
Abstract
Timing and duration of sleep are controlled by the circadian system, which keeps an ~24-h internal rhythm that entrains to environmental stimuli, and the sleep homeostat, which rises as a function of time awake. There is a normal distribution across the population in how the circadian system aligns with typical day and night resulting in varying circadian preferences called chronotypes. A portion of the variation in the population is controlled by genetics as shown by the single-gene mutations that confer extreme early or late chronotypes. Similarly, there is a normal distribution across the population in sleep duration. Genetic variations have been identified that lead to a short sleep phenotype in which individuals sleep only 4–6.5 h nightly. Negative health consequences have been identified when individuals do not sleep at their ideal circadian timing or are sleep deprived relative to intrinsic sleep need. Whether familial natural short sleepers are at risk of the health consequences associated with a short sleep duration based on population data is not known. More work needs to be done to better assess for an individual’s chronotype and degree of sleep deprivation to answer these questions.
Similar content being viewed by others
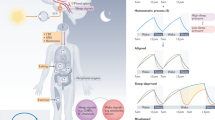

Introduction
Circadian rhythms
Nearly all living animals, from mammals to prokaryotes, have an internal clock adapted to the Earth’s rotational schedule of 24 h [1]. This internal circadian rhythmicity allows the body to predict the external environment, including when the sun will rise and set, and to anticipate the best times to sleep, wake, eat, and be active. The suprachiasmatic nucleus (SCN), located in the hypothalamus, is the central circadian pacemaker. Bilateral lesions of the SCN in hypothalami of rodents result in elimination of nocturnal and circadian rhythms in drinking, locomotive behavior, and hormone release [2, 3]. The SCN helps coordinate these rhythms throughout the body down to the cellular level, and SCN outputs target many tissues including the liver, adrenal glands, and pineal gland [4].
The human circadian pacemaker keeps an average rhythm that is slightly longer than 24 h with interindividual variability [5, 6]. Therefore it requires external cues, called zeitgebers (time givers), to continually synchronize to the environment. The strongest entraining signal for the SCN is light. The retina contains specialized nonimage forming cells, called melanopsin-expressing retinal ganglion cells or intrinsically photosensitive retinal ganglion cells (ipRGCs), that project to the SCN directly through the retino-hypothalamic tract [7, 8]. These ipRGCs are primarily responsive to blue wavelengths of light. The impact of light on sleep timing follows a phase response curve (Fig. 1) in which light in the 8–10 h prior to the minimum core body temperature, which typically occurs about 2 h before habitual wake time, will delay the circadian clock. Light in the ~9 h following core body minimum temperature will advance the clock [9, 10]. This effect is maximal at about 4 h before and after core body temperature minimum, thus artificial light in the evening and overnight will delay the circadian clock, resulting in the urge to sleep at a later hour, and light in the first 2 h of the day will advance it, resulting in an urge to sleep at an earlier hour [9, 10]. Other factors including exercise and eating also serve as zeitgebers. While research and treatments have focused primarily on light, which provides the major input to the SCN, a phase response curve has been described for exercise too, in which exercise at 7 a.m. and 1–4 p.m. advances the circadian clock and exercise from 7 to 10 p.m. delays it [11]. Interestingly, the timing of sleep and wake do not have a clear influence on the circadian clock.

a Phase response curve to a pulse of light. b The impact of light on circadian phase based on time of delivery throughout the 24-h day relative to timing of dim light melatonin onset and core body temperature
Chronotype
Chronotype describes the timing of behaviors that manifest from one’s circadian rhythm, most notably the tendency to sleep and eat at certain hours of the day. The variance in chronotype throughout the population follows a normal distribution [12, 13]. It is linked with light exposure, therefore population norms varying by longitude, latitude, and city- and rural-dwelling. The mid-sleep time on free days corrected for work-week sleep loss (MSFSC) is 4 min earlier (on average) for each degree of longitude within a time zone, accounting for the earlier sunrise [14]. The person-to-person variation is based on several factors including the duration of an individual’s internal period of oscillation, termed τ or tau. While averaging 24.2 h [1, 15], τ is unique to each individual and may be just over 23 h or extend beyond 25 h [16, 17]. Response to zeitgebers, angle of entrainment, and amplitude of τ also have an impact on an individual’s chronotype.
The two-process model: the interaction of circadian forces and sleep homeostasis
The two-process model, first described by Borbely in 1982, puts forth a description of sleep regulation that relies on both the circadian system (termed process C) and sleep homeostasis (termed process S) [18]. Process C is dependent on the ~24-h rhythmic variation of propensity to sleep, and this is balanced with process S, which increases as a function of time awake. Process S is estimated by EEG slow-wave activity and has an exponential decline during sleep. The model posits that it is the interaction of process C and process S that determines when we wake and when we sleep. It explains that circadian factors help us stay awake throughout the day as sleep pressure, modeled by process S, builds up and also help us stay asleep in the latter part of the night once this sleep pressure has largely declined. Sleep pressure also explains why more time awake can lead to more and deeper sleep.
Modern stressors on the two-process model
Modern society’s reliance on artificial lighting and reduced reliance on the rising and setting of the sun has put increased stress on this balance between circadian and homeostatic pressures. Indoor lighting is typically around 400 Lux, far weaker than the 10,000 to over 100,000 Lux of sunlight. Given that light is the main zeitgeber for our circadian system, and most of the light we experience day to day is artificial light, the overall input to the circadian system is far weaker than it was for our ancestors exposed to even cloudy skies on a daily basis [19]. In addition, light suppresses melatonin release by the pineal gland which can further influence the timing of sleep [20].
Social and occupational obligations can also alter sleep patterns and contribute to desynchrony between the internal rhythm and the timing of sleep for an individual. With reduced external input and increased evening obligations, the majority of people trend towards later bedtimes and later wake times. Under natural light conditions, such as when camping without artificial light, individuals have been shown to have a 2 h advance in dim light melatonin onset (DLMO), a marker of circadian phase, compared with typical conditions with artificial light [21]. There is greater variation in the timing of both melatonin onset and sleep under artificial light conditions compared with natural light conditions, suggesting that the weaker zeitgebers in the artificial lighting environment allow individual circadian differences to emerge. Increased time spent outside correlates with earlier chronotypes [12]. Work and school start times have not changed with increasing indoor lighting, causing shorter sleep duration for many people and increased difficulty arising in the morning. Approximately 20% of the population reports obtaining at least 1 h less sleep than their subjective sleep need, indicating these external demands are straining the system [22].
Understanding of the mechanism and control of the circadian system has progressed significantly since Borbely’s model in 1982, yet sleep homeostasis still remains very poorly understood. Here we discuss how the circadian system can vary across the population and lead to extreme phenotypes through genetic variation, sometimes resulting in disorders of sleep and wake. We will also discuss how sleep homeostasis can vary across the population, resulting in short and long sleep need, an area with increasing evidence that merits far more study.
Variations in sleep timing and duration
Circadian variation
Variations in τ, strength and angle of entrainment, and coupling of the clock to outputs result in a range of preferred sleep timing throughout the 24-h day. Extreme early types (called advanced sleep phase (ASP) or “morning larks”) prefer to go to sleep early and awaken early. An individual with ASP may sleep 8 p.m. to 4 a.m. instead of a more conventional 11 p.m. to 7 a.m. Late types (called delayed sleep phase (DSP) or “night owls”) prefer to go to bed late and awaken late. An individual with DSP may sleep 4 a.m. to 12 p.m. The majority of people have a schedule intermediate between morning larks and night owls (Fig. 2). The exact timing of what denotes a morning lark or night owl varies by location, and an intermediate chronotype in Eastern Europe has a mid-sleep time of 4 a.m., which is a late chronotype in India where the mean is closer to 3 a.m. [23].

Typical sleep period timing compared with delayed sleep phase, advanced sleep phase, and natural short sleep
Variations in chronotype by age, sex, and gender
Chronotype is a life-long trait, though there is variation across the lifespan. Infants and small children are relative morning larks, and there is an abrupt shift later in adolescence, peaking around age 20, following which there is a slow drift back to earlier sleep–wake tendencies over the subsequent decades [24]. At each age the distribution across the population remains stable with the same number of relative morning and evening types. Across the lifespan, an individual also retains his or her relative position within the group of similar age and sex. As individuals age past 65 there becomes greater variation across the population [24].
The prevalence of morning vs. evening types depends on the age of the population surveyed. In a sample of 433,268 individuals ages 37–73, 27% identified as definite morning types, while only 9% identified as definite evening types [25, 26]. In contrast, among college students, there is greater evening preference than the general population with an even greater evening trend in freshman compared with seniors [27]. There are also differences by gender. Men are later chronotypes until around age 50 after which men and women have similar chronotypes. Nearly 50% of women self-identify as a morning person compared with only 40% of men [28]. Just as puberty tends to appear earlier for girls, eveningness peaks in woman at age 19.5 and in men age 21 [24].
There are also variations by race, though all ethnicities show an approximately normal distribution of chronotype. In a report by Eastman et al., African Americans had an average τ of 24.07 h (N = 32), with no difference between men and woman, compared with Caucasians who had an average τ of 24.33 h (N = 31) [29]. There are also differences by ethnicity in morningness, with African Americans 1.4 times more likely to identify as morning types (vs. intermediate) compared with Caucasian [26]. This may be related to differential response to light, as African Americans show a larger phase advance in response to bright morning light and smaller phase delay in response to bright evening light [30]. Short sleep is also twice as common in African Americans, which is likely multifactorial with a strong influence of social, cultural, and environmental factors.
Genetics of circadian phenotypes
Genetics of ASP
Individuals with ASP who report a family history in a first degree relative are considered to have familial ASP (FASP). While genetic and environmental factors both contribute, those whose early chronotype started at a young age are very likely to have a strong family history of this trait [31]. Many with ASP and FASP do not find earlier sleep and awakening troublesome, and these individuals do not typically come to medical attention. Among a population presenting to a sleep clinic, the prevalence of ASP, defined as onset of sleepiness by 8:30 p.m. and spontaneous awakening by 5:30 a.m. with this tendency beginning prior to age 30, is estimated to be 0.33–0.5%. The estimated prevalence of FASP in this population is at least 0.21% with the majority of ASP individuals having a family history (Table 1) [31].
The two major methods shedding light on the genetics of ASP are single-gene mutations determined from families with an autosomal dominant inheritance pattern and genome-wide association studies (GWAS). In 1999 the first paper identifying a Mendelian inheritance pattern of FASP was published [16]. The proband from the largest identified family was noted to have a τ of 23.3 h when studied in temporal isolation. At that time, genetic mutations leading to alterations in the circadian period had been generated in forward genetic screens in Drosophila and rodents but no clock gene mutations were yet identified in humans [32]. Linkage analysis led to genetic mapping of the first FASP allele to chromosome 2q and positional cloning led to identification of PER2, a homolog of the Drosophila period gene. A mutation that substitutes a serine with a glycine residue at amino acid position 662, within the casein kinase Iδ/ε (CKIδ/ε) binding region of PER2, was identified in affected members of the first FASP family [33]. This mutation was engineered into mice and led to a semidominant ASP trait, recapitulating the phenotype of the humans harboring the mutation [34]. Further study suggests this mutation, PER2S662G, is a stronger transcriptional repressor than wild-type PER2, supporting the role of PER2 in setting the speed of the molecular clock [35].
Many additional families have been identified with a similar pattern including an allelic series of PER2 mutations, but most FASP families do not have a recognized mutation in PER2. Study of these families has shown genetic heterogeneity and led to identification of multiple additional causative mutations. Mutations leading to FASP have been identified in four additional genes, CK1δ (also known as CSNK1D) (T44A), PER3 (P415A/H417R), CRY2 (A260T), and TIMELESS (R1081X) [35,36,37,38]. These mutations are present in only a small minority of a large FASP family cohort, suggesting there are novel human circadian alleles and potentially novel genes yet to be identified.
Several circadian genes have pleiotropic effects influencing other aspects of health and disease. Mutations conferring an FASP phenotype have been show to cosegregate with migraine and depression. In two of the families with a Mendelian inheritance pattern of FASP, identification of the cosegregation of migraines with the extreme morning lark trait led to the identification that a mutation in CK1δ confers both migraine and FASP in the affected individuals [39]. The two identified families have distinct missense mutations, T44A and H46R, and mice engineered to carry one of these mutations were more sensitive to pain when a typical migraine trigger was provided. They also demonstrated reduced thresholds for inducing cortical spreading depression, the physiology underlying migraine aura. FASP, depression, and seasonal mood traits have also been linked in two variants in the PER3 gene [37]. This causes seasonal affective disorder in humans and a depression-like phenotype in mice that worsens under shorter photoperiods.
GWAS of chronotype
Multiple recent GWAS, relying on large cohorts such as the UK Biobank and the “direct to consumer” genetic testing company 23andMe, have identified many loci that show an association with morningness [28, 40,41,42]. The work, compiling up to 697,828 individuals in a single study, primarily derives chronotype by asking about self-reported morning or evening preference. In a more limited sample, activity-monitor derived sleep preference is also used [43]. Through this work, morningness has been associated with lower rates of depression and with better mental health [28, 42].
Up to 351 loci have shown association with chronotype in GWAS studies. These include PER2, PER3, and Cry1, already known to cause FASP or familial DSP (FDSP) in familial studies. CK1δ, Cry2, and TIMELESS mutations, also identified as causative in individual FASP families, have not been identified through GWAS. Notably, many loci found to be associated with chronotype in GWAS have not been replicated in independent cohorts. This suggests genetic complexity and may also be related to systematic biases depending on the tools used for defining chronotype. However, several genes arise in multiple studies. Four large GWAS published in the last 4 years found loci near the PER2, FBXL13, RGS16, and AK5 genes [28, 40, 41, 43]. As PER2 is already established to be involved in the circadian clock and causative of FASP, it seems likely that this association is a direct effect via PER2. RGS16 is involved in signaling in the SCN and FBXL13 is associated with lengthened circadian period in mice [44]. Interestingly, AK5 is a gene not previously known to be involved in the circadian system prior to this work [45]. At least three studies have shown involvement of hypocretin receptor type 2, known to be involved in alertness and also associated with sleep duration via GWAS study; [46] 5-hydroxytrptamine (serotonin) receptor 6, known to be involved in sleep regulation though not circadian regulation; and trinucleotide repeat containing 6b, previously shown to have involvement in the circadian system in Drosophila and mice. A remaining and daunting challenge of this field is the elucidation of pathways relevant to chronotype in the region of an associated locus. It will not always be the case that the closest gene to an associated locus will be causative for the chronotype.
Genetics of DSP
DSP, when more extreme, is also commonly a life-long tendency, worsening during adolescence and lessening with age. When DSP exists in a first degree relative it is termed FDSP. Prevalence estimates for DSP vary by definition, location, and age, but range up from as low as 0.13 to 3.1% [47].
In twin studies, genetic factors account for about 50% of diurnal preference [48, 49]. Despite this, fewer genes have been identified that lead to DSP than ASP. This suggests DSP is likely multigenic with several genes each playing a smaller role, though nongenetic factors such as artificial evening lighting and social pressures confound the picture making the detection of genetic variants more challenging. A length polymorphism in the PER3 gene due to a variable-number tandem repeat (4 or 5 repeats) has been linked to DSP in which the longer allele is associated with morningness and the shorter allele with eveningness [50]. A missense PER2 variant, Rs35333999, is associated with longer circadian period and later chronotype [40, 51]. A single mutation in the CRY1 gene, CRY1 c.1657 + 3 A > C, has been identified that confers an autosomal dominant DSP trait [52]. This allele has a prevalence of 0.6%, so is predicted to be responsible for a fraction of FDSP in the general population. The frequency of FDSP is not known and thus, it is not clear what proportion of this trait is explained by this CRY1 variant.
Circadian misalignment
The terms ASP and DSP are descriptions of preferred sleep timing and do not describe pathology. However, society has conventional hours of operation, and if an individual is not able to sync with this schedule, it can make the regular routine of life more challenging as propensity for sleep may fall in daylight and highest alertness in darkness. When this causes complaint, distress or trouble with function, it is termed circadian misalignment.
Advanced sleep wake phase disorder
A subset of ASP individuals present to their doctor with a sleep–wake complaint such as evening sleepiness, bothersome early morning awakenings not from another cause such as depression, insomnia, or sleep apnea, or daytime sleepiness from insufficient sleep due to trying to stay up for social, family, or work obligations. This subset with a complaint related to their early circadian timing meet criteria for advanced sleep–wake phase disorder (ASWPD) in the International Classification of Sleep Disorders (ICSD-3) [53]. One in eight with ASP meet criteria for ASWPD, suggesting a prevalence of at least 0.04% or 1/2500 (Table 2) [31]. This prevalence estimate does not account for those who become advanced only in later years as part of the common phase advance associated with aging (ASP of aging). Only those individuals who complain of this trait will carry the diagnosis of ASWPD. Figure 3 details the relationship between FASP, ASWPD, and ASP of aging. The relative size of the circles in Fig. 3 does not reflect the relative prevalence of FASP, ASWPD vs. ASP of aging, as the prevalence of ASP of aging is not known.

Diagram of the relationship between familial advanced sleep phase (FASP), advanced sleep–wake phase disorder (ASWPD), and ASP of Aging. This figure is adapted from a figure that is reprinted with permission from the journal SLEEP [31]. Relatives sizes do not reflect relative prevalence as the prevalence of ASP of Aging is not known
Delayed sleep–wake phase disorder (DSWPD)
For those who have a related complaint of insomnia or daytime sleepiness and feel better when allowed to sleep on their chosen delayed sleep schedule, the ICSD-3 classification is DSWPD [53]. This is a far more common complaint than ASWPD, and it is estimated that ~10% presenting with insomnia have DSWPD [53]. The prevalence of DSWPD varies by population but ranges from 7 to 16% among adolescents and young adults [53]. It is likely that a larger proportion of those with DSP have a complaint compared with those with ASP due to several factors. It is harder to live on a “conventional” schedule (typical school or work start times) for individuals with DSP than those with ASP. The increase in evening artificial light helps ASP individuals delay, keeping them closer to a conventional schedule, while evening light only further exacerbates the misalignment for those with DSP. Also, those with DSP experience greater sleep inertia, the phenomenon of a delay to feeling fully alert after awakening, compared with those with ASP [12]. While ASP individuals commonly feel alert within seconds or minutes of waking, those with DSP may take minutes to hours [31].
Other alterations of circadian rhythms
Some individuals are not able to entrain to a 24-h day and instead of having a stably advanced or delayed schedule, their sleep timing is continually shifting, resulting in alternating insomnia and excessive daytime sleepiness. This is termed non-24 sleep–wake disorder (N24SWD) [53]. Typically this occurs with rhythms over 24 h and most who suffer from N24SWD are totally blind and cannot entrain due to lack of the entraining input from light to the SCN. While the incidence of N24SWD is not known, over half of blind individuals do complain of sleep disturbance. Less commonly, some individuals are not able to entrain due to dementia, developmental abnormalities, or psychiatric disorders. Those with developmental delay and neurodegenerative disease may also suffer from irregular sleep–wake rhythm disorder in which the affected individual sleeps throughout the 24-h day and does not have a day vs. night preference [53]. This problem has also been reported following traumatic brain injury [54].
Impact of circadian misalignment on health
Circadian misalignment has negative health impacts. Causes of misalignment include shift work and social jetlag. Epidemiologic data reveals higher incidence of ischemic heart disease, metabolic syndrome, obesity, and cancer among shift workers [55,56,57,58]. Sleeping out of phase with one’s biological circadian rhythm has been shown to decrease leptin, increase insulin resistance, and increase mean arterial pressure [59]. Misalignment may influence breast cancer progression and long-term rotating night shift workers have a higher risk of breast cancer [60, 61]. Lack of entrainment, as demonstrated by melatonin rise during nonsleep hours, correlates with impaired learning and memory [62]. Destabilization of circadian rhythmicity may be a factor in tumor progression as the circadian rhythmicity of multiple circadian genes has been noted to be lower or absent in cancer cells [63].
Misalignment can occur between an individual and the environment, such as with ASWPD, DSWPD, or shift work, and within an individual, called internal misalignment. The SCN is responsible for synching internal physiologic processes including feeding and fasting, and peripheral clocks are subservient to the SCN. The body anticipates nightly fasting and stimulates gluconeogenesis, putting lipids and glucose levels under tight circadian control [64, 65]. This is demonstrated by mice with a liver specific deletion of the essential clock gene Bmal1 who developed hypoglycemia during fasting [66]. While light is the major input to the SCN, food is the most important input to peripheral oscillators. When meals are not synchronized to the light driven SCN, peripheral clocks can become decoupled. For example, when rodents are put on a restricted feeding schedule in which the timing of food offering is not synched with expected timing by the light cycle, the SCN remains synched with light but multiple peripheral oscillators including the liver, kidney, heart, and pancreas become synched with the eating schedule [67, 68]. This desynchrony with the liver has been shown to contribute to metabolic consequences such as obesity and type 2 diabetes [69].
Impact of chronotype on health
Chronotype alone also appears to impact disease. Recent work suggests decreased well-being, shorter lifespan, and increase mortality in evening types [25, 70]. There are higher rates of a wide range of disorders including psychological disorders, diabetes, neurological, gastrointestinal, and respiratory in definite evening types compared with definite morning types [25]. Morning preference appears to have a protective effect against breast cancer using a Mendelian randomization study model to investigate the impact of chronotype on breast cancer diagnosis in the UK Biobank [71]. Within diseases, such as bipolar and chronic migraine, evening type may impact frequency of episodes [72, 73]. Whether this negative impact of evening chronotype is mediated by increased misalignment is not known.
Clinical evaluation of circadian misalignment
When patients in clinic complain of trouble falling asleep, staying asleep, early morning awakenings, or daytime sleepiness, chronotype is an important factor to consider in devising optimal treatment. However, it can be difficult to determine an individual’s chronotype. There are many tools used for this purpose currently, but all have limitations. The timing of sleep is one tool. To characterize chronotype, it is important to ask about timing of first falling asleep and final awakening on long weekends or vacations without obligations. It is important to focus on prolonged time off because weekday and weekend obligations often shape sleep timing and rebound sleep on the weekend can prolong sleep. Two questionnaires, the Munich ChronoType Questionnaire, which generates a mean sleep on free days (MSF) time, and the morningness–eveningness questionnaire are used to screen for chronotype. Objective measures include DLMO, which is considered the most reliable measure of central circadian timing. Melatonin levels are very low during the day and start to rise in the evening. The timing of this rise represents the action of the central circadian pacemaker, the SCN, which signals the pineal gland to release melatonin [74]. However, light suppresses melatonin release [20, 75]. Therefore, this collection must be done at frequent intervals in the hours before habitual bedtime in dim light conditions. It is not widely available clinically and is cumbersome to collect and therefore is not routinely used in clinical practice. Core body temperature minimum is also a strong indicator of the timing of the central pacemaker. However, this is difficult to obtain clinically and is not typically done outside of a research setting. In determining how to treat circadian disorders, it is also useful to understand the τ of an individual, but this is difficult to determine as optimal testing requires a challenging protocol called the forced desynchrony protocol, carried out only in research settings. This procedure requires an individual to follow a cycle of rest and activity outside the range of entrainment of the circadian pacemaker, commonly a 28 h cycle, and measures circadian changes during this desynchrony [1].
Promising future approaches to identify intrinsic rhythm rely on the fact that transcription and metabolism are under circadian regulation in mammals [76, 77]. The transcriptome for mammals has been characterized [78] and work is underway to utilize this to better define circadian phase. Nearly half of protein-coding genes express rhythmicity at some site in the body [79]. Thus, the transcriptome or metabolome of cells in blood promises to be useful as a marker for circadian phase. Multiple efforts have been made on that front. Liang et al. report a whole blood mRNA based predictor looking at 100 biomarkers, primarily involved in glucocorticoid signaling and immune function, which can predict melatonin cycle timing when sampling at two time points 12 h apart [80]. Braun et al. have created a machine-learning algorithm that can be trained to use gene expression to predict circadian rhythms [81]. Wittenbrink and colleagues report a monocyte NanoString-based gene expression profile based on 12 genes that can predict circadian timing with a single daytime sample [82]. This approach has the potential to improve detection of chronotype in routine clinical practice, however no tool is currently clinically validated and available.
The sleep homeostat
Sleep need
Similar to chronotype, habitual sleep duration varies across the population and follows a normal distribution [83]. In 2015, the National Sleep Foundation released a recommendation that adults obtain 7–9 h of sleep [84]. This has led to an important national conversation about the value of adequate sleep. However, there is also increased anxiety from those whose biological sleep “need” does not match these recommendations as not everyone with short or long sleep need has pathology.
Sleep need: epidemiologic data
The majority of information regarding adequate sleep duration comes from epidemiologic data and sleep deprivation experiments. Epidemiologic data shows an association between short and long sleep and increased mortality. The lowest mortality is for ~7 h of sleep, and deviation from this in both directions is associated with increased mortality in approximately a u-shaped distribution [85]. This relationship has been shown in numerous cultures across the world [86]. There is a similar relationship between short and long sleep duration and glucose tolerance, type 2 diabetes, elevated BMI, obesity, and metabolic syndrome [87,88,89]. It is important to note that these studies measure an association in a population of people. It is likely that the optimal amount of sleep for an individual varies dramatically across the population.
Epidemiologic data is based on self-reported sleep duration and does not incorporate a reason for short sleep hours. Thus, such studies do not differentiate between those who have short sleep accompanied by a sleep-related complaint, such as insomnia or excessive daytime sleepiness, and those who do not. In fact, a u-shaped curve of sleep complaints has also been reported, indicating many of those reporting short sleep also complain about trouble with nocturnal sleep or have daytime sleep-related symptoms [90]. The 5% who report the shortest sleep duration have a high incidence of insomnia suggesting that this u-shaped curve may be picking up the comorbidity of insomnia [91].
Sleep need: sleep deprivation data
The other major factor driving recommendations on sleep duration is sleep deprivation experiments. Numerous laboratory studies show that sleep deprivation has negative consequences for health. Metabolic consequences of acute sleep deprivation include insulin resistance, endocrine dysfunction, decreased leptin, increases ghrelin, and increased blood pressure [92,93,94,95]. Sleepiness and reduced sleep time have been linked to motor vehicle accidents [96,97,98]. Short sleep has been associated with increased incidence of depression [99]. Acute sleep deprivation causes sleepiness and deficits in alertness, attention, learning, and memory [100]. Notably, however, cognitive function tests during sleep deprivation experiments reveal a range of responses following sleep loss, with some individuals having minimal negative consequences. These interindividual differences in vulnerability to the cognitive effects of sleep loss appear trait-like, remaining stable over time [101].
Genetic variation in sleep need
We hypothesize that variation in vulnerability to sleep loss may be related to differences in an individual’s biological requirement for sleep—ranging from natural short sleep to natural long sleep.
Variable response to sleep loss is, at least in part, genetic. EEG delta power recorded during non-REM sleep, which increases the longer an individual has been awake prior to sleeping and dissipates during non-REM sleep, is a marker of homeostatic sleep pressure, and interindividual variation around delta power likely reflects a central factor in sleep need variation. Studies in multiple inbred strains of mice show that delta power varies by strain with delta power depending on both prior wake time and genotype [102, 103]. Mice with a mutation in the clock gene showed decreased sleep time over 24 h with ~1 h reduction in heterozygotes and a 2 h reduction in homozygotes [104].
In humans, we have known since the 1930s through twin studies that sleep length is heritable [105, 106]. GWAS data show 10–21% heritability of sleep duration with several clock genes implicated. A relatively early GWAS published in 2007 with 749 participants shows a SNP in NPSR1 at rs324981 to be associated with a 15 min later bedtime for each allele present [107]. Two larger studies compiling data from 47,180 and 446,118 individuals both show associations of multiple loci with sleep duration, however the genes and mechanisms remain to be elucidated [46, 108]. A polymorphism at rs4753426 in a melatonin receptor gene, MTNR1B, correlates with more time in bed on weekends, suggesting a potential role in sleep duration, and a separate variant, rs7942988, has been shown to impact duration of melatonin secretion [109, 110].
The sleep EEG is remarkably stable for an individual and follows a trait-like pattern with repeated assessment at baseline and following sleep deprivation showing reproducible characteristics [111, 112]. Non-REM EEG delta power was among the most stable of the sleep parameters for an individual over multiple assessments with the greatest variability between individuals. Much of this interindividual variability in the sleep EEG is thought to be driven by genetics [113, 114]. For example, carriers of a common PER2 variant have 22%, or 20 min, less slow wave sleep (SWS) compared with noncarriers [110].
There are multiple genes, including PER2 and DEC2, identified as clock genes that have also been demonstrated to influence the sleep homeostat and vice versa [33, 110, 115, 116]. This implies a deep interdependence between the systems. Given the systems coevolved, this interdependence is not surprising. However the dynamics of this interplay remain unknown and more study is needed to better understand this complex relationship.
Physiologic differences in sleep between short and long sleepers
There has been limited investigation on the physiologic changes in sleep of habitual short and long sleepers. Familial natural short sleep (FNSS) is a subgroup within those reporting habitual short sleep. Thus, this research does not describe FNSS specifically. Short sleepers have a shorter duration of melatonin release and earlier times of cortisol peak and temperature nadir compared with long sleepers [117]. Increased time awake in the short sleepers leads to higher waking levels of theta and low-frequency activity. Short sleepers appear to have more spectral power in the 5.25–9.0 Hz and 17.25–18.0 Hz range than long sleepers [118]. Aeschbach et al. investigated how to apply Borbely’s two-process model to short and long sleepers by completing sleep EEG under baseline and sleep deprivation conditions [119]. At baseline, short sleepers slept over 3 h less than long sleepers. This was primarily made up by differences in REM and stage N2 sleep; SWS did not significantly differ between groups on any night. With sleep deprivation, sleep latency and REM density decreased more in long sleepers than in short sleepers. Short sleepers did have a shorter sleep latency and a larger amount of SWS when matching the initial portion of the long sleepers’ nights to the total night for the short sleepers, suggesting that short sleepers are under higher sleep pressure. Short sleepers also appeared to awaken with a higher sleep pressure, suggesting that short sleepers tolerate a higher homeostatic sleep pressure [119].
Familial natural short sleep
In 2009 the first human genetic variant leading to a short sleeping phenotype was described in the DEC2 gene, P385R [115]. Fu and colleagues termed this FNSS. FNSS is defined as a stable trait of sleeping 4–6.5 h/night without daytime sleepiness or clear impairments (Table 1). These individuals are sometimes seen in sleep clinics because they are told they “need” more sleep—they are sometimes considered insomniacs. However, when these individuals are asked how they feel after sleeping 4–6.5 h, they often report feeling “great” and “well rested.” In the first described family, the DEC2 mutation leads to reduced sleep duration in the affected individuals. Engineering this mutation in Drosophila and transgenic mice produces a similar phenotype. DEC2 is a transcriptional repressor and increases expression of hypocretin [120]. Hypocretin, also known as orexin, is wake promoting and involved in sleep–wake transitions. Loss of hypocretin producing neurons in the lateral hypothalamus leads to narcolepsy with cataplexy. A novel mutation in DEC2 has also been identified in a pair of dizygotic twins with the same BMI but in which one twin had reduced sleep duration, less recovery sleep following sleep deprivation, and fewer performance lapses following sleep deprivation [121]. Another mutation causing FNSS has been identified in the beta 1 adrenergic receptor gene (ADRB1) [122]. ADRB1 is highly expressed in the dorsal pons and the neurons are active during REM and wake. Multiple additional families with an autosomal dominant pattern of decreased sleep need have been collected, and two additional genes have been identified with the short sleep phenotype recapitulated in mouse models [122].
Individuals with FNSS report decreased sleep need throughout adult life, some dating to childhood. They report sleep durations ranging from 4 to 6.5 h per night without daytime sleepiness or reported deficits from sleep deprivation. FNSS individuals are in part distinguished from facultative short sleepers by their lack of catch-up sleep on weekends and free days. They report greater flexibility around sleep timing and less subjective deficit after sleep deprivation. They often deny experiencing jetlag (unpublished data). However, it is not just sleep duration that characterizes this group of individuals. There is a high behavioral drive among those with FNSS, and individuals report a need to always be mentally active resulting commonly in high profile, high pressure jobs or holding multiple jobs. FNSS individuals also appear to have high pain thresholds and relative resilience to life stressors (unpublished data). Thus, the phenotype does not only encompass a sleep pattern but also daytime behavior. Individuals with short sleep and increased behavioral drive have been described in the literature as early as 1972 when Hartmann et al., while studying sleep need variation, described those who sleep less than 6 h as “smooth [and] efficient” and with greater tolerance and flexibility and less depression compared with long sleepers [123]. It is possible that DEC2, ADRB1 and other causative mutations all lead to a higher behavioral drive, which allows those with FNSS to overcome increased homeostatic sleep pressure though much work needs to be done to test this hypothesis.
Familial natural long sleep (FNLS)
Similar to short sleep, there is likely a group of individuals who need a greater amount of sleep. However there are not yet any identified genetic variants causing FNLS. Comorbidities such as depression and lifestyle factors complicate the picture making this group harder to detect.
Despite these knowledge gaps, it is crucial to recognize genetic and biological differences among people with regard to biological “need” for sleep to avoid labeling individuals as having pathology when none exists and to offer appropriate care when there is a mismatch between sleep need and sleep duration. Additional understanding about how to define and measure sleep need is in order.
Future research directions
Across the population, there is a normal distribution of chronotype and habitual sleep duration. Genetics accounts for a lot of this variation in the population, most clearly at the extremes for circadian rhythm and short sleep duration. Single-gene mutations have been found that lead to extreme chronotypes in FASP and FDSP. Single-gene mutations also cause extremes of sleep duration in FNSS families and likely FNLS though this is not yet described in the literature. There are many FASP and FNSS families with an autosomal dominant pattern of inheritance without an identified gene, suggesting extension beyond known clock genes. Forward genetic screens in flies and mice have focused on mutations with large effects on period or rhythmicity. We postulate that some of the FASP genes will affect entrainment or coupling of clock to physiologic outputs. FNSS genes likely impact behavioral drive, delta power, and sleep efficiency.
There are many unanswered questions regarding variations in sleep need and FNSS. These include whether those with FNSS are resistant to any of the reported metabolic and cognitive consequences of sleep deprivation and whether they are susceptible to the epidemiologically described long-term health consequences. We hypothesize they are resistant to many of these given the observation that they are often high achieving without any clear medical comorbidities. Better individual and epidemiologic studies of this population are merited. Further, understanding the possible tolerance to higher sleep pressure and behavioral drive may have implications for a wide range of occupations requiring long durations of wakefulness including pilots, military personnel, and physicians and for treatment of disorders of sleep and arousal.
We need a conceptual framework for genetic and biologic contributions to sleep in individuals vs. populations. The current model of circadian factors and sleep homeostasis does not explain this third dimension of behavioral drive observed in FNSS. The circadian clock, sleep homeostat, behavioral drive, and environmental factors such as light exposure and daily obligations together impact the ultimate timing and duration of sleep (Fig. 4). Future work should focus on circadian factors affecting entrainment and clock-output coupling and sleep homeostatic factors affecting biologic sleep need, tolerance of homeostatic sleep pressure, and sleep efficiency. Finally, genes and proteins impacting behavioral drive merit direct investigation.

A conceptual framework for the relationship among different aspects of biology leading to innate traits of sleep timing and duration. These genetic factors interact with environmental factors such as electric lights, chemicals (ethanol, caffeine, medications), and familial and societal responsibilities which together may manifest as sleep duration and timing different from that programed by genetics. The relative contribution of each component is likely variable and remains under investigation
It is imperative that we did not impose rigid recommendations for what constitutes a “healthy” amount of sleep or “healthy” timing of sleep for an individual based on population averages. It is clear that an individual who does not sleep at his or her ideal circadian timing or for his or her ideal duration will suffer consequences. However, given the interindividual variation in the ideal timing or duration, we need to better understand and measure an individual’s need. This means we need better tools to assess chronotype and sleep deprivation, which may ultimately come from measurements of the transcriptome and proteome. The identification of Mendelian families with FASP, FDSP, and FNSS makes clear that there are strong genetic factors affecting sleep timing and quantity. The variation between the extremes is certain to result, in part, from complex genetic factors. Identification and study of additional human circadian and sleep genes causing extreme phenotypes will lead to better understanding of genes and pathways regulating sleep timing and quantity in humans.
Funding and disclosure
This work was supported by NIH grant NS099333 to LJP, NS072360 and NS104782 to Y-HF, and by the William Bowes Neurogenetics Fund to LJP and Y-HF.
References
Czeisler CA, et al. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284:2177–81.
Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci USA. 1972;69:1583–6.
Moore RY, Eichler VB. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972;42:201–6.
Kalsbeek A, et al. SCN outputs and the hypothalamic balance of life. J Biol Rhythms. 2006;21:458–69.
Czeisler CA. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284:2177–81.
Wright KP, Hughes RJ, Kronauer RE, Dijk DJ, Czeisler CA. Intrinsic near-24-h pacemaker period determines limits of circadian entrainment to a weak synchronizer in humans. Proc Natl Acad Sci USA. 2001;98:14027–32.
Hattar S, Liao HW, Takao M, Berson DM, Yau KW. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295:1065–70.
Lockley SW, Brainard GC, Czeisler CA. High sensitivity of the human circadian melatonin rhythm to resetting by short wavelength light. J Clin Endocrinol Metab. 2003;88:4502–5.
Minors DS, Waterhouse JM, Wirz-Justice A. A human phase-response curve to light. Neurosci Lett. 1991;133:36–40.
Khalsa SBS, Jewett ME, Cajochen C, Czeisler CA. A phase response curve to single bright light pulses in human subjects. J Physiol. 2003;549:945–52.
Youngstedt SD, Elliott JA, Kripke DF. Human circadian phase-response curves for exercise. J Physiol. (2019). https://doi.org/10.1113/JP276943.
Roenneberg T, Wirz-Justice A, Merrow M. Life between clocks: daily temporal patterns of human chronotypes. J Biol Rhythms. 2003;18:80–90.
Fischer D, Lombardi DA, Marucci-Wellman H, Roenneberg T. Chronotypes in the US – influence of age and sex. PLoS ONE. 2017;12:e0178782.
Roenneberg T, Kumar CJ, Merrow M. The human circadian clock entrains to sun time. Curr Biol. 2007;17:R44–5.
Hiddinga AE, Beersma DG, Van den Hoofdakker RH. Endogenous and exogenous components in the circadian variation of core body temperature in humans. J Sleep Res. 1997;6:156–63.
Jones CR, et al. Familial advanced sleep-phase syndrome: a short-period circadian rhythm variant in humans. Nat Med. 1999;5:1062–5.
Micic G, et al. The endogenous circadian temperature period length (tau) in delayed sleep phase disorder compared to good sleepers. J Sleep Res. 2013;22:617–24.
Borbély AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204.
Foster RG. Biological clocks: who in this place set up a sundial? Curr Biol. 2012;22:R405–7.
Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Markey SP. Light suppresses melatonin secretion in humans. Science. 1980;210:1267–9.
Wright KP, et al. Entrainment of the human circadian clock to the natural light-dark cycle. Curr Biol. 2013;23:1554–8.
Ursin R, Bjorvatn B, Holsten F. Sleep duration, subjective sleep need, and sleep habits of 40- to 45-year-olds in the Hordaland Health Study. Sleep. 2005;28:1260–9.
ROENNEBERG T. What is chronotype? Sleep Biol Rhythms. 2012;10:75–76.
Roenneberg T, et al. A marker for the end of adolescence. Curr Biol. 2004;14:R1038–9.
Knutson KL, von Schantz, M. Associations between chronotype, morbidity and mortality in the UK Biobank cohort. Chronobiol Int. 1–9 (2018). https://doi.org/10.1080/07420528.2018.1454458
Malone SK, Patterson F, Lu Y, Lozano A, Hanlon A. Ethnic differences in sleep duration and morning–evening type in a population sample. Chronobiol Int. 2016;33:10–21.
Taylor DJ, Clay KC, Bramoweth AD, Sethi K, Roane BM. Circadian phase preference in college students: relationships with psychological functioning and academics. Chronobiol Int. 2011;28:541–7.
Hu Y, et al. GWAS of 89,283 individuals identifies genetic variants associated with self-reporting of being a morning person. Nat Commun. 2016;7:10448.
Eastman CI, Tomaka VA, Crowley SJ. Sex and ancestry determine the free-running circadian period. J Sleep Res. 2017;26:547–50.
Smith MR, Burgess HJ, Fogg LF, Eastman CI. Racial differences in the human endogenous circadian period. PLoS ONE. 2009;4:e6014.
Curtis BJ, Ashbrook LH, Young T, Finn LA, Fu YH, Ptáček LJ, Jones CR. Extreme morning chronotypes are often familial and not exceedingly rare: the estimated prevalence of advanced sleep phase (ASP), familial advanced sleep phase (FASP), and advanced sleep-wake phase disorder (ASWPD) in a sleep clinic population. Sleep. (2019).
Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–90.
Toh KL, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–3.
Xu Y, et al. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell. 2007;128:59–70.
Kurien P, et al. TIMELESS mutation alters phase responsiveness and causes advanced sleep phase. Proc Natl Acad Sci USA. 2019;116:12045–53.
Xu Y, et al. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–4.
Zhang L, et al. A PERIOD3 variant causes a circadian phenotype and is associated with a seasonal mood trait. Proc Natl Acad Sci USA. 2016;113:E1536–44.
Hirano A, et al. A cryptochrome 2 mutation yields advanced sleep phase in humans. Elife. 2016;5:e16695.
Brennan KC, et al. Casein kinase iδ mutations in familial migraine and advanced sleep phase. Sci Transl Med. 2013;5:183ra56. 1–11.
Lane JM, et al. Genome-wide association analysis identifies novel loci for chronotype in 100,420 individuals from the UK Biobank. Nat Commun. 2016;7:10889.
Jones SE, et al. Genome-wide association analyses in 128,266 individuals identifies new morningness and sleep duration loci. PLoS Genet. 2016;12:e1006125.
Jones SE, et al. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat Commun. 2019;10:343.
Jones SE, et al. Genetic studies of accelerometer-based sleep measures yield new insights into human sleep behaviour. Nat Commun. 2019;10:1585.
Kalmbach DA et al. Genetic basis of chronotype in humans: insights from three landmark GWAS. Sleep. 2017;40:zsw048.
von Schantz M. Natural Variation in Human Clocks. in. Adv Genet. 2017;99:73–96.
Dashti HS, et al. Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nat Commun. 2019;10:1100.
Wyatt JK. Delayed Sleep Phase Syndrome: Pathophysiology and Treatment Options. Sleep. 2004;27:1195–203.
Koskenvuo M, Hublin C, Partinen M, Heikkilä K, Kaprio J. Heritability of diurnal type: a nationwide study of 8753 adult twin pairs. J Sleep Res. 2007;16:156–62.
Barclay NL, Eley TC, Buysse DJ, Archer SN, Gregory AM. Diurnal Preference and Sleep Quality: Same Genes? A Study of Young Adult Twins. Chronobiol Int. 2010;27:278–96.
Archer SN, et al. A length polymorphism in the circadian clock gene Per3 is linked to delayed sleep phase syndrome and extreme diurnal preference. Sleep. 2003;26:413–5.
Chang A-M, et al. Chronotype Genetic Variant in PER2 is Associated with Intrinsic Circadian Period in Humans. Sci Rep. 2019;9:5350.
Patke A, et al. Mutation of the Human Circadian Clock Gene CRY1 in Familial Delayed Sleep Phase Disorder. Cell. 2017;169:203–15.e13.
American Academy of Sleep Medicine. International classification of sleep disorders. 3rd ed. (ICSD-3). (American Academy of Sleep Medicine, Darien, IL, 2014).
Ayalon L, Borodkin K, Dishon L, Kanety H, Dagan Y. Circadian rhythm sleep disorders following mild traumatic brain injury. Neurology. 2007;68:1136–40.
Knutsson A, Akerstedt T, Jonsson BG, Orth-Gomer K. Increased risk of ischaemic heart disease in shift workers. Lancet. 1986;2:89–92.
Karlsson B, Knutsson A, Lindahl B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med. 2001;58:747–52.
Tüchsen F, Hannerz H, Burr H. A 12 year prospective study of circulatory disease among Danish shift workers. Occup Environ Med. 2006;63:451–5.
Lahti T, Merikanto I, Partonen T. Circadian clock disruptions and the risk of cancer. Ann Med. 2012;44:847–53.
Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. PNAS. 209;17:453–8.
Hahm B-J, et al. Bedtime misalignment and progression of breast cancer. Chronobiol Int. 2014;31:214–21.
Wegrzyn LR, et al. Rotating night-shift work and the risk of breast cancer in the Nurses’ Health Studies. Am J Epidemiol. 2017;186:532–40.
Wright KP, Hull JT, Hughes RJ, Ronda JM, Czeisler CA. Sleep and wakefulness out of phase with internal biological time impairs learning in humans. J Cogn Neurosci. 2006;18:508–21.
Soták M, Polidarová L, Ergang P, Sumová A, Pácha J. An association between clock genes and clock-controlled cell cycle genes in murine colorectal tumors. Int J Cancer. 2013;132:1032–41.
Zhang EE, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat Med. 2010;16:1152–6.
Panda S, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–20.
Lamia KA, Storch K-F, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA. 2008;105:15172–7.
Stokkan K-A, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the circadian clock in the liver by feeding. Science. 2001;291:490–3.
Damiola F, et al. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14:2950–61.
Oosterman JE, Kalsbeek A, la Fleur SE, Belsham DD. Impact of nutrients on circadian rhythmicity. Am J Physiol Integr Comp Physiol. 2015;308:R337–50.
Didikoglu A, Maharani A, Payton A, Pendleton N, Canal MM. Longitudinal change of sleep timing: association between chronotype and longevity in older adults. Chronobiol. Int. 2019; 1–16. https://doi.org/10.1080/07420528.2019.1641111.
Richmond RC, et al. Investigating causal relations between sleep traits and risk of breast cancer in women: Mendelian randomisation study. BMJ. 2019;365:l2327.
Melo MCA, Abreu RLC, Linhares Neto VB, de Bruin PFC, de Bruin VMS. Chronotype and circadian rhythm in bipolar disorder: a systematic review. Sleep Med Rev. 2017;34:46–58.
Viticchi, G et al. Influence of chronotype on migraine characteristics. Neurol. Sci. (2019). https://doi.org/10.1007/s10072-019-03886-4.
Lewy AJ, Cutler NL, Sack RL. The endogenous melatonin profile as a marker for circadian phase position. J Biol Rhythms. 1999;14:227–36.
Aoki H, Yamada N, Ozeki Y, Yamane H, Kato N. Minimum light intensity required to suppress nocturnal melatonin concentration in human saliva. Neurosci Lett. 1998;252:91–4.
Duffield GE. DNA microarray analyses of circadian timing: the genomic basis of biological time. J Neuroendocr. 2003;15:991–1002.
Archer SN, Oster H. How sleep and wakefulness influence circadian rhythmicity: effects of insufficient and mistimed sleep on the animal and human transcriptome. J Sleep Res. 2015;24:476–93.
Mure LS, et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science. 2018;359:eaao0318.
Ruben MD, et al. A database of tissue-specific rhythmically expressed human genes has potential applications in circadian medicine. Sci Transl Med. 2018;10:eaat8806.
Laing, EE et al. Blood transcriptome based biomarkers for human circadian phase. Elife. 2017;6:e20214.
Braun R, et al. Universal method for robust detection of circadian state from gene expression. Proc Natl Acad Sci USA. 2018;115:E9247–56.
Wittenbrink N, et al. High-accuracy determination of internal circadian time from a single blood sample. J Clin Investig. 2018;128:3826–39.
Groeger JA, Zijlstra FRH, Dijk D-J. Sleep quantity, sleep difficulties and their perceived consequences in a representative sample of some 2000 British adults. J Sleep Res. 2004;13:359–71.
Hirshkowitz M, et al. National Sleep Foundation’s sleep time duration recommendations: methodology and results summary. Sleep Health. 2015;1:40–43.
Itani O, Jike M, Watanabe N, Kaneita Y. Short sleep duration and health outcomes: a systematic review, meta-analysis, and meta-regression. Sleep Med. 2017;32:246–56.
Grandner MA, Hale L, Moore M, Patel NP. Mortality associated with short sleep duration: the evidence, the possible mechanisms, and the future. Sleep Med Rev. 2010;14:191–203.
Bjorvatn B, et al. The association between sleep duration, body mass index and metabolic measures in the Hordaland Health Study. J Sleep Res. 2007;16:66–76.
Chaput J-P, Després J-P, Bouchard C, Tremblay A. Association of sleep duration with type 2 diabetes and impaired glucose tolerance. Diabetologia. 2007;50:2298–304.
Hall MH, et al. Self-reported sleep duration is associated with the metabolic syndrome in midlife adults. Sleep. 2008;31:635–43.
Grandner MA, Kripke DF. Self-reported sleep complaints with long and short sleep: a nationally representative sample. Psychosom. Med. 2004;66:239–41.
Ohayon MM, Vecchierini M-F. Normative sleep data, cognitive function and daily living activities in older adults in the community. Sleep. 2005;28:981–9.
Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–9.
Van Cauter E, Spiegel K, Tasali E, Leproult R. Metabolic consequences of sleep and sleep loss. Sleep Med. 2008;9:S23–8.
Spiegel K, Tasali E, Penev P, Van Cauter E. Brief communication: sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med. 2004;141:846–50.
Lusardi P, et al. Effects of insufficient sleep on blood pressure in hypertensive patients: a 24-h study. Am J Hypertens. 1999;12:63–8.
Ingre M, et al. Subjective sleepiness and accident risk avoiding the ecological fallacy. J Sleep Res. 2006;15:142–8.
Varughese, Allen. Fatal accidents following changes in daylight savings time: the American experience. Sleep Med. 2001;2:31–36.
Philip P, Akerstedt T. Transport and industrial safety, how are they affected by sleepiness and sleep restriction? Sleep Med Rev. 2006;10:347–56.
Zhai L, Zhang H, Zhang D. Sleep duration and depression among adults: a meta-analysis of prospective studies. Depress Anxiety. 2015;32:664–70.
Van Dongen HPA, Maislin G, Mullington JM, Dinges DF. the cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26:117–26.
Tkachenko O, Dinges DF. Interindividual variability in neurobehavioral response to sleep loss: a comprehensive review. Neurosci Biobehav Rev. 2018;89:29–48.
Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–21.
Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–69.
Naylor E, et al. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci. 2000;20:8138–43.
Partinen M, Kaprio J, Koskenvuo M, Putkonen P, Langinvainio H. Genetic and environmental determination of human sleep. Sleep. 1983;6:179–85.
Dauvilliers Y, Maret S, Tafti M. Genetics of normal and pathological sleep in humans. Sleep Med Rev. 2005;9:91–100.
Gottlieb DJ, O’Connor GT, Wilk JB. Genome-wide association of sleep and circadian phenotypes. BMC Med Genet. 2007;8:S9.
Gottlieb DJ, et al. Novel loci associated with usual sleep duration: the CHARGE Consortium Genome-Wide Association Study. Mol Psychiatry. 2015;20:1232–9.
Silva ACPe, et al. Melatonin receptor 1B −1193T>C polymorphism is associated with diurnal preference and sleep habits. Sleep Med. 2019;53:106–14.
Chang A-M, et al. Circadian gene variants influence sleep and the sleep electroencephalogram in humans. Chronobiol Int. 2016;33:561–73.
Tucker AM, Dinges DF, Van Dongen HPA. Trait interindividual differences in the sleep physiology of healthy young adults. J Sleep Res. 2007;16:170–80.
Buckelmüller J, Landolt H-P, Stassen HH, Achermann P. Trait-like individual differences in the human sleep electroencephalogram. Neuroscience. 2006;138:351–6.
Landolt H-P. Genetic determination of sleep EEG profiles in healthy humans. Prog Brain Res. 2011;193:51–61.
Friess E, et al. Heritability of sleep electroencephalogram. Biol Psychiatry. 2008;64:344–8.
He Y, et al. The transcriptional repressor DEC2 regulates sleep length in mammals. Science. 2009;325:866–70.
Honma S, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841–4.
Aeschbach D, et al. A longer biological night in long sleepers than in short sleepers. J Clin Endocrinol Metab. 2003;88:26–30.
Aeschbach D, et al. Evidence from the waking electroencephalogram that short sleepers live under higher homeostatic sleep pressure than long sleepers. Neuroscience. 2001;102:493–502.
Aeschbach D, Cajochen C, Landolt H, Borbély AA. Homeostatic sleep regulation in habitual short sleepers and long sleepers. Am J Physiol. 1996;270:R41–53.
Hirano A, et al. DEC2 modulates orexin expression and regulates sleep. Proc Natl Acad Sci USA. 2018;115:3434–9.
Pellegrino R, et al. A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans. Sleep. 2014;37:1327–36.
Shi G, et al. β1adrenergic receptors participate in Sleep Regulation. Neuron 2019 (in press).
Hartmann E, Baekeland F, Zwilling GR. Psychological differences between long and short sleepers. Arch Gen Psychiatry. 1972;26:463–8.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ashbrook, L.H., Krystal, A.D., Fu, YH. et al. Genetics of the human circadian clock and sleep homeostat. Neuropsychopharmacol. 45, 45–54 (2020). https://doi.org/10.1038/s41386-019-0476-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0476-7
This article is cited by
-
A prospective study of the association of weekend catch-up sleep and sleep duration with mortality in middle-aged adults
Sleep and Biological Rhythms (2023)
-
Mapping the associations of daily pain, sleep, and psychological distress in a U.S. sample
Journal of Behavioral Medicine (2023)
-
Sleep and neuropsychiatric illness
Neuropsychopharmacology (2020)
