
A common belief among statistical geneticists is that genome-wide association studiesFootnote 1 (GWAS) rendered candidate gene studiesFootnote 2 obsolete. It is also widely believed that nearly all (if not all) candidate gene hypotheses were incorrect (for psychiatric disordersFootnote 3). The goal of this article is to summarize findings that support these conclusions and further: to highlight a recent surge of fundamental discoveries in psychiatric genetics research. Indeed, many robust genetic risk factors for psychiatric disorders have been discovered, providing novel clues about disease etiology, which can fuel future discoveries.
Reviews of psychiatric GWAS findings are available elsewhere [1, 2]. Briefly, all major psychiatric disorders have now been shown to be polygenic, and for most disorders, specific loci have been identified via GWAS. The critical element for success, for each of the major psychiatric disorders, was increased statistical power, which was almost exclusively achieved via massive increases in sample sizes (i.e. encompassing tens of thousands of participants, or more). Indeed, many early and underpowered GWAS yielded false positives. The Psychiatric Genomics Consortium (PGC) is the world’s largest consortium of psychiatric genetics researchers, and the discoveries described here are largely attributable to the substantial and sustained efforts of the PGC [1, 3]. More recently, biorepositories from the UK Biobank [4], the Million Veterans Program [5], and 23&Me have boosted the discovery of genetic risk variants for many psychiatric disorders. Leading results for specific disorders include schizophrenia* [6], depression* [7, 8], bipolar disorder* [9], substance use disorder [10, 11], PTSD [12,13,14], anorexia [15], ADHD [16], and autism [17, 18] (asterisks denoting GWAS with at least 20 significant loci). In sum, there are already many highly credible risk loci for psychiatric disorders, and discoveries are continuing at a rapid pace.
This article contextualizes the findings that have – in recent years – convinced human genetics researchers that traditional candidate gene studies should no longer be trusted. Indeed, it was only via systematic examination of the genome (via GWAS) that a robust understanding of genetic risk for psychiatric disorders emerged. Results from studies of schizophrenia will be used to illustrate these findings. Schizophrenia [6] is representative of other psychiatric disorders [7,8,9,10,11,12, 15,16,17,18,19] with respect to the genetic effects being described, and – of all the psychiatric disorders – it is best suited for illustrating these points because genetics research progress is most advanced for schizophrenia. Seeing the results (in the plots below) is critical because it explains why genetics researchers have changed their practices so dramatically in the last ten years. It is important that psychiatric and neuroscientific researchers, as well as clinicians, are aware of these developments, given that many of the most commonly referenced genetics findings are not considered to be credible by geneticists [20,21,22,23,24]. For example, candidate gene and gene-environment interaction findings about serotonin-related genes (including 5-HTTLPR, the serotonin transporter linked polymorphic region) are inconsistent with modern genetics findings and highly likely to be incorrect [24].
This article addresses two critical questions:
-
1.
Why are genetic researchers so confident about genetic findings emerging from genome-wide association studies (GWAS)?
-
2.
What are the key genetic findings about psychiatric disorders that have emerged from GWAS? In particular, the text and figures below will clarify the following key points.
-
a.
Large effect variants are rare or non-existent. If they exist, they are easily detected.
-
b.
Genetic risk factors are numerous and diffusely distributed across the genome.
-
c.
Robust genetic risk variants are far more likely to be located in poorly understood regions of the genome than in previously implicated candidate genes.
-
a.
-
1.
GWAS results are reproducible and they surpass an even higher bar: They enable valid predictions in new datasets
Whereas many branches of science are beset with concerns about the reliability of results, this is not one of the limitations of well-powered GWAS. For one, across all of medicine, scientists have observed that most GWAS results are reproducible over time. In other words, the same genetic variants identified by GWAS in one sample, are identified by GWAS in a different sample, assuming that sample sizes are adequate to enable detection, and that ancestry is similar across samples. As an illustrative example, we examine the strongest common-variant association to schizophrenia. As shown below, as larger sample sizes afforded greater power, the statistical significance of this locus increased:
This pattern of results is expected for true results. More broadly, strengthening of statistical evidence for associated loci is the rule and not the exception for large GWAS, as sample sizes increase. In contrast, results from candidate gene studies became less clear, not more clear, as additional studies accumulated.
GWAS results are reliable (as described above), but they also meet an even a higher standard of evidence: they afford correct predictions in novel datasets. Specifically, one can use the results from a well-powered GWAS to predict phenotypes in a new study (i.e. among different individuals), using only genotypes and no phenotypic information. This practice is referred to as polygenic risk scoring (PRS; also risk profile scoring = RPS, polygenic scoring, and genetic scoring). Polygenic scoring was first introduced in 2007 by Wray and colleagues [27]. In 2009, Purcell and colleagues [25] used polygenic scoring to demonstrate polygenic influences on schizophrenia. Now polygenic scores are widely used by researchers [28, 29] and companies like 23&Me.
To be clear: polygenic predictions are very far from perfect; they are not diagnostic and (at present) they explain relatively small amounts of phenotypic variance. The modest predictive ability of polygenic predictors notwithstanding, successful demonstrations of polygenic prediction in hundreds of studies have bolstered the genetics community’s trust in GWAS results. As noted by Dudbridge [30] and others, apparent failures of polygenic prediction often occur when sample sizes are too small to expect statistically significant polygenic predictions. In sum, the ability of GWAS to yield reliable results, which enable correct predictions in novel datasets, is uncontroversial in the genetics community.
-
2.
Properties of genetic risk factors for psychiatric disorders
-
2a.
Large effect variants are rare or non-existent. If they exist, they are easily detected
Based on available GWAS of psychiatric disorders, one important take home message is already clear: large effect variants – meaning variants that explain a substantial amount of phenotypic variance in the population – do not exist. In contrast, some very rare genetic variants have large per-allele effects on schizophrenia (e.g. odds ratio > 10) [31]. Importantly, both of these observations are consistent with evolutionary theory, and with one another. See Box 1 for an important distinction regarding effect size terminology.
Some readers may wonder how geneticists can be sure that large effect sizesFootnote 4 do not exist for psychiatric disorders, but there is a straightforward explanation: large effect sizes are easy to detect. If variants explained large amounts of phenotypic variance in the population, they would have been discovered decades ago using linkage studies, and easily replicated. Indeed, some conditions, like Huntington’s disease and cystic fibrosis are caused by large effect variants, and some complex genetic phenotypes like Alzheimer’s disease have large effect variants (APOE4), and consequently detection of relevant risk loci for these conditions was comparatively easy. The fact that large effect variants have not been found for psychiatric disorders suggests that such variants do not existFootnote 5.
Figure 1 shows that large effect, common variants do not exist for schizophrenia, and it illustrates a number of additional principles about genetic risk factors for psychiatric disorders. Points in Fig. 1 represent the 128 known common-variant risk loci for schizophrenia [6]. For each variant, minor allele frequency is depicted on the x-axis and effect size, in odds ratio (OR), is on the y-axis. As the shaded blue region depicts, there is an apparent upper bound on effect sizes of schizophrenia risk variants, which varies according to minor allele frequency: the largest observed effect sizes decrease, as frequency increases. Presumably, natural selection ensures that large per-allele effects are not permissible at even modest allele frequencies. For example, the largest effect common variant has an odds ratio of ~1.32 and a minor allele frequency of 5%, see left-most point.
Fig. 1 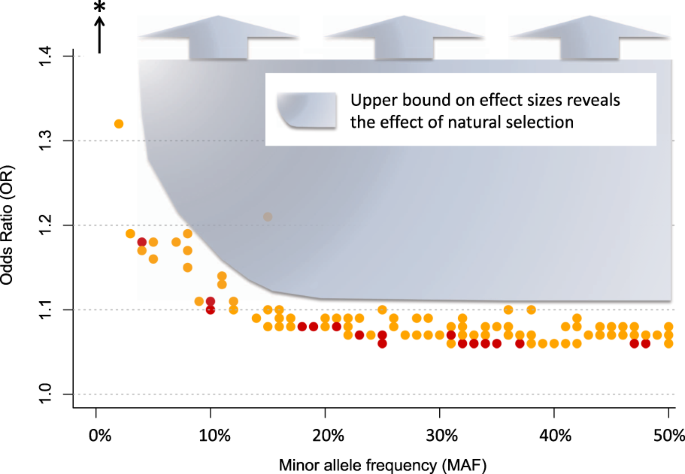
There is an upper bound on the effect sizes of common schizophrenia risk variants, which becomes increasingly stringent as minor allele frequency increases. The shaded region denotes coordinate space in which schizophrenia risk loci have not been not detected (e.g for variants with minor allele frequency greater than 20%, no risk variant has an effect size greater than 1.11). The bottom of the shaded region follows the curved upper bound of effect sizes for schizophrenia risk variants. Note that point color (yellow and red) denotes the sample size used to detect the locus (approximately 80,000 and 150,000, respectively). The black arrow and asterisk represent rare ( <1%) schizophrenia risk variants with larger per-allele effects (e.g. 22q11.2 deletions)
To make the point another way, statistical power in multiple psychiatric GWAS for large effect variants has been nearly 100%, and therefore large effect, common variants would have been easy to detect. This means that there is no shortcut for identification of risk loci. Large samples are needed to detect rare variants (because they are rare), and large samples are needed to detect common variants (because the per-allele effects are always small).
Another important observation about the detection of risk loci is demonstrated by point color in Figs. 1 and 2. Specifically, larger effect variants are easier to detect, and therefore they can (on average) be detected with smaller samples. In Fig. 2, boxplots are used to compare effect sizes of variants according to the sample size of detection [6]. Per expectations, the first variants detected (yellow) had larger effect sizes than those that were detected with the larger sample size (red) (Welch t test p = .03, Wilcoxon rank-sum test p = .001). An extension of these findings is that more risk variants will be detected with increasing sample sizes.
Fig. 2 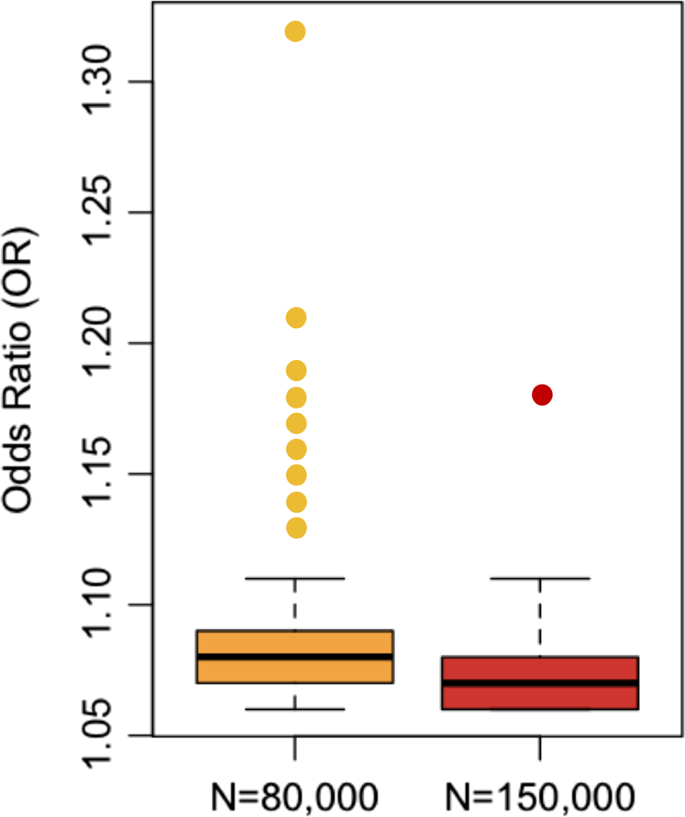
On average, the largest effect loci are detected first in GWAS. As power increases (typically through increased sample size), smaller effect variants are discovered. Each point represents one of the 128 variants from the 2014 publication of the Psychiatric Genomics Consortium (PGC) Schizophrenia Group. Of the 128 loci, 105 variants were significant associated with schizophrenia (i.e. p < 5 × 10−8) in the smaller sample size of approximately 80,000 (yellow). An additional 23 variants reached genome-wide significance using the larger sample size of approximately 150,000 (red, mostly additional control samples)
In Fig. 3, the shaded orange region reflects the coordinate space of schizophrenia risk alleles that are likely to be detected in the future. Specifically, evidence suggests that additional risk alleles will be found across the range of minor allele frequency (from ~0 to 50%), and they will have effect sizes, on average, smaller than the risk variants that have already been detected (i.e. the shaded region is below variants that have already been identified).
Fig. 3 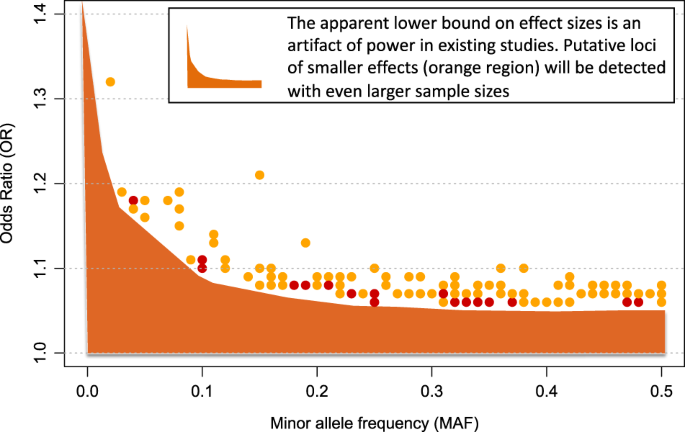
Additional risk variants for schizophrenia will be detected with larger sample sizes. The shaded orange region represents the combination of effect sizes and allele frequencies (of risk variants) that will be detectable with larger sample sizes
-
2b.
Genetic effects are numerous and diffusely distributed across the genome.
A second important principle that has emerged from GWAS is that genetic effects on complex genetic phenotypes are numerous and diffusely distributed across the genome. Current estimates for the true number of schizophrenia risk variants number in the thousands, and over 200 loci have been identifed in unpublished work. GWAS of height identified 697 specific loci [32] with many more not yet exceeding genome-wide significance. For complex genetic phenotypes such as height, body mass index (BMI), and all psychiatric disorders, polygenicity has been convincingly demonstrated.
Regarding the distribution of genetic risk variants, polygenic effects are distributed across the genomeFootnote 6. Yang and colleagues [33] provided a common-sense demonstration of this point, by showing that chromosome length predicts the amount of phenotypic variance explained. In other words, they used natural categories (chromosomes) for partitioning the genome into discrete sections, and they observed that longer chromosomes also explain more phenotypic variance (on average). These and other findings suggest that researchers should expect that risk variants for psychiatric disorders are distributed across the genome.
Thus, it is well established that genetic risk for complex genetic phenotypes is diffusely distributed across the genome. With this established, important new questions are being asked about the biological effects of genetic risk variants, together and individually. For example, multiple methods are designed to quantify the importance of particular regions of the genome (e.g. a particular set of genes) for a given phenotype, and other methods can be used to determine which brain regions and cell types are most relevant [34,35,36,37]. Additional studies are needed to determine the biological relevance of GWAS-nominated candidate genes and loci (as distinguished from traditional candidate genes), an excellent example of which is provided by Sekar et al. regarding C4 alleles at the chromosome 6 MHC locus for schizophrenia [38]. In sum, it is known that genetic risk for psychiatric disorders is distributed across the genome, and an important next step is to determine the biological relevance of GWAS.
-
2c.
Risk variants for psychiatric disorders nearly always implicate regions of the genome that are poorly understood, rather than “expected” regions like candidate genes
Having established that effect sizes for schizophrenia and other psychiatric disorders are small, and that there are many – likely thousands – of risk variants, we can also ask: where are the risk variants located? In particular, we can categorize the locations (e.g. within genes, between genes) and whether or not robust GWAS loci are located in candidate genes. For schizophrenia, sufficient evidence is already available to address these questions, as shown below and previously reported [23]. In Fig. 4, all of the genetic variants in the top 128 schizophrenia loci [6] are plotted by category of functional location in the genome [39]. Note that there are many thousands of variants in this analysis because all variants in linkage disequilibrium with the top variant at each locus are included in this analysis. For schizophrenia, it is clear that few variants are located in exons (i.e. approximately 1% in exons), a finding that is consistent with systematic studies about GWAS loci in the broader genetics literature [40].
Fig. 4 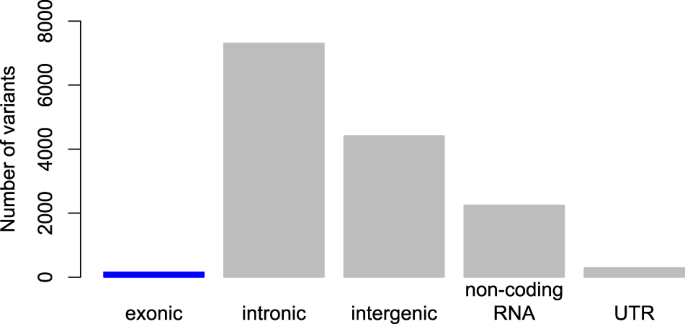
Location of schizophrenia risk variants reveals that very few variants (only 1.1%) are in the most strongly hypothesized regions of the genome, exons, which code for proteins. This demonstrates that current biological knowledge is insufficient to correctly specify most candidate genes/variants given that nearly all known schizophrenia variants fall in relatively poorly understood regions of the genome. This pattern of findings is typical for complex genetic phenotypes
-
2a.




As shown in Fig. 4, empirical schizophrenia data reveal that the most strongly associated variants are not where scientists originally expected them to be. A priori, researchers expected that risk loci would be identified in genes. This figure also demonstrates why exome sequencing is not expected to be a “magic bullet” for psychiatric gene discovery. Indeed, an exome sequencing study with over five thousand participants failed to identify any statistically significant individual risk loci for schizophrenia [41].
Performance of candidate genes, and candidate polymorphisms, in GWAS
Regarding the status of classic candidate genes, it is now possible to determine how well candidate genes have fared in GWAS. The simple answer is that they have fared poorly, as reported already for schizophrenia [23] and depression [24]. Indeed, very well-powered GWAS (e.g. with sample size > 100,000) are available for schizophrenia [23, 42] and depression [7, 8], so we use these two disorders as examples to illustrate the poor performance of candidate gene studies. Preliminary evidence suggests that the same conclusions will be true for other psychiatric disorders (though note a small number of exceptions for substance use phenotypes, for which correct candidate genes, if not the correct candidate polymorphisms, were hypothesized previously [10]).
For depression, the top 44 loci do not implicate any candidate genes [7]. For example, the much-studied serotonin transporter linked polymorphic region (5-HTTLPR, a polymorphism in the promoter region of the serotonin transporter gene) was not associated with depression [24]. Further, the brain derived neurotrophic factor (BDNF) gene, which has been included in many candidate gene studies of depression (including one [43] authored by the first author of this paper) was not supported in the best powered GWAS of depression [7]. Put another way, the sum of available evidence suggests that findings from Wray et al (2018) should entirely supersede Duncan et al (2009) and all other candidate gene studies of depression.
Similarly, Johnson and colleagues systematically examined schizophrenia candidate genes and reported, “as a group, variants in the most-studied candidate genes were no more associated with schizophrenia than were variants in control sets of non-candidate genes” [23]. For most schizophrenia candidate genes, this conclusion is easy to see: the 128 known schizophrenia loci do not overlap any portion of most candidate genesFootnote 7. For a few genes, however, a more nuanced explanation is needed, as explained here for the D2 dopamine receptor (DRD2) gene. DRD2 was a natural candidate gene for schizophrenia because it is a target of all effective antipsychotic medications, and it has been examined numerous times using candidate gene methodology. Notably, genetic variants spanning a portion of the DRD2 gene exceeded genome-wide significance in the best powered GWAS of schizophrenia [6] (see Fig. 5, orange line denotes genome-wide significance).

demonstrates that one seemingly true result from the candidate gene era (about DRD2 and schizophrenia) was not actually supported by GWAS results. The orange line denotes genome-wide significance (−log10(5 × 10−8) = 7.3), and multiple variants on the right side of the figure exceed genome-wide significance. Each point in the figure denotes one genetic variant on chromosome 11 in the region around the D2 dopamine receptor gene (DRD2). The fact that multiple variants exceed genome-wide significance reflects linkage disequilibrium (i.e. correlated alleles). Linkage disequilibrium does not extend, however, to the candidate polymorphism on the left, denoted by the long arrow
In Fig. 5, the right arrow indicates the most significant variants in the DRD2 locus, from the best powered GWAS of schizophrenia [6]. In contrast, the DRD2 polymorphism often studied in schizophrenia candidate gene studies (left arrow) is not associated with schizophrenia in the best powered data (i.e., the relevant point is well below genome-wide significance). Thus, even though this candidate gene hypothesis was generally correct (i.e. genetic variation in DRD2 is associated with schizophrenia), the candidate gene polymorphism (left arrow) from candidate gene studies is not associated with schizophrenia. Precisely defined hypotheses are necessary in science, as the gene map of DRD2 in Fig. 5 demonstrates, wherein specification of correct polymorphisms is necessary to accurately describe a link between the DRD2 gene and schizophrenia.
Figure 5 also demonstrates another limitation of the candidate gene era (i.e. drastically limited scope of analysis compared to GWAS). Indeed, readers can appreciate the size of the genome by observing how many common genetic variations (i.e. points in Fig. 5) are found in just this one small portion of chromosome 11 around DRD2 (positions 113,250,000 to 113,380,000 on chromosome 11). It is therefore understandable that a big data approach was necessary to systematically determine which genetic variants are associated with schizophrenia (and the same is true for other psychiatric disorders).
In sum, available data suggest that candidate genes – or at least the specific polymorphisms studied in the candidate gene era – are not supported by GWAS [23]. Large-scale GWAS are well powered to detect genetic effects in or near candidate genes, and their failure to implicate candidate genes – while implicating many other loci – is informative and should be sufficient to reject candidate gene hypotheses. Most promisingly, what has emerged (and is still emerging) from GWAS is a set of novel variants that provide clues about psychiatric disease etiology.
Conclusion
In this article, we sought to answer two key questions: 1) Why are genetic researchers so confident about genetic findings emerging from GWAS?; and 2) What are the key genetic findings about psychiatric disorders that have emerged from GWAS? In brief, the reason that geneticists trust GWAS results is because the results have proven to be reliable indicators of psychiatric disease risk [6, 8, 44, 45]. The key genetic findings that have emerged include novel risk variants [6, 7, 9, 10, 16], the fact that large-effect variants are rare or non-existent for psychiatric disorders, that true genetic risk for psychiatric disorders is diffusely distributed across the genome, and the fact the robust risk variants for psychiatric disorders typically fall in poorly understood regions of the genome, and not in the places originally expected (i.e. candidate genes). These findings, which were only made possible by large-scale genome-wide approaches, provide entirely novel clues about the etiology of psychiatric disorders. These modern genetic findings also demonstrate why candidate gene studies are no longer trusted by geneticists. Specifically, candidate gene studies nearly always hypothesized the wrong portions of the genome (and they may have always hypothesized the wrong variants), they hypothesized effect sizes larger than those that exist in nature. Further, candidate gene studies are methodologically inadequate in their ability to account for subtle differences in ancestry and relatedness, which can confound results. In sum, it is time to abandon candidate gene studies, and the results that they produced, in favor of the numerous highly reliable results that have emerged from GWAS.
Proponents of candidate gene studies may note that we have not considered endophenotypes or more precise phenotyping. In brief, while it is theoretically possible that alternative phenotypes will reveal simpler genetic architecture involving larger effect variants, in practice this has not been observed. More careful phenotyping can increase power, but evidence suggests that power gains are modest; the CONVERGE results for depression may be the best psychiatric example [46]. Perhaps a more informative way of viewing the evidence is to consider GWAS results for height, a trait which can be measured with high reliability and validity. Notably, the genetic architecture of height is very similar to that of schizophrenia, in that there are no large effect variants for height (that is, there are no common variants with large effects), and height is highly polygenic. Thus, GWAS results for schizophrenia and other psychiatric disorders are entirely consistent with the broader body of modern genetics findings.
For those that are interested in the best-available GWAS results for any psychiatric disorder, there are a few tips that can aid identification of appropriate resources. A good rule of thumb is that the largest GWAS available to date, for any given psychiatric disorder, is typically the best powered study of common variant contributions to disease risk. Indeed, “small” GWAS with under 20,000 participants may yield few or no genome-wide significant loci, and those that are reported are less likely to be replicated. For such studies, polygenic results are often the most informative findings. One excellent web-resource for GWAS results is the PGC’s website: http://www.med.unc.edu/pgc/.
At this juncture it is arguably time to declare that the candidate gene era has ended. Approximately $250 million was spent [1, 23]. Graduate students, postdocs, and professors devoted portions of their lives to these investigations. Yet, a more comprehensive and more statistically robust approach has emerged, and the results from GWAS have rendered old ideas and technology obsolete. Risk loci from large-scale genomic approaches provide new avenues for research, and GWAS results ground psychiatric phenotypes more firmly in biology than ever before. Ideally, these results will advance personalized medicine, particularly for response to drugs. The novel and robust clues about biological processes underlying psychiatric disorders – that have emerged from GWAS – will almost certainly underpin our future understanding of psychiatric disease etiology.
Funding and disclosure
In the past 36 months: Dr. Ostacher has been a consultant to Alkermes, Johnson & Johnson (Janssen), Lundbeck, Otsuka, Sage Therapeutics, and Supernus Pharmaceuticals, and he has received research funding from Palo Alto Health Sciences, Inc. Dr. Ballon has served as a consultant to Alkermes, Pear Therapeutics, and Alto, and he has received research funding from Otsuka, Alkermes, Janssen, and Roche.
Notes
It is common to use the abbreviation “GWAS” for both the singular and plural forms of genome-wide association study/studies. “GWASs” is sometimes used to denote the plural (i.e. more than one GWAS).
Throughout the remainder of this manuscript, the term “candidate gene study” refers to traditional candidate gene studies, meaning studies that test for an association between one or a small number of polymorphisms and a phenotype of interest (e.g. depression), without examining genome-wide data. These “traditional” candidate gene studies should be distinguished from a different class of studies, focused on candidate genes identified through genome-wide searches. The latter is an important area of ongoing research.
There are potential exceptions for substance use disorders.
Experts have discrepant opinions about whether or not 22q11.2-linked schizophrenia is the same as “typical” schizophrenia, and broader questions about potential genetic subtypes of schizophrenia are not yet settled.
Meaning variants that explain a large fraction of phenotypic variance in the population; see Box 1.
Here we state the common-sense conclusion, that “such variants do not exist.” Technically: hypotheses about large effect variants have been repeatedly rejected, in large, high-quality studies.
Efforts to determine the portions of the genome most likely to harbor GWAS loci are ongoing, and the most consistent finding to date is that GWAS loci are concentrated in portions of the genome that are evolutionarily conserved across species.
Note that in practice, merely observing presence/absence of overlap of a GWAS locus with a candidate gene is insufficient to include or exclude that gene’s putative relevance to disease, but discussion of this is beyond the scope of this paper. For this paper, it is sufficient to ask the question of whether or not the specific hypotheses put forward by candidate gene studies are supported or not by GWAS results.
References
Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Børglum AD, Breen G, et al. Psychiatric genomics: an update and an agenda. Am J Psychiatry. 2018;175:15–27.
Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13:537–51.
Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012. https://doi.org/10.1038/nrg3240.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203.
Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J Clin Epidemiol. 2016;70:214–23.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 Loci Associated with Bipolar Disorder. BioRxiv. 2018:173062.
Walters RK, Polimanti R, Johnson EC, McClintick JN, Adams MJ, Adkins AE, et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat Neurosci. 2018;21:1656.
Sanchez-Roige S, Palmer AA, Fontanillas P, Elson SL, 23andMe Research Team, the Substance Use Disorder Working Group of the Psychiatric Genomics Consortium, Adams MJ, et al. Genome-wide association study meta-analysis of the alcohol use disorders identification test (AUDIT) in two population-based cohorts. Am J Psychiatry. 2019;176:107–18.
Duncan LE, Ratanatharathorn A, Aiello AE, Almli LM, Amstadter AB, Ashley-Koch AE, et al. Largest GWAS of PTSD (N=20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry. 2018;23:666–73.
Nievergelt C, Maihofer A, Dalvie S, Duncan L, Ratanatharathorn A, Ressler K, et al. 157. large-scale genetic characterization of PTSD: addressing heterogeneity across ancestry, sex, and trauma. Biol Psychiatry. 2018;83:S64.
Stein M, Gelernter J, Zhao H, Sun N, Pietrzak R, Harrington K, et al. 159. GWAS of PTSD re-experiencing symptoms in the VA million veteran program. Biol Psychiatry. 2018;83:S64–5.
Duncan LE, Yilmaz Z, Gaspar H, Walters R, Goldstein J, Anttila V, et al. Significant locus and metabolic genetic correlations revealed in genome-wide association study of anorexia nervosa. Am J Psychiatry. 2017;174:850–8.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431.
Anney RJL, Ripke S, Anttila V, Grove J, Holmans P, Huang H, et al. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 2017;8:21.
Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–94.
Colhoun HM, McKeigue PM, Smith GD. Problems of reporting genetic associations with complex outcomes. Lancet. 2003;361:865–72.
Sullivan PF. Spurious genetic associations. Biol Psychiatry. 2007;61:1121–6.
Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry. 2011;168:1041–9.
Johnson EC, Border R, Melroy-Greif WE, de Leeuw CA, Ehringer MA, Keller MC. No evidence that schizophrenia candidate genes are more associated with schizophrenia than noncandidate genes. Biol Psychiatry. 2017;82:702–8.
Border R, Johnson EC, Evans LM, Smolen A, Berley N, Sullivan PF, et al. No support for historical candidate gene or candidate gene-by-interaction hypotheses for major depression across multiple large samples. Am J Psychiatry. 2019;appi.ajp.2018:18070881.
Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52.
Psychiatric GWAS Consortium - PGC - Schizophrenia. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–76.
Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–8.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–24.
Wray NR, Lee SH, Mehta D, Vinkhuyzen AAE, Dudbridge F, Middeldorp CM. Research review: polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry. 2014;55:1068–87.
Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013;9:e1003348.
Rees E, Walters JTR, Georgieva L, Isles AR, Chambert KD, Richards AL, et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br J Psychiatry. 2014;204:108–14.
Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–86.
Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43:519–25.
Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–35.
Pers TH, Timshel P, Ripke S, Lent S, Sullivan PF, O’Donovan MC, et al. Comprehensive analysis of schizophrenia-associated loci highlights ion channel pathways and biologically plausible candidate causal genes. Hum Mol Genet. 2016;25:1247–54.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. Genetic identification of brain cell types underlying schizophrenia. Nat Genet. 2018;50:825–33.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83.
Watanabe K, Taskesen E, Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–7.
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–90.
Sullivan PF. How good were candidate gene guesses in schizophrenia genetics? Biol Psychiatry. 2017;82:696–7.
Duncan LE, Hutchison KE, Carey G, Craighead WE. Variation in brain-derived neurotrophic factor (BDNF) gene is associated with symptoms of depression. J Affect Disord. 2009;115:215–9.
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22.
Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24.
CONVERGE Consortium. Sparse swhole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–91.
Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R, Borrow J, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci USA. 1995;92:7612–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Duncan, L.E., Ostacher, M. & Ballon, J. How genome-wide association studies (GWAS) made traditional candidate gene studies obsolete. Neuropsychopharmacol. 44, 1518–1523 (2019). https://doi.org/10.1038/s41386-019-0389-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0389-5
This article is cited by
-
Dimensional and transdiagnostic phenotypes in psychiatric genome-wide association studies
Molecular Psychiatry (2023)
-
Genome-wide association study of traumatic brain injury in U.S. military veterans enrolled in the VA million veteran program
Molecular Psychiatry (2023)
-
Clinical characteristics indexing genetic differences in schizophrenia: a systematic review
Molecular Psychiatry (2023)
-
Biobank-scale methods and projections for sparse polygenic prediction from machine learning
Scientific Reports (2023)
-
AmelHap: Leveraging drone whole-genome sequence data to create a honey bee HapMap
Scientific Data (2023)
