoms and behaviors. As the field matures, it will be important to refine and replicate these models and optimize them for prediction: i.e. optimizing model parameters to allow for accurate predictions of symptoms, behaviors, or clinical outcomes in held-out data and in prospective replication samples. Well validated models would be reproducible in new datasets contingent on predetermined criteria related to data quality, subject samples, and other issues (see Box 1), and they should either advance our understanding of depression neurobiology or have some clinical utility. Clinically useful models might facilitate treatment decisions (e.g. selecting between two antidepressants or predicting treatment response); enable the identification of a biomarker that could be used in diagnosis; or inform prognostic predictions about the course of illness in a given individual.
Personalized medicine and treatment prediction
Investigating individual differences in pathophysiology will be critical for developing personalized treatments. Existing treatments are effective, but not for everyone [14]. To the extent that distinct pathophysiological processes are at play in subsets of depressed patients, heterogeneity may contribute to treatment resistance and treatment failures. In the long term, parsing diagnostic heterogeneity in depression and defining subtype-specific mechanisms could give rise to individualized treatment strategies targeting personalized molecular signaling pathways, brain circuits, and behaviors. Multiple groups are already investigating how neuroimaging tools could be used to facilitate personalized treatment selection [83, 182,183,184,185] and for identifying the optimal target area for brain stimulation interventions [98, 171, 186]. As always, efforts to identify treatment predictive biomarkers must be followed up with rigorous prospective tests not only of their accuracy but also of their utility for improving clinical outcomes, preferably in the setting of a randomized control trial.
Funding and disclosure
This work was supported by grants from the National Institute of Mental Health (MH118388,MH114976, MH123154, MH118451, MH109685, MH109685-04S1), the National Institute onDrug Abuse (DA047851), the Hope for Depression Research Foundation, the PritzkerNeuropsychiatric Disorders Research Consortium, the Klingenstein–Simons Foundation Fund,the One Mind Institute, the Rita Allen Foundation, the Dana Foundation, the Foundation for OCD Research, and the Brain and Behavior Research Foundation (NARSAD). CL is listed asan inventor for Cornell University patent applications on neuroimaging biomarkers for depression that are pending or in preparation. The authors declare no competing interests.
References
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). American Psychiatric Publishing 2013.
Freeman HL. Historical and nosological aspects of dysthymia. Acta Psychiatr Scand Suppl. 1994;383:7–11.
Evans KM. ‘Interrupted by fits of weeping’: Cicero’s major depressive disorder and the death of Tullia. Hist Psychiatry 2007;18:81–102.
Burton R. The Anatomy of Melancholy. Thomas Tegg; 1845.
Labonté B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med 2017;23:1102–11.
Blazer DG, Kessler RC, McGonagle KA, Swartz MS. The prevalence and distribution of major depression in a national community sample: the National Comorbidity Survey. Am J Psychiatry. 1994;151:979–86.
Kessler RC, Nelson CB, McGonagle KA, Liu J, Swartz M, Blazer DG. Comorbidity of DSM–III–R major depressive disorder in the general population: results from the US national comorbidity survey. Br J Psychiatry. 1996;168:17–30.
Post RM, Denicoff KD, Leverich GS, Altshuler LL, Frye MA, Suppes TM, et al. Morbidity in 258 bipolar outpatients followed for 1 year with daily prospective ratings on the NIMH life chart method. J Clin Psychiatry. 2003;64:680–90. quiz 738–739.
Casey BJ, Craddock N, Cuthbert BN, Hyman SE, Lee FS, Ressler KJDSM-5. and RDoC: progress in psychiatry research? Nat Rev Neurosci. 2013;14:810–4.
Insel TR, Cuthbert BN. Brain disorders? Precisely. Science 2015;348:499–500.
Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research Domain Criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–51.
Wang PS, Berglund PA, Olfson M, Kessler RC. Delays in initial treatment contact after first onset of a mental disorder. Health Serv Res. 2004;39:393–415.
Lee S, Fung SC, Tsang A, Zhang MY, Huang YQ, He YL, et al. Delay in initial treatment contact after first onset of mental disorders in metropolitan China. Acta Psychiatr Scand. 2007;116:10–16.
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17.
Thase M, Connolly KR. Unipolar depression in adults: Choosing treatment for resistant depression. In: Post TW, editor. Waltham: UpToDate; 2019.
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003;289:3095–105.
Substance Abuse and Mental Health Services Administration. Key substance use and mental health indicators in the United States: Results from the 2018 National Survey on Drug Use and Health. Rockville: Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration; 2019.
Chisholm D, Sweeny K, Sheehan P, Rasmussen B, Smit F, Cuijpers P, et al. Scaling-up treatment of depression and anxiety: a global return on investment analysis. Lancet Psychiatry 2016;3:415–24.
James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1789–858.
U.S. Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. National Survey on Drug Use and Health (NSDUH-2018); 2018.
Freedland KE, Carney RM, Rich MW. Effect of depression on prognosis in heart failure. Heart Fail. Clin. 2011;7:11–21.
Pan A, Sun Q, Okereke OI, Rexrode KM, Hu FB. Depression and risk of stroke morbidity and mortality: a meta-analysis and systematic review. JAMA 2011;306:1241–9.
Frasure-Smith N, Lespérance F, Talajic M. The impact of negative emotions on prognosis following myocardial infarction: is it more than depression? Health Psychol 1995;14:388–98.
MacMahon KMA, Lip GYH. Psychological factors in heart failure: a review of the literature. Arch Intern Med. 2002;162:509–16.
Centers for Disease Control and Prevention. Fatal Injury and Nonfatal Injury. Web-based Injury Statistics Query and Reporting System (WISQARS) [Online]. National Center for Injury Prevention and Control, Centers for Disease Control and Prevention (producer); 2018.
Takahashi Y. Depression and suicide. Jpn Med Assoc J 2001;44:359–63.
Cavanagh JTO, Carson AJ, Sharpe M, Lawrie SM. Psychological autopsy studies of suicide: a systematic review. Psychol Med 2003;33:395–405.
American Association of Suicidology. Facts about suicide and depression based on 2010 data. 2012.
McIntosh AM, Sullivan PF, Lewis CM. Uncovering the Genetic Architecture of Major Depression. Neuron 2019;102:91–103.
Sullivan PF, Daly MJ, Ripke S, Lewis CM, Lin D-Y, Wray NR, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013;18:497–511.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 2018;50:668–81.
Flint J, Kendler KS. The genetics of major depression. Neuron 2014;81:484–503.
Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci 2019;22:343–52.
Lynch CJ, Gunning FM, Liston C. Causes and consequences of diagnostic heterogeneity in depression: paths to discovering novel biological depression subtypes. Biol Psychiatry. 2020. https://doi.org/10.1016/j.biopsych.2020.01.012.
Feczko E, Miranda-Dominguez O, Marr M, Graham AM, Nigg JT, Fair DA. The heterogeneity problem: approaches to identify psychiatric subtypes. Trends Cogn Sci. 2019;23:584–601.
Beijers L, Wardenaar KJ, van Loo HM, Schoevers RA. Data-driven biological subtypes of depression: systematic review of biological approaches to depression subtyping. Mol Psychiatry 2019;24:888–900.
Telles-Correia D, Marques JG. Melancholia before the twentieth century: fear and sorrow or partial insanity? Front Psychol 2015;6:81.
Healy D. From mania to bipolar disorder. In: Yatham LN, Maj M, editors. Bipolar Disorder: Clinical and Neurobiological Foundations. Chichester: John Wiley & Sons, Ltd; 2010. p. 1–7
Kraepelin E Manic-Depressive Insanity and Paranoia (1921). Translated by Barclay RM. In: Robertson GM, editor. Salem: Reprinted in facsimile by the Ayer Company; 1987.
Pichot P. [DSM-III: the 3d edition of the Diagnostic and Statistical Manual of Mental Disorders from the American Psychiatric Association]. Rev Neurol. 1986;142:489–99.
Blashfield RK, Keeley JW, Flanagan EH, Miles SR. The cycle of classification: DSM-I through DSM-5. Annu Rev Clin Psychol. 2014;10:25–51.
Caspi A, Houts RM, Ambler A, Danese A, Elliott ML, Hariri A, et al. Longitudinal Assessment of Mental Health Disorders and Comorbidities Across 4 Decades Among Participants in the Dunedin Birth Cohort Study. JAMA Netw Open. 2020;3:e203221.
Post RM. Kindling and sensitization as models for affective episode recurrence, cyclicity, and tolerance phenomena. Neurosci Biobehav Rev. 2007;31:858–73.
Lebowitz BD, Pearson JL, Schneider LS, Reynolds CF 3rd, Alexopoulos GS, Bruce ML, et al. Diagnosis and treatment of depression in late life. Consensus statement update. JAMA 1997;278:1186–90.
Alexopoulos GS, Meyers BS, Young RC, Campbell S, Silbersweig D, Charlson M. ‘Vascular depression’ hypothesis. Arch Gen Psychiatry 1997;54:915–22.
Toenders YJ, van Velzen LS, Heideman IZ, Harrison BJ, Davey CG, Schmaal L. Neuroimaging predictors of onset and course of depression in childhood and adolescence: a systematic review of longitudinal studies. Dev Cogn Neurosci. 2019;39:100700.
Dohm K, Redlich R, Zwitserlood P, Dannlowski U. Trajectories of major depression disorders: a systematic review of longitudinal neuroimaging findings. Aust N. Z J Psychiatry. 2017;51:441–54.
Zisook S, Rush AJ, Lesser I, Wisniewski SR, Trivedi M, Husain MM, et al. Preadult onset vs. adult onset of major depressive disorder: a replication study. Acta Psychiatr Scand. 2007;115:196–205.
Zisook S, Rush AJ, Albala A, Alpert J, Balasubramani GK, Fava M, et al. Factors that differentiate early vs. later onset of major depression disorder. Psychiatry Res 2004;129:127–40.
Sung SC, Wisniewski SR, Balasubramani GK, Zisook S, Kurian B, Warden D, et al. Does early-onset chronic or recurrent major depression impact outcomes with antidepressant medications? A CO-MED trial report. Psychol Med 2013;43:945–60.
Kozel FA, Trivedi MH, Wisniewski SR, Miyahara S, Husain MM, Fava M, et al. Treatment Outcomes for Older Depressed Patients With Earlier Versus Late Onset of First Depressive Episode: A Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Report. Am J Geriatr Psychiatry. 2008;16:58–64.
Perlis RH, Dennehy EB, Miklowitz DJ, DelBello MP, Ostacher M, Calabrese JR, et al. Retrospective age at onset of bipolar disorder and outcome during two-year follow-up: results from the STEP-BD study. Bipolar Disord 2009;11:391–400.
Weissman MM. Cross-National epidemiology of major depression and bipolar disorder. JAMA: J Am Med Assoc. 1996;276:293.
Lewinsohn PM, Clarke GN, Seeley JR, Rohde P. Major depression in community adolescents: age at onset, episode duration, and time to recurrence. J Am Acad Child Adolesc Psychiatry. 1994;33:809–18.
Kobrosly RW, van Wijngaarden E, Seplaki CL, Cory-Slechta DA, Moynihan J. Depressive symptoms are associated with allostatic load among community-dwelling older adults. Physiol Behav 2014;123:223–30.
Strain JJ. The psychobiology of stress, depression, adjustment disorders and resilience. World J Biol Psychiatry. 2018;19:S14–S20.
McEwen BS, Rasgon NL. The Brain and Body on Stress: Allostatic Load and Mechanisms for Depression and Dementia. In: Strain JJ, Blumenfield M, editors. Depression as a Systemic Illness. Oxford University Press; 2018.
Patel V, Burns JK, Dhingra M, Tarver L, Kohrt BA, Lund C. Income inequality and depression: a systematic review and meta-analysis of the association and a scoping review of mechanisms. World Psychiatry 2018;17:76–89.
Kendler KS, Gardner CO. Sex differences in the pathways to major depression: a study of opposite-sex twin pairs. Am J Psychiatry. 2014;171:426–35.
Akincigil A, Olfson M, Siegel M, Zurlo KA, Walkup JT, Crystal S. Racial and ethnic disparities in depression care in community-dwelling elderly in the United States. Am J Public Health. 2012;102:319–28.
Alegría M, Chatterji P, Wells K, Cao Z, Chen C-N, Takeuchi D, et al. Disparity in depression treatment among racial and ethnic minority populations in the United States. Psychiatr Serv 2008;59:1264–72.
Chireh B, D’Arcy C. Shared and unique risk factors for depression and diabetes mellitus in a longitudinal study, implications for prevention: an analysis of a longitudinal population sample aged ⩾45 years. Ther Adv Endocrinol Metab. 2019;10:2042018819865828.
Harald B, Gordon P. Meta-review of depressive subtyping models. J Affect Disord. 2012;139:126–40.
Lichtenberg P, Belmaker RH. Subtyping major depressive disorder. Psychother Psychosom. 2010;79:131–5.
Goodwin FK, Jamison KR. Manic-depressive illness. Oxford University Press; 1990. p. 938.
Day CV, Rush AJ, Harris AWF, Boyce PM, Rekshan W, Etkin A, et al. Impairment and distress patterns distinguishing the melancholic depression subtype: an iSPOT-D report. J Affect Disord. 2015;174:493–502.
Spanemberg L, Caldieraro MA, Vares EA, Wollenhaupt-Aguiar B, Kauer-Sant’Anna M, Kawamoto SY, et al. Biological differences between melancholic and nonmelancholic depression subtyped by the CORE measure. Neuropsychiatr Dis Treat. 2014;10:1523–31.
Stewart JW, McGrath PJ, Rabkin JG, Quitkin FM. Atypical depression. A valid clinical entity? Psychiatr Clin North Am. 1993;16:479–95.
Lewy AJ, Lefler BJ, Emens JS, Bauer VK. The circadian basis of winter depression. Proc Natl Acad Sci USA. 2006;103:7414–9.
Lewy AJ, Sack RL, Miller LS, Hoban TM. Antidepressant and circadian phase-shifting effects of light. Science 1987;235:352–4.
de Bodinat C, Guardiola-Lemaitre B, Mocaër E, Renard P, Muñoz C, Millan MJ. Agomelatine, the first melatonergic antidepressant: discovery, characterization and development. Nat Rev Drug Disco. 2010;9:628–42.
Gaudiano BA, Dalrymple KL, Zimmerman M. Prevalence and clinical characteristics of psychotic versus nonpsychotic major depression in a general psychiatric outpatient clinic. Depress Anxiety 2009;26:54–64.
Schatzberg AF, Posener JA, DeBattista C, Kalehzan BM, Rothschild AJ, Shear PK. Neuropsychological deficits in psychotic versus nonpsychotic major depression and no mental illness. Am J Psychiatry. 2000;157:1095–1100.
Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu E-E, Schold C, et al. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry. 2002;52:386–92.
Keller J, Flores B, Gomez RG, Solvason HB, Kenna H, Williams GH, et al. Cortisol circadian rhythm alterations in psychotic major depression. Biol Psychiatry 2006;60:275–81.
Nelson JC, Bickford D, Delucchi K, Fiedorowicz JG, Coryell WH. Risk of psychosis in recurrent episodes of psychotic and nonpsychotic major depressive disorder: a systematic review and meta-analysis. Am J Psychiatry. 2018;175:897–904.
Lamers F, Rhebergen D, Merikangas KR, de Jonge P, Beekman ATF, B W J. Stability and transitions of depressive subtypes over a 2-year follow-up. Psychol Med 2012;42:2083–93.
Coryell W, Winokur G, Shea T, Maser JD, Endicott J, Akiskal HS. The long-term stability of depressive subtypes. Am J Psychiatry. 1994;151:199–204
Melartin T, Leskelä U, Rytsälä H, Sokero P, Lestelä-Mielonen P, Isometsä E. Co-morbidity and stability of melancholic features in DSM-IV major depressive disorder. Psychol Med 2004;34:1443–52.
Lovibond PF. Long-term stability of depression, anxiety, and stress syndromes. J Abnorm Psychol. 1998;107:520–6.
Zuroff DC, Blatt SJ, Sanislow CA III, Bondi CM, Pilkonis PA. Vulnerability to depression: Reexamining state dependence and relative stability. J Abnorm Psychol. 1999;108:76–89.
Musil R, Seemüller F, Meyer S, Spellmann I, Adli M, Bauer M, et al. Subtypes of depression and their overlap in a naturalistic inpatient sample of major depressive disorder. Int J Methods Psychiatr Res. 2018;27:e1569.
Arnow BA, Blasey C, Williams LM, Palmer DM, Rekshan W, Schatzberg AF, et al. Depression subtypes in predicting antidepressant response: a report from the iSPOT-D trial. Am J Psychiatry. 2015;172:743–50.
Chekroud AM, Gueorguieva R, Krumholz HM, Trivedi MH, Krystal JH, McCarthy G. Reevaluating the efficacy and predictability of antidepressant treatments: a symptom clustering approach. JAMA Psychiatry. 2017;74:370–8.
Paul R, Andlauer TFM, Czamara D, Hoehn D, Lucae S, Pütz B, et al. Treatment response classes in major depressive disorder identified by model-based clustering and validated by clinical prediction models. Transl Psychiatry 2019;9:187.
Ingram WM, Baker AM, Bauer CR, Brown JP, Goes FS, Larson S. et al. Defining major depressive disorder cohorts using the EHR: multiple phenotypes based on ICD-9 codes and medication orders. neurol psychiatry. Neurol Psychiatry Brain Res. 2020;36:18–26.
Huang SH, LePendu P, Iyer SV, Tai-Seale M, Carrell D, Shah NH. Toward personalizing treatment for depression: predicting diagnosis and severity. J Am Med Inf Assoc. 2014;21:1069–75.
Xia CH, Ma Z, Ciric R, Gu S, Betzel RF, Kaczkurkin AN, et al. Linked dimensions of psychopathology and connectivity in functional brain networks. Nat Commun 2018;9:3003.
Mihalik A, Ferreira FS, Rosa MJ, Moutoussis M, Ziegler G, Monteiro JM, et al. Brain-behaviour modes of covariation in healthy and clinically depressed young people. Sci Rep. 2019;9:11536.
Zhang B, Lin P, Shi H, Öngür D, Auerbach RP, Wang X, et al. Mapping anhedonia-specific dysfunction in a transdiagnostic approach: an ALE meta-analysis. Brain Imaging Behav. 2016;10:920–39.
McTeague LM, Huemer J, Carreon DM, Jiang Y, Eickhoff SB, Etkin A. Identification of common neural circuit disruptions in cognitive control across psychiatric disorders. Am J Psychiatry. 2017;174:676–85.
Zilverstand A, Parvaz MA, Goldstein RZ. Neuroimaging cognitive reappraisal in clinical populations to define neural targets for enhancing emotion regulation. A systematic review. Neuroimage 2017;151:105–16.
Schäfer JÖ, Naumann E, Holmes EA, Tuschen-Caffier B, Samson AC. Emotion regulation strategies in depressive and anxiety symptoms in youth: a meta-analytic review. J Youth Adolesc. 2017;46:261–76.
Sun X, Zhu C, So SHW. Dysfunctional metacognition across psychopathologies: a meta-analytic review. Eur Psychiatry 2017;45:139–53.
Price RB, Lane S, Gates K, Kraynak TE, Horner MS, Thase ME, et al. Parsing heterogeneity in the brain connectivity of depressed and healthy adults during positive mood. Biol Psychiatry 2017;81:347–57.
Price RB, Gates K, Kraynak TE, Thase ME, Siegle GJ. Data-driven subgroups in depression derived from directed functional connectivity paths at rest. Neuropsychopharmacology 2017;42:2623–32.
Feder S, Sundermann B, Wersching H, Teuber A, Kugel H, Teismann H, et al. Sample heterogeneity in unipolar depression as assessed by functional connectivity analyses is dominated by general disease effects. J Affect Disord. 2017;222:79–87.
Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, Meng Y, et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med 2016;23:28–38.
Dinga R, Schmaal L, Penninx BWJH, van Tol MJ, Veltman DJ, van Velzen L, et al. Evaluating the evidence for biotypes of depression: Methodological replication and extension of Drysdale et al. (2017). Neuroimage Clin. 2019:101796.
Grosenick L, Shi TC, Gunning FM, Dubin MJ, Downar J, Liston C. Functional and optogenetic approaches to discovering stable subtype-specific circuit mechanisms in depression. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4:554–66.
Tokuda T, Yoshimoto J, Shimizu Y, Okada G, Takamura M, Okamoto Y, et al. Identification of depression subtypes and relevant brain regions using a data-driven approach. Sci Rep. 2018;8:14082.
Van Dijk KRA, Sabuncu MR, Buckner RL. The influence of head motion on intrinsic functional connectivity MRI. Neuroimage 2012;59:431–8.
Satterthwaite TD, Wolf DH, Loughead J, Ruparel K, Elliott MA, Hakonarson H, et al. Impact of in-scanner head motion on multiple measures of functional connectivity: relevance for studies of neurodevelopment in youth. Neuroimage 2012;60:623–32.
Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 2012;59:2142–54.
Power JD, Mitra A, Laumann TO, Snyder AZ, Schlaggar BL, Petersen SE. Methods to detect, characterize, and remove motion artifact in resting state fMRI. Neuroimage 2014;84:320–41.
Power JD, Lynch CJ, Dubin MJ, Silver BM, Martin A, Jones RM. Characteristics of respiratory measures in young adults scanned at rest, including systematic changes and ‘missed’ deep breaths. Neuroimage. 2020;204:116234.
Power JD, Lynch CJ, Silver BM, Dubin MJ, Martin A, Jones RM. Distinctions among real and apparent respiratory motions in human fMRI data. Neuroimage. 2019;201:116041.
Friedman L, Glover GH. Report on a multicenter fMRI quality assurance protocol. J Magn Reson Imaging. 2006;23:827–39.
Friedman L, Stern H, Brown GG, Mathalon DH, Turner J, Glover GH, et al. Test-retest and between-site reliability in a multicenter fMRI study. Hum Brain Mapp. 2008;29:958–72.
Biswal BB, Mennes M, Zuo X-N, Gohel S, Kelly C, Smith SM, et al. Toward discovery science of human brain function. Proc Natl Acad Sci USA. 2010;107:4734–9.
Prathikanti S, Weinberger DR. Psychiatric genetics-the new era: genetic research and some clinical implications. Br Med Bull. 2005;73-74:107–22.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Ormel J, Hartman CA, Snieder H. The genetics of depression: successful genome-wide association studies introduce new challenges. Transl Psychiatry 2019;9:114.
Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet C Semin Med Genet. 2003;123C:48–58.
Halldorsdottir T, Binder EB. Gene × environment interactions: from molecular mechanisms to behavior. Annu Rev Psychol. 2017;68:215–41.
Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004;7:847–54.
Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–92.
Howard DM, Adams MJ, Shirali M, Clarke T-K, Marioni RE, Davies G, et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat Commun 2018;9:1470.
Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 2016;48:1031–6.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511:421–7.
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–92.
Duncan LE, Ostacher M, Ballon J. How genome-wide association studies (GWAS) made traditional candidate gene studies obsolete. Neuropsychopharmacology 2019;44:1518–23.
Canli T, Lesch K-P. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci 2007;10:1103–9.
Carter CS, Bearden CE, Bullmore ET, Geschwind DH, Glahn DC, Gur RE, et al. Enhancing the informativeness and replicability of imaging genomics studies. Biol Psychiatry 2017;82:157–64.
Pereira LP, Köhler CA, de Sousa RT, Solmi M, de Freitas BP, Fornaro M, et al. The relationship between genetic risk variants with brain structure and function in bipolar disorder: a systematic review of genetic-neuroimaging studies. Neurosci Biobehav Rev. 2017;79:87–109.
Pereira LP, Köhler CA, Stubbs B, Miskowiak KW, Morris G, de Freitas BP, et al. Imaging genetics paradigms in depression research: Systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2018;86:102–13.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996;274:1527–31.
Sen S, Burmeister M, Ghosh D. Meta-analysis of the association between a serotonin transporter promoter polymorphism (5-HTTLPR) and anxiety-related personality traits. Am J Med Genet B Neuropsychiatr Genet. 2004;127B:85–89.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003;301:386–9.
Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry. 2011;68:444–54.
Risch N, Herrell R, Lehner T, Liang K-Y, Eaves L, Hoh J, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA 2009;301:2462–71.
Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci 2005;8:828–34.
Hariri AR, Drabant EM, Weinberger DR. Imaging genetics: perspectives from studies of genetically driven variation in serotonin function and corticolimbic affective processing. Biol Psychiatry 2006;59:888–97.
Hariri AR, Mattay VS, Tessitore A, Fera F, Weinberger DR. Neocortical modulation of the amygdala response to fearful stimuli. Biol Psychiatry 2003;53:494–501.
Alexopoulos GS, Murphy CF, Gunning-Dixon FM, Glatt CE, Latoussakis V, Kelly RE Jr, et al. Serotonin transporter polymorphisms, microstructural white matter abnormalities and remission of geriatric depression. J Affect Disord. 2009;119:132–41.
Taylor WD, Steffens DC, Payne ME, MacFall JR, Marchuk DA, Svenson IK, et al. Influence of serotonin transporter promoter region polymorphisms on hippocampal volumes in late-life depression. Arch Gen Psychiatry. 2005;62:537–44.
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003;112:257–69.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P-F, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011;475:91–95.
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 2006;311:864–8.
Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry 2006;59:1116–27.
Chen Z-Y, Jing D, Bath KG, Ieraci A, Khan T, Siao C-J, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 2006;314:140–3.
Frodl T, Skokauskas N, Frey E-M, Morris D, Gill M, Carballedo A. BDNF Val66Met genotype interacts with childhood adversity and influences the formation of hippocampal subfields. Hum Brain Mapp. 2014;35:5776–83.
Cole J, Weinberger DR, Mattay VS, Cheng X, Toga AW, Thompson PM, et al. No effect of 5HTTLPR or BDNF Val66Met polymorphism on hippocampal morphology in major depression. Genes Brain Behav. 2011;10:756–64.
Border R, Johnson EC, Evans LM, Smolen A, Berley N, Sullivan PF, et al. No support for historical candidate gene or candidate gene-by-interaction hypotheses for major depression across multiple large samples. Am J Psychiatry. 2019;176:376–87.
Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet. 2015;97:576–92.
Maier RM, Visscher PM, Robinson MR, Wray NR. Embracing polygenicity: a review of methods and tools for psychiatric genetics research. Psychol Med 2018;48:1055–67.
Halldorsdottir T, Piechaczek C, Soares de Matos AP, Czamara D, Pehl V, Wagenbuechler P, et al. Polygenic risk: predicting depression outcomes in clinical and epidemiological cohorts of youths. Am J Psychiatry. 2019;176:615–25.
Choi SW, Mak TS-H, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15:2759–72.
Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 2009;41:56–65.
Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N. Engl J Med. 2016;375:2349–58.
Khera AV, Chaffin M, Aragam KG. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–24.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013;381:1371–9.
Yüksel D, Dietsche B, Forstner AJ, Witt SH, Maier R, Rietschel M, et al. Polygenic risk for depression and the neural correlates of working memory in healthy subjects. Prog Neuropsychopharmacol Biol Psychiatry. 2017;79:67–76.
Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–45.
Ward J, Lyall LM, Bethlehem RAI, Ferguson A, Strawbridge RJ, Lyall DM, et al. Novel genome-wide associations for anhedonia, genetic correlation with psychiatric disorders, and polygenic association with brain structure. Transl Psychiatry. 2019;9:327. https://doi.org/10.1038/s41398-019-0635-y.
Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015;47:291–5.
Hawrylycz M, Miller JA, Menon V, Feng D, Dolbeare T, Guillozet-Bongaarts AL, et al. Canonical genetic signatures of the adult human brain. Nat Neurosci 2015;18:1832–44.
Diez I, Larson AG, Nakhate V, Dunn EC, Fricchione GL, Nicholson TR, et al. Early-life trauma endophenotypes and brain circuit-gene expression relationships in functional neurological (conversion) disorder. Mol Psychiatry. 2020. https://doi.org/10.1038/s41380-020-0665-0.
Morgan SE, Seidlitz J, Whitaker KJ, Romero-Garcia R, Clifton NE, Scarpazza C, et al. Cortical patterning of abnormal morphometric similarity in psychosis is associated with brain expression of schizophrenia-related genes. Proc Natl Acad Sci USA. 2019;116:9604–9.
Romero-Garcia R, Seidlitz J, Whitaker KJ, Morgan SE, Fonagy P, Dolan RJ, et al. Schizotypy-related magnetization of cortex in healthy adolescence is colocated with expression of schizophrenia-related genes. Biol Psychiatry. 2019. https://doi.org/10.1016/j.biopsych.2019.12.005.
Whitaker KJ, Vértes PE, Romero-Garcia R, Váša F, Moutoussis M, Prabhu G, et al. Adolescence is associated with genomically patterned consolidation of the hubs of the human brain connectome. Proc Natl Acad Sci USA. 2016;113:9105–10.
Vértes PE, Rittman T, Whitaker KJ, Rafael R-G, Váša F, Kitzbichler MG, et al. Gene transcription profiles associated with inter-modular hubs and connection distance in human functional magnetic resonance imaging networks. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150362.
Romero-Garcia R, Warrier V, Bullmore ET, Baron-Cohen S, Bethlehem RAI. Synaptic and transcriptionally downregulated genes are associated with cortical thickness differences in autism. Mol Psychiatry 2019;24:1053–64.
Smith DJ, Nicholl BI, Cullen B, Martin D, Ul-Haq Z, Evans J, et al. Prevalence and characteristics of probable major depression and bipolar disorder within UK biobank: cross-sectional study of 172,751 participants. PLoS One 2013;8:e75362.
Cheng W, Rolls ET, Ruan H, Feng J. Functional connectivities in the brain that mediate the association between depressive problems and sleep quality. JAMA Psychiatry. 2018;75:1052–61.
Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature 2014;508:199–206.
Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013;41:D996–D1008.
Shmueli G. To explain or to predict? Stat Sci 2010;25:289–310.
Williams NR, Sudheimer KD, Bentzley BS, Pannu J, Stimpson KH, Duvio D, et al. High-dose spaced theta-burst TMS as a rapid-acting antidepressant in highly refractory depression. Brain 2018;141:e18.
Chen AC, Oathes DJ, Chang C, Bradley T, Zhou Z-W, Williams LM, et al. Causal interactions between fronto-parietal central executive and default-mode networks in humans. Proc Natl Acad Sci USA. 2013;110:19944–9.
Padmanabhan JL, Cooke D, Joutsa J, Siddiqi SH, Ferguson M, Darby RR, et al. A human depression circuit derived from focal brain lesions. Biol Psychiatry 2019;86:749–58.
Fox MD. Mapping symptoms to brain networks with the human connectome. N. Engl J Med. 2018;379:2237–45.
Salk RH, Hyde JS, Abramson LY. Gender differences in depression in representative national samples: meta-analyses of diagnoses and symptoms. Psychol Bull 2017;143:783–822.
Eid RS, Gobinath AR, Galea LAM. Sex differences in depression: Insights from clinical and preclinical studies. Prog Neurobiol 2019;176:86–102.
Gratton C, Laumann TO, Nielsen AN, Greene DJ, Gordon EM, Gilmore AW, et al. Functional brain networks are dominated by stable group and individual factors, not cognitive or daily variation. Neuron 2018;98:439–.e5.
Braga RM, Buckner RL. Parallel interdigitated distributed networks within the individual estimated by intrinsic functional connectivity. Neuron 2017;95:457–.e5.
Laumann TO, Gordon EM, Adeyemo B, Snyder AZ, Joo SJ, Chen M-Y, et al. Functional system and areal organization of a highly sampled individual human brain. Neuron 2015;87:657–70.
Kirkby LA, Luongo FJ, Lee MB, Nahum M, Van Vleet TM, Rao VR, et al. An amygdala-hippocampus subnetwork that encodes variation in human mood. Cell 2018;175:1688–1700.e14.
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 2007;131:391–404.
Hultman R, Ulrich K, Sachs BD, Blount C, Carlson DE, Ndubuizu N, et al. Brain-wide electrical spatiotemporal dynamics encode depression vulnerability. Cell 2018;173:166–180.e14.
Ferenczi E, Zalocusky KA, Liston C, Katovich K, Amatya D, Warden MR, et al. Prefrontal cortical regulation of brainwide circuit dynamics and reward-related behavior. Science 2016;351:41–53.
Pizzagalli DA, Webb CA, Dillon DG, Tenke CE, Kayser J, Goer F, et al. Pretreatment rostral anterior cingulate cortex theta activity in relation to symptom improvement in depression: a randomized clinical trial. JAMA Psychiatry. 2018;75:547–54.
Schatzberg AF, DeBattista C, Lazzeroni LC, Etkin A, Murphy GM Jr, Williams LM. ABCB1 genetic effects on antidepressant outcomes: a report from the iSPOT-D trial. Am J Psychiatry. 2015;172:751–9.
Williams LM. Precision psychiatry: a neural circuit taxonomy for depression and anxiety. Lancet Psychiatry 2016;3:472–80.
Dunlop BW, Rajendra JK, Craighead WE, Kelley ME, McGrath CL, Choi KS, et al. Functional connectivity of the subcallosal cingulate cortex and differential outcomes to treatment with cognitive-behavioral therapy or antidepressant medication for major depressive disorder. Am J Psychiatry. 2017;174:533–45.
Weigand A, Horn A, Caballero R, Cooke D, Stern AP, Taylor SF, et al. Prospective validation that subgenual connectivity predicts antidepressant efficacy of transcranial magnetic stimulation sites. Biol Psychiatry 2018;84:28–37.
Meng X, Brunet A, Turecki G, Liu A, D’Arcy C, Caron J. Risk factor modifications and depression incidence: a 4-year longitudinal Canadian cohort of the Montreal Catchment Area Study. BMJ Open 2017;7:e015156.
Rudkjoebing LA, Bungum AB, Flachs EM, Eller NH, Borritz M, Aust B, et al. Work-related exposure to violence or threats and risk of mental disorders and symptoms: a systematic review and meta-analysis. Scand J Work Environ Health. 2020. https://doi.org/10.5271/sjweh.3877.
Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327:557–60.
Lewis SJ, Lawlor DA, Davey Smith G, Araya R, Timpson N, Day INM, et al. The thermolabile variant of MTHFR is associated with depression in the British Women’s Heart and Health Study and a meta-analysis. Mol Psychiatry 2006;11:352–60.
Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165:1–13.
López-León S, Janssens ACJW, González-Zuloeta Ladd AM, Del-Favero J, Claes SJ, Oostra BA, et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry. 2008;13:772–85.
Samaan Z, Gaysina D, Cohen-Woods S, Craddock N, Jones L, Korszun A, et al. Methylenetetrahydrofolate reductase gene variant (MTHFR C677T) and migraine: a case control study and meta-analysis. BMC Neurol 2011;11:66.
Saha T, Chatterjee M, Sinha S, Rajamma U, Mukhopadhyay K. Components of the folate metabolic pathway and ADHD core traits: an exploration in eastern Indian probands. J Hum Genet. 2017;62:687–95.
Lok A, Bockting CLH, Koeter MWJ, Snieder H, Assies J, Mocking RJT, et al. Interaction between the MTHFR C677T polymorphism and traumatic childhood events predicts depression. Transl Psychiatry 2013;3:e288.
Noh K, Lee H, Choi T-Y, Joo Y, Kim S-J, Kim H, et al. Negr1 controls adult hippocampal neurogenesis and affective behaviors. Mol Psychiatry 2019;24:1189–205.
Wang X, Cheng W, Zhu J, Yin H, Chang S, Yue W, et al. Integrating genome-wide association study and expression quantitative trait loci data identifies NEGR1 as a causal risk gene of major depression disorder. J Affect Disord. 2020;265:679–86.
Zandoná MR, Sangalli CN, Campagnolo PDB, Vitolo MR, Almeida S, Mattevi VS. Validation of obesity susceptibility loci identified by genome-wide association studies in early childhood in South Brazilian children. Pediatr Obes. 2017;12:85–92.
Kuc K, Bielecki M, Racicka-Pawlukiewicz E, Czerwinski MB, Cybulska-Klosowicz A. The SLC6A3 gene polymorphism is related to the development of attentional functions but not to ADHD. Sci Rep. 2020;10:6176.
Marinho FVC, Pinto GR, Oliveira T, Gomes A, Lima V, Ferreira-Fernandes H, et al. The SLC6A3 3′-UTR VNTR and intron 8 VNTR polymorphisms association in the time estimation. Brain Struct Funct. 2019;224:253–62.
Marinho V, Oliveira T, Bandeira J, Pinto GR, Gomes A, Lima V, et al. Genetic influence alters the brain synchronism in perception and timing. J Biomed Sci. 2018;25:61.
Zahavi AY, Sabbagh MA, Washburn D, Mazurka R, Bagby RM, Strauss J, et al. Serotonin and dopamine gene variation and theory of mind decoding accuracy in major depression: a preliminary investigation. PLoS ONE 2016;11:e0150872.
Kishi T, Tsunoka T, Ikeda M, Kawashima K, Okochi T, Kitajima T, et al. Serotonin 1A receptor gene and major depressive disorder: an association study and meta-analysis. J Hum Genet. 2009;54:629–33.
Kishi T, Yoshimura R, Fukuo Y, Okochi T, Matsunaga S, Umene-Nakano W, et al. The serotonin 1A receptor gene confer susceptibility to mood disorders: results from an extended meta-analysis of patients with major depression and bipolar disorder. Eur Arch Psychiatry Clin Neurosci. 2013;263:105–18.
Zhang K, Xu Q, Xu Y, Yang H, Luo J, Sun Y, et al. The combined effects of the 5-HTTLPR and 5-HTR1A genes modulates the relationship between negative life events and major depressive disorder in a Chinese population. J Affect Disord. 2009;114:224–31.
López León S, Croes EA, Sayed-Tabatabaei FA, Claes S, Van Broeckhoven C, van Duijn CM. The dopamine D4 receptor gene 48-base-pair-repeat polymorphism and mood disorders: a meta-analysis. Biol Psychiatry 2005;57:999–1003.
Bircher J, Kotyuk E, Fulop M, Vereczkei A, Ronai Z, Varga K, et al. Gene-sex interaction in hypercompetitive attitude suggests beneficial effect of the DRD4 7-repeat allele in adaptation. Neuropsychopharmacol Hung 2019;21:47–58.
Ji H, Xu X, Liu G, Liu H, Wang Q, Shen W, et al. Dopamine receptor D4 promoter hypermethylation increases the risk of drug addiction. Exp Ther Med. 2018;15:2128–33.
Green CG, Babineau V, Jolicoeur-Martineau A, Bouvette-Turcot A-A, Minde K, Sassi R, et al. Prenatal maternal depression and child serotonin transporter linked polymorphic region (5-HTTLPR) and dopamine receptor D4 (DRD4) genotype predict negative emotionality from 3 to 36 months. Dev Psychopathol 2017;29:901–17.
Badamasi IM, Lye MS, Ibrahim N, Stanslas J. Genetic endophenotypes for insomnia of major depressive disorder and treatment-induced insomnia. J Neural Transm. 2019;126:711–22.
Keers R, Bonvicini C, Scassellati C, Uher R, Placentino A, Giovannini C, et al. Variation in GNB3 predicts response and adverse reactions to antidepressants. J Psychopharmacol 2011;25:867–74.
Lin E, Chen PS, Chang HH, Gean P-W, Tsai HC, Yang YK, et al. Interaction of serotonin-related genes affects short-term antidepressant response in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1167–72.
Hu Q, Zhang S-Y, Liu F, Zhang XJ, Cui G-C, Yu E-Q, et al. Influence of GNB3 C825T polymorphism on the efficacy of antidepressants in the treatment of major depressive disorder: a meta-analysis. J Affect Disord. 2015;172:103–9.
Krishnan M, Shelling AN, Wall CR, Mitchell EA, Murphy R, McCowan LME, et al. Gene-by-activity interactions on obesity traits of 6-year-old New Zealand European children: a children of SCOPE Study. Pediatr Exerc Sci. 2018;30:69–80.
Johnston KJA, Adams MJ, Nicholl BI, Ward J, Strawbridge RJ, McIntosh AM, et al. Identification of novel common variants associated with chronic pain using conditional false discovery rate analysis with major depressive disorder and assessment of pleiotropic effects of LRFN5. Transl Psychiatry 2019;9:310.
Nho K, Ramanan VK, Horgusluoglu E, Kim S, Inlow MH, Risacher SL, et al. Comprehensive gene- and pathway-based analysis of depressive symptoms in older adults. J Alzheimers Dis. 2015;45:1197–206.
Choi Y, Nam J, Whitcomb DJ, Song YS, Kim D, Jeon S, et al. SALM5 trans-synaptically interacts with LAR-RPTPs in a splicing-dependent manner to regulate synapse development. Sci Rep. 2016;6:26676.
de Bruijn DRH, van Dijk AHA, Pfundt R, Hoischen A, Merkx GFM, Gradek GA, et al. Severe progressive autism associated with two de novo changes: a 2.6-Mb 2q31.1 deletion and a balanced t(14;21)(q21.1;p11.2) translocation with long-range epigenetic silencing of LRFN5 expression. Mol Syndromol 2010;1:46–57.
Cappuccio G, Attanasio S, Alagia M, Mutarelli M, Borzone R, Karali M, et al. Microdeletion of pseudogene chr14.232.a affects LRFN5 expression in cells of a patient with autism spectrum disorder. Eur J Hum Genet. 2019;27:1475–80.
Wamsley B, Jaglin XH, Favuzzi E, Quattrocolo G, Nigro MJ, Yusuf N, et al. Rbfox1 mediates cell-type-specific splicing in cortical interneurons. Neuron 2018;100:846–.e7.
Kong L-L, Miao D, Tan L, Liu S-L, Li J-Q, Cao X-P, et al. Genome-wide association study identifies RBFOX1 locus influencing brain glucose metabolism. Ann Transl Med. 2018;6:436.
Fernàndez-Castillo N, Gan G, van Donkelaar MMJ, Vaht M, Weber H, Retz W, et al. RBFOX1, encoding a splicing regulator, is a candidate gene for aggressive behavior. Eur Neuropsychopharmacol 2020;30:44–55.
Zhao W-W. Intragenic deletion of RBFOX1 associated with neurodevelopmental/neuropsychiatric disorders and possibly other clinical presentations. Mol Cytogenet 2013;6:26.
Clarke H, Flint J, Attwood AS, Munafò MR. Association of the 5- HTTLPR genotype and unipolar depression: a meta-analysis. Psychol Med 2010;40:1767–78.
Furlong RA, Ho L, Walsh C, Rubinsztein JS, Jain S, Paykel ES, et al. Analysis and meta-analysis of two serotonin transporter gene polymorphisms in bipolar and unipolar affective disorders. Am J Med Genet 1998;81:58–63.
Dosenbach NUF, Koller JM, Earl EA, Miranda-Dominguez O, Klein RL, Van AN, et al. Real-time motion analytics during brain MRI improve data quality and reduce costs. Neuroimage 2017;161:80–93.
Power JD, Silver BM, Silverman MR, Ajodan EL, Bos DJ, Jones RM. Customized head molds reduce motion during resting state fMRI scans. Neuroimage 2019;189:141–9.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8:118–27.
Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc Ser B-Stat Methodol. 2005;67:301–20.
Sui J, Adali T, Pearlson G, Yang H, Sponheim SR, White T, et al. A CCA+ICA based model for multi-task brain imaging data fusion and its application to schizophrenia. Neuroimage 2010;51:123–34.
Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012;489:391–9.
Arloth J, Bader DM, Röh S, Altmann A. Re-annotator: annotation pipeline for microarray probe sequences. PLoS ONE 2015;10:e0139516.
Braschi B, Denny P, Gray K, Jones T, Seal R, Tweedie S, et al. Genenames.org: the HGNC and VGNC resources in 2019. Nucleic Acids Res. 2019;47:D786–D792.
Arnatkevic Iūtė A, Fulcher BD, Fornito A. A practical guide to linking brain-wide gene expression and neuroimaging data. Neuroimage. 2019. https://doi.org/10.1016/j.neuroimage.2019.01.011.
Rohart F, Gautier B, Singh A, Lê Cao K-A. mixOmics: an R package for ’omics feature selection and multiple data integration. PLoS Comput Biol. 2017;13:e1005752.
Monteiro JM, Rao A, Shawe-Taylor J, Mourão-Miranda J, Initiative AD. A multiple hold-out framework for sparse partial least squares. J Neurosci Methods. 2016;271:182–94.
Ressa A, Fitzpatrick M, van den Toorn H, Heck AJR, Altelaar M. PaDuA: a python library for high-throughput (Phospho)proteomics data analysis. J Proteome Res. 2019;18:576–84.
Krishnan A, Williams LJ, McIntosh AR, Abdi H. Partial least squares (PLS) methods for neuroimaging: a tutorial and review. Neuroimage 2011;56:455–75.
Abdi H, Williams LJ. Partial least squares methods: partial least squares correlation and partial least square regression. Methods Mol Biol. 2013;930:549–79.
Boulesteix A-L, Strimmer K. Partial least squares: a versatile tool for the analysis of high-dimensional genomic data. Brief Bioinform 2007;8:32–44.
Bennett KP, Embrechts MJ. An optimization perspective on kernel partial least squares regression Nato Science Series sub series III computer and systems sciences. 2003;190:227–50.
Wong E, Palande S, Wang B, Zielinski B, Anderson J, Fletcher PT. Kernel partial least squares regression for relating functional brain network topology to clinical measures of behavior. In Proc 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI), Prague: 2016. p. 1303–6. https://doi.org/10.1109/ISBI.2016.7493506.
Alexander-Bloch AF, Shou H, Liu S, Satterthwaite TD, Glahn DC, Shinohara RT, et al. On testing for spatial correspondence between maps of human brain structure and function. Neuroimage 2018;178:540–51.
Kock N. Should bootstrapping be used in PLS-SEM? Toward stable P-Value calculation methods. J Appl Struct Equ Modeling. 2018;2:1–12.
Eriksson L, Johansson E, Kettaneh-Wold N, Trygg J, Wikström C, Wold S. Multi-and megavariate data analysis, Vol 1. Sweden: Umetrics Sweden; 2006.
Kvalheim OM. Interpretation of partial least squares regression models by means of target projection and selectivity ratio plots. J Chemom 2010;24:496–504.
Rajalahti T, Arneberg R, Kroksveen AC, Berle M, Myhr K-M, Kvalheim OM. Discriminating variable test and selectivity ratio plot: quantitative tools for interpretation and variable (Biomarker) selection in complex spectral or chromatographic profiles. Anal Chem 2009;81:2581–90.
Webber W, Moffat A, Zobel J. A similarity measure for indefinite rankings. ACM Trans Inf Syst (TOIS). 2010;28:20.
The Gene Ontology Consortium. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–D338.
Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613.
Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:W234–W241.
Gaspar HA, Hübel C, Breen G. Drug Targetor: a web interface to investigate the human druggome for over 500 phenotypes. Bioinformatics 2019;35:2515–7.
Author information
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Buch, A.M., Liston, C. Dissecting diagnostic heterogeneity in depression by integrating neuroimaging and genetics. Neuropsychopharmacol. 46, 156–175 (2021). https://doi.org/10.1038/s41386-020-00789-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-00789-3
This article is cited by
-
Polygenic risk for major depression, attention deficit hyperactivity disorder, neuroticism, and schizophrenia are correlated with experience of intimate partner violence
Translational Psychiatry (2024)
-
Gene–brain–behavior mechanisms underlying autism spectrum disorder: implications for precision psychiatry
Neuropsychopharmacology (2024)
-
Integration of whole-exome sequencing and structural neuroimaging analysis in major depressive disorder: a joint study
Translational Psychiatry (2024)
-
Behind the Screen: A Narrative Review on the Translational Capacity of Passive Sensing for Mental Health Assessment
Biomedical Materials & Devices (2024)
-
Opportunities and challenges for the use of deep brain stimulation in the treatment of refractory major depression
Discover Mental Health (2024)
