
Abstract
Sleep is essential for memory formation. Active systems consolidation maintains that memory traces that are initially stored in a transient store such as the hippocampus are gradually redistributed towards more permanent storage sites such as the cortex during sleep replay. The complementary synaptic homeostasis theory posits that weak memory traces are erased during sleep through a competitive down-selection mechanism, ensuring the brain’s capability to learn new information. We discuss evidence from neuropharmacological experiments in humans to show how major neurotransmitters and neuromodulators are implicated in these memory processes. As to the major excitatory neurotransmitter glutamate that plays a prominent role in inducing synaptic consolidation, we show that these processes, while strengthening cortical memory traces during sleep, are insufficient to explain the consolidation of hippocampus-dependent declarative memories. In the inhibitory GABAergic system, we will offer insights how drugs may alter the intricate interplay of sleep oscillations that have been identified to be crucial for strengthening memories during sleep. Regarding the dopaminergic reward system, we will show how it is engaged during sleep replay, but that dopaminergic neuromodulation likely plays a side role for enhancing relevant memories during sleep. Also, we briefly go into basic evidence on acetylcholine and cortisol whose low tone during slow wave sleep (SWS) is crucial in supporting hippocampal-to-neocortical memory transmission. Finally, we will outline how these insights can be used to improve treatment of neuropsychiatric disorders focusing mainly on anxiety disorders, depression, and addiction that are strongly related to memory processing.
Similar content being viewed by others

Introduction
Sleep benefits the long-term storage of memories [1,2,3,4]. However, it is important to note that successful memory relies on at least three distinct processes that are influenced by sleep in different ways. Encoding or learning is the process that initially establishes the memory trace, whereas retrieval is the process of recalling a trace from memory. The third process is an active process of stabilizing the memory trace during the retention period between the two and has been coined as consolidation [5, 6]. Consolidation is thought to profit most profoundly from sleep. At the beginning of the twentieth century one of the first experimental accounts of sleep’s memory effect was published by Jenkins and Dallenbach [7]. Their work extended Hermann Ebbinghaus’ seminal work on forgetting curves [8] that showed a pronounced improvement after 1 day and has since been replicated twice more [9]. Their approach compared declarative memory performance (see Box 1 for an introduction to declarative and procedural memory) after a retention interval of sleep with that after a retention interval of wakefulness and found less forgetting in the sleep condition. Importantly, this improvement was not attributed to an active (consolidation) process, but rather explained through an absence of interference, since during sleep encoding of new stimuli is reduced. Limiting sleep’s role for memory to a passive protection rather than to an active consolidation process may explain why researchers initially did not dwell on the topic much longer.
A major breakthrough in sleep research further contributed to this. In 1929, Hans Berger discovered that brain activity can readily be measured on the surface of the scalp. Applying this technique during sleep led to the discovery of the distinct sleep stages (see Box 2 for a brief introduction of the different sleep stages and associated hallmark oscillations) rapid eye movement (REM) sleep and non-rapid eye movement (NonREM) sleep [10, 11]. While the importance of this discovery for the field of sleep cannot be overstated, it was unfortunate that sleep and memory researchers mainly focussed on REM sleep rather than NonREM sleep for most of the rest of the twentieth century due to its similarity to waking brain activity. However, a second breakthrough occurred in the field of animal electrophysiology, based on the discovery of neurons (place cells) in the hippocampus that fired only at specific places in an open field [12], which was early on identified as a potential code for memory [13]. Interestingly, it was soon discovered that hippocampal place cell firing during wake exploration influenced firing during subsequent sleep [14] and that coincident firing patterns of place cells that developed during awake learning re-emerged during subsequent sleep, and specifically during slow wave sleep (SWS) [15]. This finding of potentially active memory processing during sleep rekindled the field and research-efforts have been accelerating ever since.
Importantly, this finding was extended to human research using a unique experimental approach, where memory contents are associated with sensory stimuli that can be used to target this content specifically during sleep. In their experiments, Rasch et al. [16] used a card-pair location task similar to the game Concentration. During learning, participants were exposed to the smell of roses and this smell was then used during sleep for targeted reactivation of the associated memory task. Strikingly, this memory cueing procedure was only effective during SWS, which in humans is the deepest form of NonREM sleep, but not during REM sleep or wakefulness. In fact, later extensions showed that targeted reactivation during wakefulness—similar to that used during sleep—can even have a detrimental effect on memory [17] providing evidence for the unique properties SWS has for consolidation. This technique has since been firmly established as a standard methodology to probe sleep and memory consolidation. It has been shown that individual memories can be targeted [18, 19], that procedural motor memories can also be boosted [20, 21], and targeted memory activation can greatly enhance the effectivity of sleep to consolidate [22]. In fact, there are now first attempts to use it as an augmentation for psychotherapy (e.g., ref. [23]).
In the following, the main two theories that try to explain sleep-dependent memory consolidation, active systems consolidation and the synaptic homeostasis hypothesis will be introduced. Next, we will run through human neuropharmacological work that has scrutinized the four major neurotransmitter systems glutamate, gamma-amino-butyric-acid (GABA), dopamine, and acetylcholine as well as the stress-hormone cortisol to identify their role during sleep-dependent memory processing. Focussing on work in humans, it is important to acknowledge that a significant portion of the work referenced in this review was performed by the authors’ lab and that the field of sleep and memory research would benefit from more labs undertaking the burdensome work of running pharmacological studies in humans. There is a dire need of an attempt at independently replicating most of the findings. Neuropharmacological work in humans is important as it can shed light on mechanisms without the need of subsequent translation. However, it comes at the expense of somewhat limited experimental control, e.g., drugs cannot be directly infused into specific brain areas. Finally, we discuss how basic findings in the area of sleep and memory research might inform therapeutic strategies for neuropsychiatric disorders giving details for anxiety disorders, depression, and addiction. We conclude by naming some of the most pressing challenges to current neuropharmacological sleep research. Although we do mention findings in other transmitter systems in passing, we do not aim to be comprehensive.
Theoretical frameworks
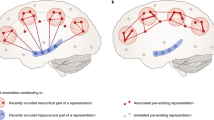
The field of sleep and memory is in the fortunate position of having two strong theories that have been developed to accommodate its findings. The first is active systems consolidation (Fig. 1), which has been most elaborately tested in the declarative domain [2, 24], but is formulated as a general principle that is applicable also to non-declarative forms of memory [25] and even in non-neuronal immunological memory [26]. Systems consolidation was formulated as a solution to the plasticity-stability dilemma [27, 28], i.e., that in a sequential learning process a memory system must hold useful information stable and protect it from being overwritten by incoming new information, while at the same time allowing plastic changes for new learning [29]. During learning, memories traces are initially encoded into a fast changing transient store and are then gradually transferred into a slow changing permanent store [30]. In the declarative domain, new associations are stored in the hippocampus that acts as a hub binding together cortical information [31] and these connections are later redistributed to the neocortex [30, 32, 33]. Systems consolidation uses synaptic consolidation (including forms of synaptic long-term potentiation and transcription of immediate early genes) as a subroutine in neuronal assemblies to strengthen representations locally. Active systems consolidation suggests that the gradual redistribution of synaptic connections moving the representation towards neocortical networks is most readily achieved during NonREM sleep, specifically SWS [24]. It has been suggested that, during active wakefulness, a high tone of acetylcholine enables encoding into the hippocampus, whereas, during SWS, a low tone allows information to flow from the hippocampus to the neocortex [34]. Essentially, during wakefulness, sensory information is able to flow from the cortex to the hippocampus to encode associations, but this neocortico-hippocampal dialog is reversed during SWS [35]. Mechanistically, replay in the hippocampus is thought to be initiated by the neocortical slow oscillation [36] to co-occur with its rising flank and associated sleep spindle activity [37, 38]. Replay information coincides with sharp wave-ripples, which themselves nest into the excitable troughs of spindles [37, 39], and thus are ideally suited to induce plastic processes [40, 41] thereby redistributing the memory trace towards the cortex. Coordinated replay in the hippocampus and the cortex occurring in the same sequence as during wake activity has been suggested to crucially support this redistribution [42]. Whereas most of this work was based on recordings of place cell activity during spatial experience, more recent work shows that sleep-dependent consolidation likewise occurs after fear context learning, where no clear sequence of events is evident, and is supported by other forms of coordinated network activity [43]. In fact, when place cell firing was not restricted by a linear track, replay from a freely explorable environment was not limited to trajectories the animal had experienced but covered Brownian diffusion like random trajectories [44]. These findings and the fact that novel environments elicit much longer lasting replay [45], cast doubt on the necessity of strictly sequential replay for sleep-dependent memory consolidation.

Three stages of memory formation. Memory performance is affected by the efficient execution of learning the information by encoding the memory trace, consolidating the trace for long-term storage, and subsequently successfully retrieving the trace. Active systems consolidation ascribes sleep a vital role for the consolidation step. In a nutshell, a memory is learned during wakefulness, consolidated during sleep, and retrieved again during wakefulness. For example, if you meet Jane at the beach and she shows off her new red car, neocortical sites process this information, however, the hippocampus associates all four items of this memory acting as a hub that binds the information into an episodic representation. Therefore, the hippocampus does not hold the actual information, but rather its unique combination. During subsequent sleep, the hippocampus replays the associative trace allowing the slow learning cortical networks to form an associative trace of their own (this may include recruitment of the prefrontal cortex to succeed the hippocampus as a hub, which is not depicted here for simplicity). To form representations outside the hippocampus, systems consolidation relies on processes of synaptic consolidation as a subroutine (including long-term potentiation) to strengthen connections of the representation locally. With ongoing systems consolidation, the cortical trace becomes sufficient to retrieve the information without recruitment of the hippocampus and you will know that Jane drives a red car and likes going to the beach. Within this framework, there is a complementary role for homeostatic processes as proposed by the synaptic homeostasis hypothesis, inasmuch as the hippocampal trace may be removed to make space for new learning, when the cortical trace has been established and it is no longer needed. Additionally, there is room for further pruning of the cortical trace making it more efficient after it has been firmly established. Importantly, the described process likely does not complete within one single night of sleep, but will take several nights. In addition, the cortical trace loses most of the episodic features of your meeting with Jane leaving only the semantic content
The second theory explaining sleep’s beneficial effect on memory is down-selection theory that developed from the synaptic homeostasis hypothesis. This theory originates from a different perspective on sleep and memory that focusses on homeostatic needs of the brain. It states that most learning in the brain relies on some form of Hebbian plasticity, i.e., an increase in connection strength between neurons during learning [46]. This potentiating of connections is thought to come at the price of increased demands for energy and space that cannot be sustained indefinitely [47, 48]. Evidence from experiments comparing glutamatergic (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-AMPA) receptor density at the active sites of synapses have supported this view, as excitatory receptors are upregulated after longer phases of wakefulness and downregulated after sleep [49]. Similarly, dendritic spines, morphological correlates of potentiation, show net upregulation during wakefulness and downregulation during sleep in mice [50, 51]. Importantly, this process has been suggested to be specific leaving stronger synapses intact [52]. In humans, it was shown using transcranial magnetic stimulation that sleep restores the capability of the cortex for long-term potentiation-like plasticity [53]. Importantly, these results show that synaptic up- and downregulation can occur in both sleep and wakefulness, but that the absolute amount of each is different, leading to a net downscaling during sleep. In humans, the most convincing evidence for a physiological homeostatic process active during sleep comes from studies of new learning after sleep. Here, it was shown that participants are able to encode information better, if they are allowed to sleep before learning [54]. This effect seems to be mediated by SWS, as manipulations of this sleep stage led to altered encoding capabilities in young and old participants [55, 56]. The latest iteration of this theory accounts for the abundance of memory enhancing and replay findings related to sleep, by suggesting a mechanism that chooses connections that should be downregulated, sparing those connections engaging in newly formed memory representations [4]. Intriguingly, the sharp wave-ripples that have been shown to be strong markers of reactivation have been shown to also support downregulation of synapses in the hippocampus [57]. In the motor cortex, synaptic pruning during REM sleep that balances excessive plasticity during learning has been shown to improve subsequent learning [58, 59]. However, to support a direct involvement of downregulation in memory consolidation more empirical evidence linking selective synaptic downscaling to improved memory function should be produced in future (for a more in depths critique of the synaptic homeostasis hypothesis, see refs. [60, 61]).
Integrating active systems consolidation and the synaptic homeostasis hypothesis leads to a seeming contradiction of the same mechanisms being related both to the strengthening and weakening of memory traces. However, the fate of a memory trace may be mediated by its relevance for the future. For example, the instruction that a list of word-pairs will be retrieved after sleep enables sleep’s beneficial effect [62]. Similarly, rewards and emotions can modulate the strengthening effect of sleep on memory [63,64,65,66,67,68]. Therefore, while sleep has been clearly linked to the consolidation of relevant memories, it may also serve the function of erasing irrelevant ones. In addition, sleep likely homeostatically restores hippocampal function for encoding after systems consolidation has made the traces held there redundant, thereby honing the memory store for ideal function [69]. Ideally, future research will break down which sleep phenomena link to which function individually for each memory domain and/or brain region. For example, there seems to be some evidence that NonREM sleep strengthens synapses within the cortex [59, 70,71,72], while both NonREM and REM sleep act together to downregulate them in the hippocampus [57, 73], which would be ideally suited for a well-functioning declarative memory system. While active systems consolidation argues that on the long run memories can become independent of the hippocampus, there is also evidence from animal and human studies showing increased firing rates as well as a time-limited increase in blood oxygen level-dependent (BOLD) activity in the hippocampus, respectively, caused by processes during initial sleep after learning [74, 75]. Delineating the time course and specificity of potentiation and downregulation during sleep are important future research objectives.
Glutamatergic neurotransmission
Learning in the brain is thought to mainly rely on Hebbian plasticity where neurons that are active together get connected more tightly, so that activating one neuron will soon activate the other, i.e., what fires together wires together [46]. Glutamatergic long-term potentiation (LTP) is the most studied mechanism of such synaptic potentiation, and this form of synaptic consolidation is indeed viewed by many as the main substrate of high-level learning in the brain (for details [76, 77]). In this framework, the AMPA receptor transmits neuronal activation from one neuron to the other and the N-methyl-d-aspartate (NMDA) receptor enables plastic processes, i.e., the strengthening of the connection between the neurons through AMPA receptors.
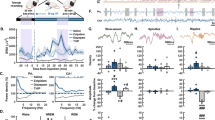
In an attempt to shed light on the involvement of AMPA and NMDA receptors during sleep-dependent memory consolidation, we ran a series of pharmacological experiments in humans (Fig. 2). First, we tested our hypothesis that sleep reactivation of memory traces leads to memory strengthening via glutamatergic processes in the cortex, as there is evidence from cats that sleep-dependent ocular dominance plasticity in the visual cortex is mediated by NMDA-receptor activation [78]. The first experiment investigated the effect of pharmacologically blocking AMPA and NMDA receptors during post-learning sleep [79]. Participants learned a visual texture discrimination task that mainly relies on plastic changes to the visual cortex for encoding [80] and is strongly dependent on NonREM sleep for synaptic consolidation of those traces [81]. In two different groups, caroverine was used to block AMPA receptors and ketamine to block NMDA receptors during sleep. Caroverine is a quinoxaline-derivative that at low doses can be used in humans to block AMPA receptors. Compared to placebo, participants performed significantly worse across retention when they were infused with either of the drugs during sleep. In line with our hypothesis, this result highlights that memory consolidation that relies on reactivation of the local memory trace is susceptible to blocking neuronal information transduction via the AMPA receptor, which coincidentally blocks plastic processes, as well as blocking coincidence detection via the NMDA receptor that leaves signal transduction intact and blocks only plastic changes.

Glutamatergic system neuroplasticity during sleep. a Memory reactivation seems ideally suited to support Hebbian plasticity (i.e., a kind of synaptic consolidation), where coincidentally activated excitatory neurons form stronger connections. b Glutamatergic LTP is the most well-studied form of such plasticity. Broadly speaking, in response to glutamate signaling AMPA receptors are responsible for maintaining signal transduction, whereas, NMDA receptors initiate plastic processes. Plasticity goes along with an increase in AMPA receptor availability at the active zone, but it also acts via pre-synaptic increases in transmitter release and structural changes. c Blocking the AMPA receptor prevents signal transduction leaving the post-synaptic neuron’s membrane potential at hyperpolarized levels and thus preventing plasticity signals via the NMDA receptor. If reactivation spreads via glutamatergic neurotransmission, this should block memory consolidation, which is true for cortical memories. However, for hippocampus-dependent declarative memories no such effect is evident. d Blocking the NMDA receptor directly with Ketamine leaves glutamatergic signal transduction intact, but blocks plastic processes. Again, this drug blocks consolidation of cortical memories but not hippocampal memories. e Enhancing NDMA receptor function with D-cycloserine that acts via the glycine-binding site and thereby boosting plasticity, on the other hand, greatly improved sleep-dependent consolidation of hippocampal memories
Importantly, performance of the visual texture discrimination task above does not essentially rely on the hippocampus [80]. Therefore, sleep-dependent performance gains might primarily reflect synaptic consolidation within the cortical storage sites rather than systems consolidation from the hippocampus to the neocortex [29]. Although, this does not exclude that hippocampus-dependent systems consolidation during sleep can also contribute to more gradually emerging gains in perceptual skill [25]. Against this backdrop, we expanded these findings to the declarative domain, where the hippocampus plays a crucial role already in the initial expression of the memory that is later replaced by neocortical structures [33]. We ran a follow-up study using a word-pair task that has been shown to recruit the hippocampus during learning [82] and robustly depends on NonREM sleep for consolidation [83,84,85]. Within the active systems consolidation framework, we hypothesized that blocking AMPA and NMDA receptors would impair hippocampal memory reactivations and the transmission of reactivated information to the cortex, similarly, as it impairs cortical memory consolidation. We again used caroverine and ketamine to block AMPA and NMDA receptors during early NonREM sleep, after learning [86]. Participants retrieved the word-pairs after sleep and performance was compared to placebo nights. Surprisingly, retention performance, i.e., the difference in word-pairs remembered at retrieval minus those remembered at learning, was affected by neither of the drugs indicating that these important receptors do not contribute to declarative memory consolidation during sleep. This difference to the initial study is likely due to the recruitment of hippocampus-dependent systems consolidation processes during sleep that affect word-pair memory directly, but may change visual texture discrimination memory, if at all, only with substantial delay.
However, we ruled out the possibility of complete disengagement from consolidation during sleep for the NMDA-receptor, in these experiments, by using D-cycloserine in a similar paradigm [86]. d-cycloserine acts as co-agonist at the glycine-binding site of the NMDA-receptor and thereby positively modulates its function [87]. It has been shown to enhance retention, if given acutely after learning [88]. Our participants received the drug before going to sleep, after learning the word-pairs, and retrieval was tested the next evening, due to the longer half-life of D-cycloserine. Here, participants retained more word-pairs if they were given the drug rather than placebo. A control group that was not allowed to sleep during the retention interval did not benefit from the treatment. Taken together, these findings indicate that active systems consolidation does not recruit the usual form of LTP for plasticity as outlined above, but that the NMDA receptor nevertheless plays some essential role. Crucially, in the absence of AMPA receptor engagement, there must exist an alternative form of information transfer that could be recruited by ripples. Identifying such a mechanism seems all the more important since our attempts to further scrutinize the glutamatergic system by applying the metabotropic glutamate receptor 5 blocker fenobam did not show any effects on sleep-dependent memory consolidation or forgetting (Feld, Bergmann, Alizadeh-Asfestani, Stuke, Wriede, Soekadar and Born, unpublished data). This rules out that our report of a lack of effect of blocking NMDA and AMPA receptors can be explained by a functional shift within the glutamatergic system itself during sleep [89, 90].
In an attempt to scrutinize whether active systems consolidation and associated NMDA receptor-mediated processes leads to diminished encoding capacity after sleep, we performed an additional study that focussed on retro-active interference rather than consolidation [91]. Participants again learned word-pairs in the evening and retained them until the next evening, but were then asked to learn a new set of word-pairs that partially overlapped with the original word-pairs. Thereby allowing us to investigate the retro-active interference elicited by the originally learned information [92], after it is consolidated by sleep. We predicted that enhanced consolidation during sleep—elicited by a D-cycloserine-induced enhancement of NMDA-receptor activity—would be accompanied by an increase in retro-active interference and, thus, reduce new learning. However, this was not the case, and D-cycloserine rather generally increased the amount of learning regardless of sleep occurring during the retention interval. These results can be taken to suggest that some forms of synaptic renormalization as predicted by synaptic homeostasis theory [47] occur independent of sleep, possibly via a type of sleep-independent active decay [93]. In fact, NMDA-receptor-dependent weakening of synaptic connections during ripples [57] provides such a mechanism. It seems that precise timing of reactivation in hippocampal sub-regions during ripples plays an important role determining whether a connection is potentiated or not [40], whereby, these findings suggest potential mechanisms for simultaneous selective strengthening of relevant traces and weakening of irrelevant traces. However, currently it remains unclear what factors determine the direction of plasticity.
This set of studies demonstrates that active systems consolidation of declarative memory during sleep likely recruits a mode of plasticity not essentially relying on glutamatergic transmission in hippocampo-cortical networks, and, in this way, contrasts with learning during wakefulness, where glutamatergic LTP in the hippocampus has been established as the major correlate of encoding traces. This might be due to the unique neuromodulatory and neurooscillatory environment offered by deep NonREM sleep. It is therefore important to acknowledge that sleep does not simply augment processes involved in the formation of memory traces but transforms them. In this sense, the recent finding that sharp wave-ripples depotentiate synaptic connections in the hippocampus and that blocking NMDA receptors impairs this process [57] and the broad evidence for a transfer of memory traces to the cortex during sharp wave-ripples (e.g., ref. [41]) offers the intriguing possibility that replay supports both active systems consolidation and homeostatic needs in the hippocampus. In this framework, memories are being wiped little by little from the hippocampus at the rate at which they are being established in the neocortical networks [69]. To shed light onto this, studying the intracellular signaling cascades that translate neurotransmitter signals into plastic changes will be essential [1, 94]. For example, eliciting LTP at a synapse primes downstream processes to simultaneously prevent long-term depression at that synapse via a glycogen synthase kinase 3 beta-related pathway within the post-synaptic neuron [95]. Such a metaplastic process could be used by replay in a switch like manner to tag individual synapses for increased or reduced downscaling during sleep.
GABAergic neuromodulation
GABA, the main inhibitory neurotransmitter of the brain, contributes to all of the major sleep EEG rhythms and GABAergic neurons, mainly in the ventrolateral preoptic area, that inhibit arousal centers in the brainstem, hypothalamus, and basal forebrain have been identified as crucial for sleep induction and maintenance [96]. Correspondingly, GABA A receptors have early on been targeted to enhance sleep in disorders such as insomnia, however, so far none of the drugs has been without drawbacks such as un-physiological sleep [97]. In this regard, it is possible to differentiate between GABA A-positive modulators and GABA A agonists, the former enhancing more shallow NonREM sleep and sleep spindle activity at the expense of SWS and REM sleep. Whereas, the latter enhance SWS and associated slow wave activity at the cost of other sleep stages [98, 99].
In active systems consolidation, the primary oscillations of NonREM sleep take an important role of coordinating the hippocampo-neocortical dialog [36, 100,101,102]. Briefly, slow oscillatory activity (<1 Hz) that consists of down-states with widespread cortical silence and up-states of wake-like activation coordinate with fast cortical spindles (12–15 Hz) that occur most frequently during the down-to-up-state transition of the slow oscillation [103] and enhance cortical plasticity through the influx of calcium into pyramidal cells ([104, 105], see Fig. 3). Sharp wave-ripple-associated replay that rides on the troughs of the sleep spindle thus arrives at the cortex in an ideal environment to exert plastic changes [37,38,39, 41, 106]. This fine-tuning of oscillations is thought to play a crucial role for delivering sleep’s beneficial effects and, correspondingly, disrupting these activity patterns by inhibiting parvalbumin-positive interneurons in hippocampal circuitry interferes with sleep-dependent memory consolidation [107, 108]. Rhythmic stimulation of these neurons can even reverse the detrimental effects of sleep-deprivation on memory [74].

GABAergic system interactions with sleep oscillations. The tightly linked cardinal oscillations observed during sleep are essential for memory consolidation. The neocortical slow oscillation plays the role of a clock that drives the hippocampal sharp-waves/ripples to occur during sleep spindle troughs. Spindle-ripple events are thus time-locked to occur during the up-state of the slow oscillation, when excitation of the cortex is facilitated. Additionally, reactivation of memories that coincides with ripples can thereby profit from plasticity promoting properties of spindles, such as increases in intracellular calcium activity, to strengthen cortical memory traces. GABAergic drugs influence the generation and the interplay of these oscillations. The GABA reuptake inhibitor tiagabine enhances activity of the system tonically and thereby strongly enhances the amount of slow wave activity, including slow oscillations observed during NonREM sleep. However, it also leads to a reduced spindle to slow oscillation coupling, which explains that this boost does not improve memory consolidation. Zolpidem, a GABA A-positive modulator, on the other hand acts in a phasic manner, since enhanced receptor action must first be activated by GABA excretion. It strongly enhances the amount of sleep spindles and, concomitantly, enhances declarative memory consolidation
Sleep-dependent declarative memory consolidation can be enhanced by applying transcranial direct current or alternating current electric stimulation that mimics slow oscillatory activity [109, 110]. Therefore, to pharmacologically boost memory consolidation a recent experiment used the GABA reuptake inhibitor tiagabine, which is known to enhance SWS and associated slow wave activity (0.5–2 Hz) by tonically activating GABA receptors [99, 111, 112]. During learning, participants encoded a declarative word-pair task as well as a procedural finger sequence tapping task and then received the drug orally before going to bed [113]. The next evening retrieval was tested, when the drug had washed out. Although we robustly replicated a stark increase in SWS and even found strongly increased slow oscillatory activity, we did not find any behavioral evidence for improved memory retention. In fact, procedural memory consolidation was even significantly impaired. This effect was likely due to a decrease in fast and slow sleep spindles and disruption of sleep spindle-to-slow oscillation coupling that was also evident in the treatment condition. In addition, the endogenous signal from inhibitory neuronal populations that drive plasticity during sleep may have been disrupted by the increased GABAergic tone [114].
Sleep spindles have been found consistently to be linked to declarative and procedural memory consolidation [115,116,117,118,119] and this link is further underlined by another experiment using a GABAergic agent. Mednick et al. [120] used the GABA A receptor-positive modulator zolpidem that is known to enhance fast spindle activity during NonREM sleep [98]. Participants learned a declarative word-pair task and a procedural finger sequence tapping task before receiving the drug. The drug positively affected the amount of word-pairs retained and this effect was directly related to the enhancement of sleep spindles induced by zolpidem. This effect is likely due to the strong memory enhancing effects of spindles [121], especially when they occur during the slow oscillation upstate [122].
These studies demonstrate that care must be taken, when developing sleep-inducing drugs not to interfere with the intricate pattern of oscillations that drive important processes during sleep.
Dopaminergic neuromodulation
Dopamine plays a central role for reward-related memory processing [123, 124], dopaminergic imbalances contribute to several neuropsychiatric disorders, including schizophrenia and depression [125], and reward-related learning mechanisms are central in the pathogenesis of addiction [126]. For declarative memory formation, the hippocampus-ventral-striatum-ventral-tegmental-area-hippocampus feedback loop plays a crucial role, where dopaminergic input from the ventral tegmental area (VTA) to the hippocampus—as a reward signal—effectively gates what enters long-term memory [127]. In their seminal paper, Adcock et al. [128] asked participants to learn a motivated learning task that promised an item specific amount of reward for successful recognition 1 day later. As learning took place in a magnetic resonance imaging (MRI) scanner, they were able to show that coordinated activity between the hippocampus and the ventral striatum, as well as functional connectivity between the two areas predicts whether items are recognized 1 day later.
It is important to consider whether this circuitry is recruited exclusively during learning and reward contingencies are no longer available to alter consolidation during sleep. Paradigms showing that rewards can affect memory retroactively over a 24 h interval, if related information is rewarded after learning, provide evidence against this idea [129,130,131]. Importantly, such a boosting effect of retro-active rewards on procedural memory can even be elicited, if the reward simply is promised after encoding as long as participants sleep during the retention interval [132] and a beneficial effect of sleep on reward-related memory is also observed in the declarative domain [67, 68]. However, there are also accounts of negative reinforcement impairing sleep-dependent memory consolidation [133], no effect of rewards over the SWS-rich first half of the night [134] and reward information generalizing across a period of sleep [135], rendering the role of reward for sleep-dependent memory consolidation less consistent.
There exist two competing accounts, how sleep may selectively benefit high-reward memories (Fig. 4). Studies using simultaneous electrophysiological recordings could show that hippocampal replay precedes and likely drives replay of motivationally relevant patterns in the ventral striatum [136, 137]. Evidence for sleep replay also occurring in the ventral tegmental area [138] offers the possibility that replay in the hippocampus accesses dopaminergic neuromodulation during sleep via a feedback loop that acts similar to wake. Alternatively, highly relevant memories may be tagged by a dopaminergic process during learning that simply enhances replay of these memories during sleep without further dopaminergic involvement. Evidence for this account comes from a study where optogenetically enhancing dopaminergic inputs to the hippocampus during maze learning did not alter initial learning performance [139]. However, replay of task-related cells was enhanced during post training sleep and crucially post sleep memory performance was enhanced by this manipulation.

Dopaminergic neuromodulation for reward-related memories. During sleep-dependent memory consolidation, relevant (i.e., rewarded) memories are consolidated better than less relevant memories. In our example, it may be highly relevant to remember that Jane likes to come to the beach, as you want to meet her again. During wake encoding, selective memory enhancement of reward-related information is thought to be established through dopaminergic neuromodulation from the VTA onto hippocampal plasticity. Extending this framework to sleep, a reward circuitry perspective explains selective consolidation by hippocampal reactivation driving reactivation in the reward centers of the brain (ventral striatum and VTA) that lead to a dopaminergic neuromodulatory signal enhancing neuroplasticity in the hippocampus via a feedback loop. However, dopamine also plays a major wake-promoting role and dopamine interventions have led to mixed results. Alternatively, relevance may lead to stronger consolidation by enhancing the reactivation probability of some memories over others through mechanisms already effective during encoding (magenta arrows from hippocampus to neocortex). This would allow the strengthening of rewarded memories without the recruitment of dopaminergic activity during sleep, but does necessitate that some form of dopamine-dependent tag is set during learning. In future, other reward-related neurotransmitter systems may be shown to also play an important role, such as the endocannabinoid system
The question of dopamine involvement during sleep was investigated in humans using a variant of the motivated learning task mentioned above [128] and pramipexole a dopamine d2-like receptor agonist [140]. Dopamine d2-like receptors are expressed post-synaptically in the hippocampus and regulate plasticity [141], but are also expressed as pre-synaptic auto-receptors regulating dopamine secretion [142]. In the task, participants had to learn pictures of interiors and landscapes, which were preceded by a symbol signaling a high or low reward for later successful retrieval. Importantly, pictures were shown for a short or a long duration thereby effectively controlling for encoding strength. Before sleeping, after learning, participants ingested pramipexole and recognition was tested the next evening. In the placebo condition, recognition was better for highly rewarded pictures than for lowly rewarded ones, as well as for long duration pictures versus short duration pictures. In the treatment condition, the advantage of high over low reward was wiped out, whereas, the benefit of encoding strength persisted. This result demonstrates that it is possible to influence the selectivity of sleep-dependent consolidation through dopaminergic interventions that can shift consolidation from highly to less relevant items.
We have recently followed up this work to investigate whether dopamine is necessary for this effect by using the dopamine d2-like receptor blocker sulpiride ([143], bioRxiv). Otherwise relying on the exact same procedures, we found that blocking dopaminergic neurotransmission with a dose of sulpiride that has been shown to block ~60% of d2-like receptors [144] did not affect the highly over lowly rewarded memory advantage of sleep. This means that using an antagonist instead of an agonist of the d2-like dopamine receptor did not reverse the effects found initially. While this does not exclude effects mediated by d1-like dopamine receptors, it may indicate that dopamine plays a less crucial role for selective consolidation of rewarded information during sleep and emphasizes the likelihood of a dopaminergic mechanism during learning that tags memories for sleep reactivation. Accordingly, it was found that wake replay in the VTA is more closely linked to reward-related task information than sleep replay in the VTA [145], offering the possibility that wake replay of dopaminergic afferents to the hippocampus is used to tag relevant memories for later dopamine-independent sleep replay. Importantly, these findings cannot be explained by replay frequency being merely affected by memory strength rather than relevance, since more weakly encoded memories are in fact more likely to be replayed during rest [146] and benefit more from sleep [147].
Overall, it seems most likely that reward-related specificity of active systems consolidation is mediated via an up- or downregulation of reactivation frequency rather than a recruitment of dopaminergic neuromodulation. In this scenario, dopaminergic activity during learning would tag high-reward memories for preferential reactivation during later active systems consolidation periods. Such a tag could be established similar to the mechanism described for synaptic potentiation in the synaptic tagging and capture hypothesis [148]. One could also speculate on the possibility that reward modulation is passed to other less well-studied neurotransmitter systems, such as the endocannabinoid system, during sleep. This lipid molecule signaling pathway is intricately linked to stress and sleep regulation [149, 150], but also is involved in processes of metaplasticity in the hippocampus [151, 152] and reward processing [153]. The role of this pathway in sleep-dependent consolidation needs to be examined.
Acetylcholine
Among the neurotransmitters of the brain, acetylcholine (ACh) plays an important and possibly the best-characterized role for active systems consolidation. It was early on speculated that high levels of ACh are critical to enable learning by allowing information to flow from the cortex to the hippocampus, where associations can be bound together [34], also based on experimental evidence in humans showing that blocking muscarinergic ACh receptors immediately after learning word lists impaired later performance [154]. Low levels during sleep on the other hand are important for consolidation, as they reverse the direction of information flow. Essentially, cholinergic tone has been suggested to determine the direction of the hippocampal to neocortical dialog that is critical for transfer of declarative memory in the active systems consolidation framework [35, 155]. However, cholinergic processes that influence plasticity and network properties are diverse [156], so that future research should attempt to more directly link acetylcholine to the direction of information flow in the hippocampo-cortical system.
For causal evidence of an involvement of cholinergic tone in sleep-dependent memory consolidation, Gais et al. used the cholinesterase inhibitor physostigmine to increase cholinergic tone during early SWS-rich retention sleep, after participants had learned a hippocampus-dependent declarative word-pair task and a procedural mirror tracing task [157]. The increased cholinergic tone specifically disrupted the retention of word-pairs while leaving procedural mirror tracing unaffected and this effect was not observed, if participants stayed awake during retention. This finding highlights the importance of low acetylcholine during the first half of the night for successful active systems consolidation.
More recent evidence of the effect of blocking muscarinergic ACh receptors with scopolamine on declarative learning showed that it impairs paired associate learning and increases effects of proactive interference [158]. Further, an experiment that simultaneously blocked nicotinergic and muscarinergic ACh receptors using mecamylamin and scopolamine, respectively, reduced participants’ ability to encode new declarative memories while sparing procedural ones, which emphasizes the importance of high-cholinergic tone for new learning of hippocampus-dependent contents [159]. This intervention also enhanced consolidation of declarative content during wakefulness, which indicates that cholinergic tone can even allow successful consolidation without recruitment of full-blown SWS and its hallmark oscillations. In light of this clear evidence for a functionally important dip of acetylcholine during the first half of the night, it seems prudent to again suggest a revision of the still widely accepted recommendation of administering cholinesterase inhibitors at bedtime in Alzheimer’s patients [160].
Interestingly, a recent study using targeted memory reactivation to boost declarative memory found no detrimental influence of increasing cholinergic tone during sleep with physostigmine on the reactivation benefit [161]. In this study, participants learned a card-pair location task while smelling an associated odor, which was presented again during subsequent SWS and compared to a sham condition. One group of participants also received the cholinesterase inhibitor physostigmine. In contrast to expectation, increased cholinergic tone did not block the benefit of cued reactivation. This may indicate that exogenously targeted memory reactivation does not enhance active systems consolidation but rather acts on processes of memory strengthening within the hippocampus, which would strongly limit any interpretation of targeted memory reactivation studies. To this end, it has recently been modeled that the hippocampus might even be able to support a form of systems consolidation within its own networks [162].
Cortisol
The adrenal gland hormone cortisol plays a similarly complex role for the different phases of memory formation. It circulates within the blood and enters the brain [163], where it acts especially in the hippocampus as a neuromodulatory transmitter that affects plasticity in an inverted u-shape manner with medium levels of corticosterone (the homolog for cortisol in the rat) eliciting maximum potentiation [164, 165]. Its plasma concentration follows a circadian rhythm of low concentration during the first half of the night, increasing concentration across the second and a peak after awakening, to then decline again across the day [166]. SWS suppresses cortisol release [167] and acute psycho-social stressors facilitate it [168, 169]. Enhanced levels of cortisol at learning or shortly thereafter can increase retention especially of emotional content [170, 171], whereas, enhanced levels at retrieval impair the recall of emotional content [172, 173]. Interestingly, rather than a linear effect of cortisol on retrieval it seems that again medium amounts are ideal and cortisol action follows an inverted u-shape [174, 175]. This effect is likely due to different cortisol affinities of the two cortisol receptors of the brain. In line with this, it has been shown that blocking mineralocorticoid receptors impairs memory retrieval in humans, whereas blocking glucocorticoid receptors enhances it [176].
Regarding retention, increased cortisol levels during learning seem to mediate the access of emotional declarative material to processes of consolidation during sleep but not during wakefulness [177] and this effect of high cortisol during learning is reversed for procedural memory consolidation during sleep [178]. During sleep, the precise level of minimal cortisol during the first SWS-rich half of the night seems to be critical. An early study administered the glucocorticoid receptor agonist dexamethasone in a split night paradigm to mimic increased levels of cortisol [179]. This treatment prevented consolidation of declarative word-pair memory but did not affect non-declarative visomotor skills. This relationship again seems to follow an inverted u-shape, as the administration of the cortisol synthesis inhibitor metyrapone, producing minimum cortisol levels, impaired the consolidation of neutral declarative memories [180]. Indicating that the precise low level of cortisol found during SWS is essential for active systems consolidation. In this regard, it is interesting to mention a pharmacological study in rats using direct infusion of corticosterone into the hippocampus during retention [181]. Mirroring findings in the cholinergic system, the infusion blocked consolidation of an object place recognition task (an equivalent of a declarative task in humans) during sleep, but enhanced consolidation during the wake state. This indicates that sleep and wake consolidation seem to be regulated by glucocorticoid-related processes with low levels of cortisol possibly facilitating the hippocampal cortical dialog ([182, 183]) critical to active systems consolidation. Overall, the diverse effects of cortisol on memory formation are relatively well understood, which can be attributed to careful consideration of effects during different arousal states, internal rhythms and memory phases. In light of the suggestion of using cortisol treatments in clinical settings, e.g., to prevent post-traumatic stress disorder by administering hydrocortisone after acute trauma [184], it should be emphasized that the exact timing and dosing of any such therapy will be of the upmost importance.
Neuropsychiatric implications
From a sleep research point of view, it is generally very interesting that neuropsychiatric disorders often come hand in hand with sleep disorders. For example, schizophrenic patients show a marked reduction in sleep spindle activity during NonREM sleep [185] and, correspondingly, boosting SWS with transcranial electric stimulation has been shown to normalize memory performance in these patients [186]. This relationship seems even more significant in disorders, where behavioral therapy that relies on new learning and thus memory formation to transform behavior is a key technique for treatment [187], such as in anxiety [188], depression [189], and addiction [190]. Therefore, in these cases, it may be sensible to treat comorbid sleep disturbances before attempting to treat the primary disorder to enhance the efficacy of cognitive behavior therapy. Improving sleep is even more relevant for Alzheimer’s disease, where a direct causal link between disturbed sleep and disease pathology is becoming more and more clear ([191], also covered in this issue of NPPR in detail). Next to this general principle, there exist further specific insights that link sleep-dependent memory consolidation to neuropsychiatric disorders.
Phobias
Specific phobias that entail being afraid of and avoiding certain objects or places are among the most prevalent anxiety disorders [192, 193]. Their conceptualization as a consequence of learned behavior goes back to John Watson and his (in)famous experiments with little Albert [194] and the two process model introduced by Orval Mowrer [195]. Briefly, this theory assumes that fearing an object or place is learned through classical conditioning and operant conditioning maintains this fear through avoidance of the fearful object or place that acts as negative reinforcement. Although there exists some debate about the precise nature of the learning procedures involved, it is commonly agreed that exposure therapy that tries to extinguish the fear memory is among the most powerful therapeutic options for this form of anxiety disorder [196]. In a landmark study, Hauner et al. [197] showed that fear memories can be extinguished during sleep. Here, participants underwent contextual fear conditioning, either while smelling the scent of oranges or pine trees. Only one of the odors was presented again during subsequent SWS and participants showed stimulus-specific fear extinction the next day, which putatively resulted from cuing the fear memory at a time, i.e., NonREM sleep, when the associated emotional arousal is diminished or even entirely suppressed. This may be a promising therapeutic option in patients that are overwhelmed by conventional exposure therapy, however, finding adequate stimuli to present during sleep poses a significant challenge. Although another study in humans found similar effects [198], there also exists some contradictory evidence from animal work regarding extinction during sleep [199, 200] and so far attempts at directly cueing exposure therapy sessions with associated odors has failed [23]. Possibly, a more promising avenue to utilize sleep’s memory processing capacity would be to use drugs such as D-cycloserine that have been shown to augment exposure therapy [201]. In fact, our finding of enhanced memory consolidation by D-cycloserine during sleep [86] may open a therapeutic window wherein only successful exposure therapy sessions could be boosted by administration during the subsequent night, leaving failures to be forgotten [202].
Depression
It has long been known that patients suffering major depression are biased towards remembering negative events [203], and it has been suggested that a competition between negative and positive memories may lie at the root of this disorder, as well as its therapeutic improvement [204]. In this regard, sleep and especially REM sleep have been suggested to play an important role for the preferential consolidation of emotional memory [205,206,207]. In an important contribution to this area, Payne et al. [64] were able to show that emotional scenes selectively profit from sleep. Participants’ memory for emotional items was enhanced at the cost of neutral backgrounds on which they were placed. Interestingly, the consolidation process seems to strip the emotional memories from their arousal and valence features, making them seem more similar to neutral memories regarding the activation of emotion processing networks in the brain [65]. This has led to the proposal that REM sleep may perform an important function that allows the brain to store information about emotional events without the emotional tone [208]. An alternative view proposes that REM sleep increases the immediate (i.e., as indicated by measures of the autonomic nervous system) emotional response, while SWS enhances top-down control over the elicited emotions, possibly by strengthening cortical traces through active systems consolidation [209]. This view fits with the REM sleep suppressive effects found in most antidepressants [210], as well as the prevalence of sleep disturbance in depressed patients especially the dysregulation of REM sleep [211], which may indicate that this process is a prominent driver of the negative memory bias in depressed patients. In any case, this opens the possibility that directly targeting memory consolidation during REM sleep pharmacologically rather than coincidentally through a side effect could be an effective adjunct therapy in depression.
Addiction
Similar to anxiety, a two process theory of drug addiction has been developed [126, 212], where the positive effects of the substance are connected to the context of consuming it through classical conditioning. The drug taking context then takes on the role of a secondary reinforcer and cues drug seeking behavior. It has been shown that exposure therapy can effectively attenuate neural cue-reactivity to substance stimuli [213], which opens the intriguing possibility of exposing addicts undergoing rehabilitation to drug-specific odor stimuli during sleep to extinguish their effect. Distinct substance-specific odors are likely easier to identify here than in anxiety disorders, e.g., the most frequently abused substance alcohol has a distinct smell, whereas spiders do not. Another possible route of therapy could use classical conditioning of unconditioned stimuli to shape behavior and some evidence has become available that this very low-level type of learning can be achieved during human sleep [214]. In fact, such a paradigm has already been used successfully in smokers to pair unpleasant smells with the smell of cigarettes during sleep, which reduced smoking behavior [215]. These therapeutic strategies could be accompanied by pharmacological interventions targeting the dopaminergic system aimed at boosting alternative behavior.
In sum, sleep’s memory reprocessing offers a so far underused window of opportunity to treat neuropsychiatric disorders. A precise knowledge of the mechanisms involved and especially the unique plastic processes governing sleep-dependent consolidation will be indispensable when attempting this.
Future directions and clinical implications
While significant progress has been made in deciphering the neurochemical mechanisms of sleep-dependent memory processing, the sheer complexity and the vast difference to wake encoding machinery is only beginning to be appreciated. Pharmacological research in humans will remain of essence to identify the mechanisms without the burdensome and often impossible work of translation. However, increasing regulatory requirements and extremely high costs may make this work less attractive than other lines of neuroscientific inquiry in the field of human sleep and memory research. In addition, animal research will have to complement the human work to make use of substances not available in humans and to rule out redundant pathways. For example, with respect to the role of glutamatergic transmission, it would be very informative to block NMDA- and mGlu5-receptors simultaneously to rule out that either takes over the function of the other. However, this requires carefully weighing whether the experiments can truly do without harnessing the complexity of human behavior. Correspondingly, animal in-vitro work will have to consider how it can model sleep’s-specific neurochemical and neurooscillatory milieu, else it risks painting a completely false picture of plasticity during sleep.
To end on a more positive note: the research presented here already offers several attractive candidates to develop applied interventions. In addition, targeted memory reactivation paradigms could be elegantly combined with pharmacological interventions to fine tune long-term memory stores, erasing unwanted maladaptive memories and replacing them with alternatives. It is important to acknowledge that sleep is not merely an idle state that can, e.g., be used to minimize the experience of cognitive side effects of pharmacological treatment, as is evidenced by the misguided recommendation to administer cholinesterase inhibitors before bedtime so that nausea and vertigo do not disturb patients during waking hours. Such a new perspective may also reinvigorate the development of novel neuropharmacological drugs that currently seems caught in a rut.
Funding and disclosure
G.B.F. is currently receiving an Emmy-Noether-Research-Grant (FE 1617/2-1) from the German Research Foundation (DFG). The research presented here was partly funded by a DFG grant to J.B. (SFB 654—Plasticity and Sleep). The authors declare no competing interests.
References
Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol. 2013;23:R774–88. https://doi.org/10.1016/j.cub.2013.07.025
Rasch B, Born J. About sleep’s role in memory. Physiol Rev. 2013;93:681–66. https://doi.org/10.1152/physrev.00032.2012
Stickgold R. Sleep-dependent memory consolidation. Nature. 2005;437:1272–8. https://doi.org/10.1038/nature04286
Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. https://doi.org/10.1016/j.neuron.2013.12.025
McGaugh JL. Memory–a century of consolidation. Science. 2000;287:248–51.
Müller GE, Pilzecker A. Experimentelle Beiträge zur Lehre vom Gedächtnis. Z für Psychol, Ergänzungsband. 1900;1:1–300.
Jenkins JG, Dallenbach KM. Obliviscence during sleep and waking. Am J Psychol. 1924;35:605–12.
Ebbinghaus H. Über das Gedächtnis. Leipzig: Duncker und Humblot; 1885.
Murre JM, Dros J. Replication and analysis of Ebbinghaus’ forgetting curve. PLoS ONE. 2015;10:e0120644. https://doi.org/10.1371/journal.pone.0120644
Dement W, Kleitman N. Cyclic variations in EEG during sleep and their relation to eye movements, body motility, and dreaming. Electro Clin Neurophysiol. 1957;9:673–90.
Aserinsky E, Kleitman N. Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science. 1953;118:273–4.
O’Keefe J. Place units in the hippocampus of the freely moving rat. Exp Neurol. 1976;51:78–109.
Eichenbaum H, Dudchenko P, Wood E, Shapiro M, Tanila H. The hippocampus, memory, and place cells: is it spatial memory or a memory space? Neuron. 1999;23:209–26.
Pavlides C, Winson J. Influences of hippocampal place cell firing in the awake state on the activity of these cells during subsequent sleep episodes. J Neurosci. 1989;9:2907–18.
Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–9.
Rasch B, Buchel C, Gais S, Born J. Odor cues during slow-wave sleep prompt declarative memory consolidation. Science. 2007;315:1426–9. https://doi.org/10.1126/science.1138581
Diekelmann S, Buchel C, Born J, Rasch B. Labile or stable: opposing consequences for memory when reactivated during waking and sleep. Nat Neurosci. 2011;14:381–6. https://doi.org/10.1038/nn.2744
Rudoy JD, Voss JL, Westerberg CE, Paller KA. Strengthening individual memories by reactivating them during sleep. Science. 2009;326:1079. https://doi.org/10.1126/science.1179013
Schreiner T, Rasch B. Boosting vocabulary learning by verbal cueing during sleep. Cereb Cortex. 2014. https://doi.org/10.1093/cercor/bhu139
Schönauer M, Geisler T, Gais S. Strengthening procedural memories by reactivation in sleep. J Cogn Neurosci. 2014;26:143–53. https://doi.org/10.1162/jocn_a_00471
Antony JW, Gobel EW, O’Hare JK, Reber PJ, Paller KA. Cued memory reactivation during sleep influences skill learning. Nat Neurosci. 2012;15:1114–6. https://doi.org/10.1038/nn.3152
Diekelmann S, Biggel S, Rasch B, Born J. Offline consolidation of memory varies with time in slow wave sleep and can be accelerated by cuing memory reactivations. Neurobiol Learn Mem. 2012;98:103–11. https://doi.org/10.1016/j.nlm.2012.07.002
Rihm JS, Sollberger SB, Soravia LM, Rasch B. Re-presentation of olfactory exposure therapy success cues during non-rapid eye movement sleep did not increase therapy outcome but increased sleep spindles. Front Hum Neurosci. 2016;10:340 https://doi.org/10.3389/fnhum.2016.00340
Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–26. https://doi.org/10.1038/nrn2762
Sawangjit A, Oyanedel CN, Niethard N, Salazar C, Born J, Inostroza M. The hippocampus is crucial for forming non-hippocampal long-term memory during sleep. Nature. 2018;564:109–13. https://doi.org/10.1038/s41586-018-0716-8
Westermann J, Lange T, Textor J, Born J. System consolidation during sleep—a common principle underlying psychological and immunological memory formation. Trends Neurosci. 2015;38:585–97. https://doi.org/10.1016/j.tins.2015.07.007
Grossberg S. How does a brain build a cognitive code? Psychol Rev. 1980;87:1–51.
Grossberg S. Competitive learning: From interactive activation to adaptive resonance. Cogn Sci. 1987;11:23–63.
Dudai Y. The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol. 2004;55:51–86. https://doi.org/10.1146/annurev.psych.55.090902.142050
McClelland JL, McNaughton BL, O’Reilly RC. Why there are complementary learning systems in the hippocampus and neocortex: insights from the successes and failures of connectionist models of learning and memory. Psychol Rev. 1995;102:419–57.
Battaglia FP, Benchenane K, Sirota A, Pennartz CM, Wiener SI. The hippocampus: hub of brain network communication for memory. Trends Cogn Sci. 2011;15:310–8. https://doi.org/10.1016/j.tics.2011.05.008
Bontempi B, Laurent-Demir C, Destrade C, Jaffard R. Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature. 1999;400:671–5. https://doi.org/10.1038/23270
Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–30. https://doi.org/10.1038/nrn1607
Hasselmo ME. Neuromodulation: acetylcholine and memory consolidation. Trends Cogn Sci. 1999;3:351–9.
Mitra A, Snyder AZ, Hacker CD, Pahwa M, Tagliazucchi E, Laufs H, et al. Human cortical-hippocampal dialogue in wake and slow-wave sleep. Proc Natl Acad Sci USA. 2016. https://doi.org/10.1073/pnas.1607289113
Sirota A, Csicsvari J, Buhl D, Buzsaki G. Communication between neocortex and hippocampus during sleep in rodents. Proc Natl Acad Sci USA. 2003;100:2065–9. https://doi.org/10.1073/pnas.0437938100
Staresina BP, Bergmann TO, Bonnefond M, van der Meij R, Jensen O, Deuker L, et al. Hierarchical nesting of slow oscillations, spindles and ripples in the human hippocampus during sleep. Nat Neurosci. 2015;18:1679–86. https://doi.org/10.1038/nn.4119
Clemens Z, Mölle M, Eross L, Barsi P, Halasz P, Born J. Temporal coupling of parahippocampal ripples, sleep spindles and slow oscillations in humans. Brain. 2007;130:2868–78. https://doi.org/10.1093/brain/awm146
Clemens Z, Mölle M, Eross L, Jakus R, Rasonyi G, Halasz P, et al. Fine-tuned coupling between human parahippocampal ripples and sleep spindles. Eur J Neurosci. 2011;33:511–20.
Sadowski JH, Jones MW, Mellor JR. Sharp-wave ripples orchestrate the induction of synaptic plasticity during reactivation of place cell firing patterns in the hippocampus. Cell Rep. 2016;14:1916–29. https://doi.org/10.1016/j.celrep.2016.01.061
Khodagholy D, Gelinas JN, Buzsaki G. Learning-enhanced coupling between ripple oscillations in association cortices and hippocampus. Science. 2017;358:369–72. https://doi.org/10.1126/science.aan6203
Ji D, Wilson MA. Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat Neurosci. 2007;10:100–7. https://doi.org/10.1126/science.8036517
Ognjanovski N, Maruyama D, Lashner N, Zochowski M, Aton SJ. CA1 hippocampal network activity changes during sleep-dependent memory consolidation. Front Syst Neurosci. 2014;8:61. https://doi.org/10.3389/fnsys.2014.00061
Stella F, Baracskay P, O’Neill J, Csicsvari J. Hippocampal reactivation of random trajectories resembling Brownian diffusion. Neuron. 2019;102:450–61 e457. https://doi.org/10.1016/j.neuron.2019.01.052
Giri B, Miyawaki H, Mizuseki K, Cheng S, Diba K. Hippocampal reactivation extends for several hours following novel experience. J Neurosci. 2019;39:866–75. https://doi.org/10.1523/JNEUROSCI.1950-18.2018
Hebb DO. The organization of behavior: a neuropsychological theory. New York, NY: Wiley; 1949.
Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. https://doi.org/10.1016/j.smrv.2005.05.002
Cirelli C, Tononi G. Is sleep essential? PLoS Biol. 2008;6:e216. https://doi.org/10.1371/journal.pbio.0060216
Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–8. https://doi.org/10.1038/nn2035
Maret S, Faraguna U, Nelson AB, Cirelli C, Tononi G. Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nat Neurosci. 2011;14:1418–20. https://doi.org/10.1038/nn.2934
Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, Huganir RL. Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science. 2017;355:511–5. https://doi.org/10.1126/science.aai8355
de Vivo L, Bellesi M, Marshall W, Bushong EA, Ellisman MH, Tononi G, et al. Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science. 2017;355:507–10. https://doi.org/10.1126/science.aah5982
Kuhn M, Wolf E, Maier JG, Mainberger F, Feige B, Schmid H, et al. Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat Commun. 2016;7:12455. https://doi.org/10.1038/ncomms12455
Mander BA, Santhanam S, Saletin JM, Walker MP. Wake deterioration and sleep restoration of human learning. Curr Biol. 2011;21:R183–4. https://doi.org/10.1016/j.cub.2011.01.019
Van Der Werf YD, Altena E, Schoonheim MM, Sanz-Arigita EJ, Vis JC, De Rijke W, et al. Sleep benefits subsequent hippocampal functioning. Nat Neurosci. 2009;12:122–3. https://doi.org/10.1038/nn.2253
Antonenko D, Diekelmann S, Olsen C, Born J, Molle M. Napping to renew learning capacity: enhanced encoding after stimulation of sleep slow oscillations. Eur J Neurosci. 2013;37:1142–52. https://doi.org/10.1111/ejn.12118
Norimoto H, Makino K, Gao M, Shikano Y, Okamoto K, Ishikawa T, et al. Hippocampal ripples down-regulate synapses. Science. 2018;359:1524–7. https://doi.org/10.1126/science.aao0702
Li W, Ma L, Yang G, Gan WB. REM sleep selectively prunes and maintains new synapses in development and learning. Nat Neurosci. 2017;20:427–37. https://doi.org/10.1038/nn.4479
Yang G, Lai CS, Cichon J, Ma L, Li W, Gan WB. Sleep promotes branch-specific formation of dendritic spines after learning. Science. 2014;344:1173–8. https://doi.org/10.1126/science.1249098
Frank MG, Erasing Synapses in Sleep: Is It Time to Be SHY? Neural Plasticity 2012;2012:1–15.
Puentes-Mestril C, Aton SJ. Linking network activity to synaptic plasticity during sleep: hypotheses and recent data. Front Neural Circuits. 2017;11:61. https://doi.org/10.3389/fncir.2017.00061
Wilhelm I, Diekelmann S, Molzow I, Ayoub A, Molle M, Born J. Sleep selectively enhances memory expected to be of future relevance. J Neurosci. 2011;31:1563–9. https://doi.org/10.1523/JNEUROSCI.3575-10.2011
Payne JD, Chambers AM, Kensinger EA. Sleep promotes lasting changes in selective memory for emotional scenes. Front Integr Neurosci. 2012;6:108. https://doi.org/10.3389/fnint.2012.00108
Payne JD, Stickgold R, Swanberg K, Kensinger EA. Sleep preferentially enhances memory for emotional components of scenes. Psychol Sci. 2008;19:781–8. https://doi.org/10.1111/j.1467-9280.2008.02157.x
Sterpenich V, Albouy G, Boly M, Vandewalle G, Darsaud A, Balteau E, et al. Sleep-related hippocampo-cortical interplay during emotional memory recollection. PLoS Biol. 2007;5:e282. https://doi.org/10.1371/journal.pbio.0050282
Sterpenich V, Albouy G, Darsaud A, Schmidt C, Vandewalle G, Dang Vu TT, et al. Sleep promotes the neural reorganization of remote emotional memory. J Neurosci. 2009;29:5143–52. https://doi.org/10.1523/JNEUROSCI.0561-09.2009
Studte S, Bridger E, Mecklinger A. Sleep spindles during a nap correlate with post sleep memory performance for highly rewarded word-pairs. Brain Lang. 2017;167:28–35. https://doi.org/10.1016/j.bandl.2016.03.003
Igloi K, Gaggioni G, Sterpenich V, Schwartz S. A nap to recap or how reward regulates hippocampal-prefrontal memory networks during daytime sleep in humans. Elife. 2015;4. ARTN e0790310.7554/eLife.07903
Feld GB, Born J. Sculpting memory during sleep: concurrent consolidation and forgetting. Curr Opin Neurobiol. 2017;44:20–7. https://doi.org/10.1016/j.conb.2017.02.012
Chauvette S, Seigneur J, Timofeev I. Sleep oscillations in the thalamocortical system induce long-term neuronal plasticity. Neuron. 2012;75:1105–13. https://doi.org/10.1016/j.neuron.2012.08.034
Durkin J, Aton SJ. Sleep-dependent potentiation in the visual system is at odds with the synaptic homeostasis hypothesis. Sleep. 2016;39:155–9. https://doi.org/10.5665/sleep.5338
Durkin J, Suresh AK, Colbath J, Broussard C, Wu J, Zochowski M, et al. Cortically coordinated NREM thalamocortical oscillations play an essential, instructive role in visual system plasticity. Proc Natl Acad Sci USA. 2017;114:10485–90. https://doi.org/10.1073/pnas.1710613114
Grosmark AD, Mizuseki K, Pastalkova E, Diba K, Buzsaki G. REM sleep reorganizes hippocampal excitability. Neuron. 2012;75:1001–7. https://doi.org/10.1016/j.neuron.2012.08.015
Ognjanovski N, Broussard C, Zochowski M, Aton SJ. Hippocampal network oscillations rescue memory consolidation deficits caused by sleep loss. Cereb Cortex. 2018;28:3711–23. https://doi.org/10.1093/cercor/bhy174
Gais S, Albouy G, Boly M, Dang-Vu TT, Darsaud A, Desseilles M, et al. Sleep transforms the cerebral trace of declarative memories. Proc Natl Acad Sci USA. 2007;104:18778–83. https://doi.org/10.1073/pnas.0705454104
Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. https://doi.org/10.1016/j.neuron.2004.09.012
Malenka RC, Nicoll RA. Long-term potentiation–a decade of progress? Science. 1999;285:1870–4.
Aton SJ, Seibt J, Dumoulin M, Jha SK, Steinmetz N, Coleman T, et al. Mechanisms of sleep-dependent consolidation of cortical plasticity. Neuron. 2009;61:454–66. https://doi.org/10.1016/j.neuron.2009.01.007
Gais S, Rasch B, Wagner U, Born J. Visual-procedural memory consolidation during sleep blocked by glutamatergic receptor antagonists. J Neurosci. 2008;28:5513–8. https://doi.org/10.1523/JNEUROSCI.5374-07.2008
Karni A, Sagi D. Where practice makes perfect in texture discrimination: evidence for primary visual cortex plasticity. Proc Natl Acad Sci USA. 1991;88:4966–70.
Gais S, Plihal W, Wagner U, Born J. Early sleep triggers memory for early visual discrimination skills. Nat Neurosci. 2000;3:1335–9. https://doi.org/10.1038/81881
Henke K, Weber B, Kneifel S, Wieser HG, Buck A. Human hippocampus associates information in memory. Proc Natl Acad Sci USA. 1999;96:5884–9.
Plihal W, Born J. Effects of early and late nocturnal sleep on declarative and procedural memory. J Cogn Neurosci. 1997;9:534–47. https://doi.org/10.1162/jocn.1997.9.4.534
Yaroush R, Sullivan MJ, Ekstrand BR. Effect of sleep on memory. II. Differential effect of the first and second half of the night. J Exp Psychol. 1971;88:361–6.
Ekstrand BR, Barrett TR, West JN, Maier WG. The effect of sleep on human long-term memory. In: Drucker-Colin R, McGaugh J, editors. Neurobiology of sleep and memory. New York, NY: Academic Press; 1977. p. 419–38.
Feld GB, Lange T, Gais S, Born J. Sleep-dependent declarative memory consolidation–unaffected after blocking NMDA or AMPA receptors but enhanced by NMDA coagonist D-cycloserine. Neuropsychopharmacology. 2013;38:2688–97. https://doi.org/10.1038/npp.2013.179
Hood WF, Compton RP, Monahan JB. D-cycloserine: a ligand for the N-methyl-D-aspartate coupled glycine receptor has partial agonist characteristics. Neurosci Lett. 1989;98:91–5.
Quartermain D, Mower J, Rafferty MF, Herting RL, Lanthorn TH. Acute but not chronic activation of the NMDA-coupled glycine receptor with D-cycloserine facilitates learning and retention. Eur J Pharm. 1994;257:7–12.
Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, et al. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology. 2009;34:2057–71. https://doi.org/10.1038/npp.2009.30
Chen HH, Liao PF, Chan MH. mGluR5 positive modulators both potentiate activation and restore inhibition in NMDA receptors by PKC dependent pathway. J Biomed Sci. 2011;18:19. https://doi.org/10.1186/1423-0127-18-19
Alizadeh Asfestani M, Braganza E, Schwidetzky J, Santiago J, Soekadar S, Born J, et al. Overnight memory consolidation facilitates rather than interferes with new learning of similar materials-a study probing NMDA receptors. Neuropsychopharmacology. 2018;43:2292–8. https://doi.org/10.1038/s41386-018-0139-0
Graf P, Schacter DL. Selective effects of interference on implicit and explicit memory for new associations. J Exp Psychol-Learn Mem Cogn. 1987;13:45–53. https://doi.org/10.1037//0278-7393.13.1.45
Hardt O, Nader K, Nadel L. Decay happens: the role of active forgetting in memory. Trends Cogn Sci. 2013;17:111–20. https://doi.org/10.1016/j.tics.2013.01.001
Havekes R, Meerlo P, Abel T. Animal studies on the role of sleep in memory: from behavioral performance to molecular mechanisms. Curr Top Behav Neurosci. 2015;25:183–206. https://doi.org/10.1007/7854_2015_369
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–17. https://doi.org/10.1016/j.neuron.2007.01.029
Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. https://doi.org/10.1038/nature04284
Gottesmann C. GABA mechanisms and sleep. Neuroscience. 2002;111:231–9.
Lancel M. Role of GABAA receptors in the regulation of sleep: initial sleep responses to peripherally administered modulators and agonists. Sleep. 1999;22:33–42.
Lancel M, Faulhaber J, Deisz RA. Effect of the GABA uptake inhibitor tiagabine on sleep and EEG power spectra in the rat. Br J Pharm. 1998;123:1471–7.
Molle M, Born J. Slow oscillations orchestrating fast oscillations and memory consolidation. Prog Brain Res. 2011;193:93–110. https://doi.org/10.1016/B978-0-444-53839-0.00007-7
Siapas AG, Wilson MA. Coordinated interactions between hippocampal ripples and cortical spindles during slow-wave sleep. Neuron. 1998;21:1123–8.
Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–106.
Mölle M, Bergmann TO, Marshall L, Born J. Fast and slow spindles during the sleep slow oscillation: disparate coalescence and engagement in memory processing. Sleep. 2011;34:1411–21.
Niethard N, Ngo HV, Ehrlich I, Born J. Cortical circuit activity underlying sleep slow oscillations and spindles. Proc Natl Acad Sci USA. 2018;115:E9220–9. https://doi.org/10.1073/pnas.1805517115
Seibt J, Richard CJ, Sigl-Glockner J, Takahashi N, Kaplan DI, Doron G, et al. Cortical dendritic activity correlates with spindle-rich oscillations during sleep in rodents. Nat Commun. 2017;8:684. https://doi.org/10.1038/s41467-017-00735-w
Latchoumane CV, Ngo HV, Born J, Shin HS. Thalamic spindles promote memory formation during sleep through triple phase-locking of cortical, thalamic, and hippocampal rhythms. Neuron. 2017;95:424–35 e426. https://doi.org/10.1016/j.neuron.2017.06.025
Ognjanovski N, Schaeffer S, Wu J, Mofakham S, Maruyama D, Zochowski M, et al. Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat Commun. 2017;8:15039. https://doi.org/10.1038/ncomms15039
Xia F, Richards BA, Tran MM, Josselyn SA, Takehara-Nishiuchi K, Frankland, PW. Parvalbumin-positive interneurons mediate neocortical-hippocampal interactions that are necessary for memory consolidation. Elife. 2017;6. https://doi.org/10.7554/eLife.27868
Marshall L, Helgadottir H, Molle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–3. https://doi.org/10.1038/nature05278
Marshall L, Molle M, Hallschmid M, Born J. Transcranial direct current stimulation during sleep improves declarative memory. J Neurosci. 2004;24:9985–92. https://doi.org/10.1523/JNEUROSCI.2725-04.2004
Mathias S, Wetter TC, Steiger A, Lancel M. The GABA uptake inhibitor tiagabine promotes slow wave sleep in normal elderly subjects. Neurobiol Aging. 2001;22:247–53.
Walsh JK, Randazzo AC, Frankowski S, Shannon K, Schweitzer PK, Roth T. Dose-response effects of tiagabine on the sleep of older adults. Sleep. 2005;28:673–6.
Feld GB, Wilhelm I, Ma Y, Groch S, Binkofski F, Molle M, et al. Slow wave sleep induced by GABA agonist tiagabine fails to benefit memory consolidation. Sleep. 2013;36:1317–26. https://doi.org/10.5665/sleep.2954
Puentes-Mestril C, Roach J, Niethard N, Zochowski M, Aton SJ. How rhythms of the sleeping brain tune memory and synaptic plasticity. Sleep. 2019;42. https://doi.org/10.1093/sleep/zsz095
Barakat M, Doyon J, Debas K, Vandewalle G, Morin A, Poirier G, et al. Fast and slow spindle involvement in the consolidation of a new motor sequence. Behav Brain Res. 2011;217:117–21. https://doi.org/10.1016/j.bbr.2010.10.019
Rasch B, Pommer J, Diekelmann S, Born J. Pharmacological REM sleep suppression paradoxically improves rather than impairs skill memory. Nat Neurosci. 2009;12:396–7.
Cairney SA, Guttesen AAV, El Marj N, Staresina BP. Memory Consolidation Is Linked to Spindle-Mediated Information Processing during Sleep. Curr Biol. 2018;28:948–54 e944. https://doi.org/10.1016/j.cub.2018.01.087
Clemens Z, Fabo D, Halasz P. Overnight verbal memory retention correlates with the number of sleep spindles. Neuroscience. 2005;132:529–35.
Cox R, Hofman WF, Talamini LM. Involvement of spindles in memory consolidation is slow wave sleep-specific. Learn Mem. 2012;19:264–7. https://doi.org/10.1101/lm.026252.112
Mednick SC, McDevitt EA, Walsh JK, Wamsley E, Paulus M, Kanady JC, et al. The critical role of sleep spindles in hippocampal-dependent memory: a pharmacology study. J Neurosci. 2013;33:4494–504. https://doi.org/10.1523/JNEUROSCI.3127-12.2013
Antony JW, Schonauer M, Staresina BP, Cairney SA. Sleep spindles and memory reprocessing. Trends Neurosci. 2019;42:1–3. https://doi.org/10.1016/j.tins.2018.09.012
Helfrich RF, Mander BA, Jagust WJ, Knight RT, Walker MP. Old brains come uncoupled in sleep: slow wave-spindle synchrony, brain atrophy, and forgetting. Neuron. 2018;97:221–30 e224. https://doi.org/10.1016/j.neuron.2017.11.020
Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–88. https://doi.org/10.1146/annurev.neuro.28.061604.135722
Schultz W. Updating dopamine reward signals. Curr Opin Neurobiol. 2013;23:229–38. https://doi.org/10.1016/j.conb.2012.11.012
Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. 2016;17:524–32. https://doi.org/10.1038/nrn.2016.57
Berridge KC, Robinson TE, Aldridge JW. Dissecting components of reward: ‘liking’, ‘wanting’, and learning. Curr Opin Pharm. 2009;9:65–73. https://doi.org/10.1016/j.coph.2008.12.014
Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–13. https://doi.org/10.1016/j.neuron.2005.05.002
Adcock RA, Thangavel A, Whitfield-Gabrieli S, Knutson B, Gabrieli JD. Reward-motivated learning: mesolimbic activation precedes memory formation. Neuron. 2006;50:507–17. https://doi.org/10.1016/j.neuron.2006.03.036
Patil A, Murty VP, Dunsmoor JE, Phelps EA, Davachi L. Reward retroactively enhances memory consolidation for related items. Learn Mem. 2017;24:65–9. https://doi.org/10.1101/lm.042978.116
Wimmer GE, Shohamy D. Preference by association: how memory mechanisms in the hippocampus bias decisions. Science. 2012;338:270–3. https://doi.org/10.1126/science.1223252
Braun EK, Wimmer GE, Shohamy D. Retroactive and graded prioritization of memory by reward. Nat Commun. 2018;9:4886. https://doi.org/10.1038/s41467-018-07280-0
Fischer S, Born J. Anticipated reward enhances offline learning during sleep. J Exp Psychol Learn Mem Cogn. 2009;35:1586–93. https://doi.org/10.1037/a0017256
Stamm AW, Nguyen ND, Seicol BJ, Fagan A, Oh A, Drumm M, et al. Negative reinforcement impairs overnight memory consolidation. Learn Mem. 2014;21:591–6. https://doi.org/10.1101/lm.035196.114
Groch S, Zinke K, Wilhelm I, Born J. Dissociating the contributions of slow-wave sleep and rapid eye movement sleep to emotional item and source memory. Neurobiol Learn Mem. 2014. https://doi.org/10.1016/j.nlm.2014.08.013
Javadi AH, Tolat A, Spiers HJ. Sleep enhances a spatially mediated generalization of learned values. Learn Mem. 2015;22:532–6. https://doi.org/10.1101/lm.038828.115
Lansink CS, Goltstein PM, Lankelma JV, Joosten RN, McNaughton BL, Pennartz CM. Preferential reactivation of motivationally relevant information in the ventral striatum. J Neurosci. 2008;28:6372–82. https://doi.org/10.1523/JNEUROSCI.1054-08.2008
Lansink CS, Goltstein PM, Lankelma JV, McNaughton BL, Pennartz CM. Hippocampus leads ventral striatum in replay of place-reward information. PLoS Biol. 2009;7:e1000173. https://doi.org/10.1371/journal.pbio.1000173
Valdes JL, McNaughton BL, Fellous JM. Offline reactivation of experience-dependent neuronal firing patterns in the rat ventral tegmental area. J Neurophysiol. 2015;114:1183–95. https://doi.org/10.1152/jn.00758.2014
McNamara CG, Tejero-Cantero A, Trouche S, Campo-Urriza N, Dupret D. Dopaminergic neurons promote hippocampal reactivation and spatial memory persistence. Nat Neurosci. 2014;17:1658–60. https://doi.org/10.1038/nn.3843
Feld GB, Besedovsky L, Kaida K, Munte TF, Born J. Dopamine D2-like receptor activation wipes out preferential consolidation of high over low reward memories during human sleep. J Cogn Neurosci. 2014. https://doi.org/10.1162/jocn_a_00629
Manahan-Vaughan D, Kulla A. Regulation of depotentiation and long-term potentiation in the dentate gyrus of freely moving rats by dopamine D2-like receptors. Cereb Cortex. 2003;13:123–35.
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225.
Alizadeh Asfestani M, Brechtmann V, Santiago JCP, Born J, Feld GB. Consolidation of reward memory during sleep does not require dopaminergic activation. bioRxiv. 2019. https://doi.org/10.1101/703132
Takano A, Suhara T, Yasuno F, Suzuki K, Takahashi H, Morimoto T, et al. The antipsychotic sultopride is overdosed–a PET study of drug-induced receptor occupancy in comparison with sulpiride. Int J Neuropsychopharmacol. 2006;9:539–45. https://doi.org/10.1017/S1461145705006103
Gomperts SN, Kloosterman F, Wilson MA. VTA neurons coordinate with the hippocampal reactivation of spatial experience. Elife. 2015;4. https://doi.org/10.7554/eLife.05360
Schapiro AC, McDevitt EA, Rogers TT, Mednick SC, Norman KA. Human hippocampal replay during rest prioritizes weakly learned information and predicts memory performance. Nat Commun. 2018;9:3920. https://doi.org/10.1038/s41467-018-06213-1
Drosopoulos S, Schulze C, Fischer S, Born J. Sleep’s function in the spontaneous recovery and consolidation of memories. J Exp Psychol Gen. 2007;136:169–83. https://doi.org/10.1037/0096-3445.136.2.169
Redondo RL, Morris RG. Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci. 2011;12:17–30. https://doi.org/10.1038/nrn2963
Murillo-Rodriguez E, Blanco-Centurion C, Sanchez C, Piomelli D, Shiromani PJ. Anandamide enhances extracellular levels of adenosine and induces sleep: an in vivo microdialysis study. Sleep. 2003;26:943–7.
Hill MN, Tasker JG. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience. 2012;204:5–16. https://doi.org/10.1016/j.neuroscience.2011.12.030
Xu J, Antion MD, Nomura T, Kraniotis S, Zhu Y, Contractor A. Hippocampal metaplasticity is required for the formation of temporal associative memories. J Neurosci. 2014;34:16762–73. https://doi.org/10.1523/JNEUROSCI.2869-13.2014
Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–81. https://doi.org/10.1016/j.neuron.2004.08.036
Solinas M, Goldberg SR, Piomelli D. The endocannabinoid system in brain reward processes. Br J Pharm. 2008;154:369–83. https://doi.org/10.1038/bjp.2008.130
Ghoneim MM, Mewaldt SP. Effects of diazepam and scopolamine on storage, retrieval and organizational processes in memory. Psychopharmacologia. 1975;44:257–62.
Rothschild G, Eban E, Frank LM. A cortical-hippocampal-cortical loop of information processing during memory consolidation. Nat Neurosci. 2017;20:251–9. https://doi.org/10.1038/nn.4457
Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76:116–29. https://doi.org/10.1016/j.neuron.2012.08.036
Gais S, Born J. Low acetylcholine during slow-wave sleep is critical for declarative memory consolidation. Proc Natl Acad Sci. 2004;101:2140–4.
Atri A, Sherman S, Norman KA, Kirchhoff BA, Nicolas MM, Greicius MD, et al. Blockade of central cholinergic receptors impairs new learning and increases proactive interference in a word paired-associate memory task. Behav Neurosci. 2004;118:223–36. https://doi.org/10.1037/0735-7044.118.1.223
Rasch B, Born J, Gais S. Combined blockade of cholinergic receptors shifts the brain from stimulus encoding to memory consolidation. J Cogn Neurosci. 2006;18:793–802. https://doi.org/10.1162/jocn.2006.18.5.793
Epperly T, Dunay MA, Boice JL. Alzheimer disease: pharmacologic and nonpharmacologic therapies for cognitive and functional symptoms. Am Fam Physician. 2017;95:771–8.
Klinzing JG, Kugler S, Soekadar SR, Rasch B, Born J, Diekelmann S. Odor cueing during slow-wave sleep benefits memory independently of low cholinergic tone. Psychopharmacol (Berl). 2018;235:291–9. https://doi.org/10.1007/s00213-017-4768-5
Schapiro AC, Turk-Browne NB, Botvinick MM, Norman KA. Complementary learning systems within the hippocampus: a neural network modelling approach to reconciling episodic memory with statistical learning. Philos Trans R Soc Lond B Biol Sci. 2017;372. https://doi.org/10.1098/rstb.2016.0049
Pardridge WM, Moeller TL, Mietus LJ, Oldendorf WH. Blood-brain barrier transport and brain sequestration of steroid hormones. Am J Physiol. 1980;239:E96–102. https://doi.org/10.1152/ajpendo.1980.239.1.E96
Bennett MC, Diamond DM, Fleshner M, Rose, GMJP. Serum corticosterone level predicts the magnitude of hippocampal primed burst potentiation and depression in urethane-anesthetized rats. Psychobiology. 1991;19:301–7.
Diamond DM, Bennett MC, Fleshner M, Rose GM. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus. 1992;2:421–30. https://doi.org/10.1002/hipo.450020409
Krieger DT, Allen W, Rizzo F, Krieger HP. Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab. 1971;32:266–84. https://doi.org/10.1210/jcem-32-2-266
Bierwolf C, Struve K, Marshall L, Born J, Fehm HL. Slow Wave Sleep Drives Inhibition of Pituitary‐AdrenalSecretion in Humans. Journal of Neuroendocrinology 2003;9(6):479–84.
Kirschbaum C, Pirke KM, Hellhammer DH. The ‘Trier Social Stress Test’–a tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology. 1993;28:76–81. https://doi.org/10.1159/000119004
Schwabe L, Haddad L, Schachinger H. HPA axis activation by a socially evaluated cold-pressor test. Psychoneuroendocrinology. 2008;33:890–5. https://doi.org/10.1016/j.psyneuen.2008.03.001
Buchanan TW, Lovallo WR. Enhanced memory for emotional material following stress-level cortisol treatment in humans. Psychoneuroendocrinology. 2001;26:307–17.
Cahill L, Gorski L, Le K. Enhanced human memory consolidation with post-learning stress: interaction with the degree of arousal at encoding. Learn Mem. 2003;10:270–4. https://doi.org/10.1101/lm.62403
Kuhlmann S, Kirschbaum C, Wolf OT. Effects of oral cortisol treatment in healthy young women on memory retrieval of negative and neutral words. Neurobiol Learn Mem. 2005;83:158–62. https://doi.org/10.1016/j.nlm.2004.09.001
Buchanan TW, Tranel D, Adolphs R. Impaired memory retrieval correlates with individual differences in cortisol response but not autonomic response. Learn Mem. 2006;13:382–7. https://doi.org/10.1101/lm.206306
Rimmele U, Besedovsky L, Lange T, Born J. Emotional memory can be persistently weakened by suppressing cortisol during retrieval. Neurobiol Learn Mem. 2015;119:102. https://doi.org/10.1016/j.nlm.2015.01.010
Schilling TM, Kolsch M, Larra MF, Zech CM, Blumenthal TD, Frings C, et al. For whom the bell (curve) tolls: cortisol rapidly affects memory retrieval by an inverted U-shaped dose-response relationship. Psychoneuroendocrinology. 2013;38:1565–72. https://doi.org/10.1016/j.psyneuen.2013.01.001
Rimmele U, Besedovsky L, Lange T, Born J. Blocking mineralocorticoid receptors impairs, blocking glucocorticoid receptors enhances memory retrieval in humans. Neuropsychopharmacology. 2013;38:884–94. https://doi.org/10.1038/npp.2012.254
Bennion KA, Mickley Steinmetz KR, Kensinger EA, Payne JD. Sleep and cortisol interact to support memory consolidation. Cereb Cortex. 2015;25:646–57. https://doi.org/10.1093/cercor/bht255
Dolfen N, King BR, Schwabe L, Swinnen S, Albouy G. Glucocorticoid response to stress induction prior to learning is negatively related to subsequent motor memory consolidation. Neurobiol Learn Mem. 2019;158:32–41. https://doi.org/10.1016/j.nlm.2019.01.009
Plihal W, Pietrowsky R, Born J. Dexamethasone blocks sleep induced improvement of declarative memory. Psychoneuroendocrinology. 1999;24:313–31.
Wagner U, Degirmenci M, Drosopoulos S, Perras B, Born J. Effects of cortisol suppression on sleep-associated consolidation of neutral and emotional memory. Biol Psychiatry. 2005;58:885–93. https://doi.org/10.1016/j.biopsych.2005.05.008
Kelemen E, Bahrendt M, Born J, Inostroza M. Hippocampal corticosterone impairs memory consolidation during sleep but improves consolidation in the wake state. Hippocampus. 2014;24:510–5. https://doi.org/10.1002/hipo.22266
Joels M, de Kloet E. Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science. 1989;245:1502–5.
Weiss EK, Krupka N, Bahner F, Both M & Draguhn A. Fast effects of glucocorticoids on memory-related network oscillations in the mouse hippocampus. J Neuroendocrinol 2008;20:549–557. https://doi.org/10.1111/j.1365-2826.2008.01699.x
Delahanty DL, Gabert-Quillen C, Ostrowski SA, Nugent NR, Fischer B, Morris A, et al. The efficacy of initial hydrocortisone administration at preventing posttraumatic distress in adult trauma patients: a randomized trial. CNS Spectr. 2013;18:103–11. https://doi.org/10.1017/S1092852913000096
Manoach DS, Pan JQ, Purcell SM, Stickgold R. Reduced sleep spindles in schizophrenia: a treatable endophenotype that links risk genes to impaired cognition? Biol Psychiatry. 2016;80:599–608. https://doi.org/10.1016/j.biopsych.2015.10.003
Göder R, Baier PC, Beith B, Baecker C, Seeck-Hirschner M, Junghanns K, et al. Effects of transcranial direct current stimulation during sleep on memory performance in patients with schizophrenia. Schizophr Res. 2013;144:153–4. https://doi.org/10.1016/j.schres.2012.12.014
Hofmann SG, Asnaani A, Vonk IJ, Sawyer AT, Fang A. The efficacy of cognitive behavioral therapy: a review of meta-analyses. Cogn Ther Res. 2012;36:427–40. https://doi.org/10.1007/s10608-012-9476-1
Hofmann SG, Smits JA. Cognitive-behavioral therapy for adult anxiety disorders: a meta-analysis of randomized placebo-controlled trials. J Clin Psychiatry. 2008;69:621–32.
Cuijpers P, Berking M, Andersson G, Quigley L, Kleiboer A, Dobson KS. A meta-analysis of cognitive-behavioural therapy for adult depression, alone and in comparison with other treatments. Can J Psychiatry. 2013;58:376–85. https://doi.org/10.1177/070674371305800702
Dutra L, Stathopoulou G, Basden SL, Leyro TM, Powers MB, Otto MW. A meta-analytic review of psychosocial interventions for substance use disorders. Am J Psychiatry. 2008;165:179–87. https://doi.org/10.1176/appi.ajp.2007.06111851
Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019;363:880–884.
Bandelow B, Michaelis S. Epidemiology of anxiety disorders in the 21st century. Dialog- Clin Neurosci. 2015;17:327–35.
Kessler RC, Ruscio AM, Shear K, Wittchen HU. Epidemiology of anxiety disorders. Curr Top Behav Neurosci. 2010;2:21–35.
Watson JB, Rayner R. Conditioned emotional reactions. J Exp Psychol. 1920;3:1–14. https://doi.org/10.1037/h0069608
Mowrer OH. On the dual nature of learning—a re-interpretation of “conditioning” and “problem-solving”. Harv Educ Rev. 1947;17:102–48.
Hofmann SG. Cognitive processes during fear acquisition and extinction in animals and humans: implications for exposure therapy of anxiety disorders. Clin Psychol Rev. 2008;28:199–210. https://doi.org/10.1016/j.cpr.2007.04.009
Hauner KK, Howard JD, Zelano C, Gottfried JA. Stimulus-specific enhancement of fear extinction during slow-wave sleep. Nat Neurosci. 2013;16:1553–5. https://doi.org/10.1038/nn.3527
He J, Sun HQ, Li SX, Zhang WH, Shi J, Ai SZ, et al. Effect of conditioned stimulus exposure during slow wave sleep on fear memory extinction in humans. Sleep. 2014;38:423–31.
Barnes DC, Wilson DA. Slow-wave sleep-imposed replay modulates both strength and precision of memory. J Neurosci. 2014;34:5134–42. https://doi.org/10.1523/JNEUROSCI.5274-13.2014
Rolls A, Makam M, Kroeger D, Colas D, de Lecea L, Heller HC. Sleep to forget: interference of fear memories during sleep. Mol Psychiatry. 2013;18:1166–70. https://doi.org/10.1038/mp.2013.121
Hofmann SG, Sawyer AT, Asnaani A. D-cycloserine as an augmentation strategy for cognitive behavioral therapy for anxiety disorders: an update. Curr Pharm Des. 2012;18:5659–62.
Hofmann SG. D-cycloserine for treating anxiety disorders: making good exposures better and bad exposures worse. Depress Anxiety. 2014;31:175–7. https://doi.org/10.1002/da.22257
Pyszczynski T, Hamilton JC, Herring FH, Greenberg J. Depression, self-focused attention, and the negative memory bias. J Personal Soc Psychol. 1989;57:351–7. https://doi.org/10.1037/0022-3514.57.2.351
Brewin CR. Understanding cognitive behaviour therapy: A retrieval competition account. Behav Res Ther. 2006;44:765–84. https://doi.org/10.1016/j.brat.2006.02.005
Nishida M, Pearsall J, Buckner RL, Walker MP. REM sleep, prefrontal theta, and the consolidation of human emotional memory. Cereb Cortex. 2009;19:1158–66. https://doi.org/10.1093/cercor/bhn155
Wagner U, Gais S, Born J. Emotional memory formation is enhanced across sleep intervals with high amounts of rapid eye movement sleep. Learn Mem. 2001;8:112–9. https://doi.org/10.1101/lm.36801
Wagner U, Hallschmid M, Rasch B, Born J. Brief sleep after learning keeps emotional memories alive for years. Biol Psychiatry. 2006;60:788–90. https://doi.org/10.1016/j.biopsych.2006.03.061
Walker MP, van der Helm E. Overnight therapy? The role of sleep in emotional brain processing. Psychol Bull. 2009;135:731–48. https://doi.org/10.1037/a0016570
Bolinger E, Born J, Zinke K. Sleep divergently affects cognitive and automatic emotional response in children. Neuropsychologia. 2018;117:84–91. https://doi.org/10.1016/j.neuropsychologia.2018.05.015
Wilson S, Argyropoulos S. Antidepressants and sleep. Drugs. 2005;65:927–47.
Palagini L, Baglioni C, Ciapparelli A, Gemignani A, Riemann D. REM sleep dysregulation in depression: state of the art. Sleep Med Rev. 2013;17:377–90. https://doi.org/10.1016/j.smrv.2012.11.001
Robinson TE, Berridge KC. The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction. 2000;95:S91–117.
Vollstadt-Klein S, Loeber S, Kirsch M, Bach P, Richter A, Buhler M, et al. Effects of cue-exposure treatment on neural cue reactivity in alcohol dependence: a randomized trial. Biol Psychiatry. 2011;69:1060–6. https://doi.org/10.1016/j.biopsych.2010.12.016
Arzi A, Shedlesky L, Ben-Shaul M, Nasser K, Oksenberg A, Hairston IS, Sobel N. Humans can learn new information during sleep. Nat Neurosci. 2012;15:1460–5.
Arzi A, Holtzman Y, Samnon P, Eshel N, Harel E, Sobel N, Olfactory Aversive Conditioning during Sleep Reduces Cigarette-Smoking Behavior. J Neurosci. 2014;34:15382–93.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Feld, G.B., Born, J. Neurochemical mechanisms for memory processing during sleep: basic findings in humans and neuropsychiatric implications. Neuropsychopharmacol. 45, 31–44 (2020). https://doi.org/10.1038/s41386-019-0490-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0490-9
This article is cited by
-
From childhood adversity to latent stress vulnerability in adulthood: the mediating roles of sleep disturbances and HPA axis dysfunction
Neuropsychopharmacology (2023)
-
Nicotine, alcohol, and caffeine use among individuals with untreated obstructive sleep apnea
Sleep and Breathing (2023)
-
Erregungs- und Regulationssysteme im System der Research Domain Criteria
Der Nervenarzt (2021)
-
Guanosine Neuroprotection of Presynaptic Mitochondrial Calcium Homeostasis in a Mouse Study with Amyloid-β Oligomers
Molecular Neurobiology (2020)
-
Modulation of Slow-Wave Sleep: Implications for Psychiatry
Current Psychiatry Reports (2020)
