
Abstract
Nicotine dependence and major depressive disorder (MDD) are highly comorbid, yet causal links between these prevalent disorders are unclear. One possible mechanism is that nicotine ameliorates MDD-related neurobiological dysfunction in specific networks. For instance, cortico-striatal circuitry is enhanced by nicotine, and such paths are disrupted in individuals with MDD. Specifically, MDD has been associated with reduced connectivity between the nucleus accumbens (NAc) and rostral anterior cingulate cortex (rACC) but enhanced connectivity between the dorsal striatum (DS) and dorsolateral prefrontal cortex (DLPFC). Determining whether nicotine normalizes these circuits in non-smokers with MDD may elucidate mechanisms underlying links between disorders. This was tested by administering placebo and a 2-mg dose of nicotine to unmedicated non-smokers with and without MDD prior to collecting resting-state functional magnetic imaging data using a cross-over design. On placebo, individuals with MDD showed significantly reduced NAc–rACC and a trend for enhanced DS–DLPFC functional connectivity relative to healthy controls. In MDD, acute nicotine administration normalized both pathways to the level of healthy controls, while having no impact on healthy controls. Nicotine’s effects on NAc–rACC connectivity was influenced by anhedonia, consistent with the role of this network in reward and nicotine’s ability to enhance reward deficiencies in MDD. These results indicate that nicotine normalizes dysfunctional cortico-striatal communication in unmedicated non-smokers with MDD. Nicotine’s influence on these circuitries highlights a possible mechanism whereby individuals with MDD are more vulnerable to develop nicotine dependence. Findings suggest that nicotinic agents may have therapeutic effects on disrupted cortico-striatal connectivity.
Similar content being viewed by others
Introduction
Individuals with major depressive disorder (MDD) are almost twice as likely to smoke tobacco relative to the general population [1, 2]. However, the causal link between these two highly comorbid disorders remains elusive with a recent meta-analysis providing mixed results as to which disorder precedes the other [3]. These mixed findings support two competing theories. Specifically, the self-medication model suggests that individuals with MDD smoke to mitigate mood-related symptoms while the alternative theory posits that nicotine use predisposes individuals to experience mood disorders. Disentangling the causality of these disorders is difficult as the majority of research is retrospective and focuses on well-established smokers, in whom neurobiological changes due to chronic use have likely already occurred. Furthermore, nicotine’s acute impact on the brain is complex, as nicotine can activate and desensitize different nicotinic acetylcholine receptors (nAChRs) suggesting that nicotine’s influence may depend on the specific neurobiological pathway studied [4].
Determining nicotine’s influence in specific neurobiological domains prior to changes due to long-term tobacco smoking is one way to clarify the link between MDD and the initiation of nicotine dependence. One possibility is that MDD-related abnormalities within cortico-striatal pathways predispose those with MDD to develop nicotine dependence. Relative to healthy controls (HC), individuals with MDD have reduced resting-state connectivity within the brain reward pathway, particularly between the nucleus accumbens (NAc) and medial prefrontal cortex (mPFC)/rostral anterior cingulate cortex (rACC) [5, 6]. Of relevance, reduced resting-state NAc–mPFC connectivity has been linked to anhedonia among HC [7] and reduced ability to sustain positive mood after rewards in those with a history of MDD [8], linking anhedonic symptoms with deficient cortico-striatal communication. Anhedonia has been associated with poor smoking cessation outcomes [9,10,11], suggesting that disrupted reward function plays a role in both disorders. Critically, preclinical work has shown that nicotine enhances reward-related pathway sensitivity [12, 13]. Such findings may explain nicotine’s ability to increase reward responsiveness in both healthy [14] and depressed [15, 16] samples and highlight a possible mechanistic explanation of links between MDD and nicotine dependence.
There also is evidence that MDD is characterized by disruptions in other cortico-striatal paths including increased connectivity between the dorsal striatum (DS) and dorsolateral prefrontal cortex (DLPFC) [5], particularly with greater disease severity [17]. Enhanced baseline DS–DLPFC connectivity predicted favorable response to transcranial magnetic stimulation (TMS) [18]. Treatment response was associated with a trend level decrease in DS–DLPFC connectivity, suggesting that reduced DS–DLPFC connectivity correlates with MDD symptom reduction.
In the current study, non-smokers with and without MDD were administered placebo or nicotine prior to resting-state functional magnetic resonance imaging (fMRI). We hypothesized that nicotine would normalize the reduced NAc–rACC and enhanced DS–DLPFC connectivity previously reported in those with MDD. Given the role of anhedonia in both disorders its putative moderating effect was considered. Finally, graph theory was used to explore nicotine’s impact on brain connectivity more broadly.
Methods
Participants
Participants included 35 non-smokers reporting <20 lifetime uses of nicotine, no nicotine use in the past year, and an expired carbon monoxide (CO) level of <5 ppm (Table 1). Eighteen participants met SCID-IV criteria for MDD while the remaining 17 were HC. Groups were matched on sex (eight women/group), age, and education (Table 1). The structured clinical interview for DSM-IV-TR [19] was used to exclude participants with the following: lifetime history or current diagnosis of any of the following psychiatric illnesses: organic mental disorder, schizophrenia, schizoaffective disorder, delusional disorder, psychotic disorders not otherwise specified, bipolar disorder, ADHD, patients with mood congruent or mood incongruent psychotic features. For the MDD group, simple phobia, social anxiety disorder and generalized anxiety disorders were allowed only if secondary to MDD. Participants also were excluded if they had a lifetime history of electroconvulsive therapy (ECT), if they failed to meet standard MRI safety requirements, used any anticholinergic drugs in the past week or had a history or current cardiac problems including known arrhythmias, acute coronary syndrome, or ischemic heart disease, or reported serious or unstable medical illness, lifetime history of seizure disorder. Pregnancy and nursing were excluded as was the use of any psychotropic medication or illicit substance. Pregnancy and drug use were ascertained by urinalysis. Participants were required to have a breath blood alcohol level of zero (Alco-Sensor IV, Intoximeters, St Louis, MO). All participants with MDD were unmedicated at the time of the study and were unable to have used the following: fluoxetine (6-weeks prior), any other antidepressant (2-weeks prior), neuroleptics (6-months prior), and benzodiazepines (2-weeks prior). Subjects provided written informed consent after receiving a complete description of the study. These participants were part of a larger study evaluating the impact of nicotine on brain function more broadly.
Questionnaires and peripheral measurements
Prior to drug administration, participants completed the Hamilton Depression Rating Scale [20], Hamilton Anxiety Rating Scale [21], and Snaith-Hamilton Pleasure Scale (SHAPS) [22] scales (Table 1). To evaluate acute changes in mood, the state version of the Positive and Negative Affect Schedule (PANAS) [23] was administered before and ~2 h after drug administration. Heart rate and blood pressure were measured at these time points to confirm nicotine’s influence on the periphery (see Supplement). Due to time constraints one MDD participant was unable to provide post-scanning mood and cardiovascular data on the nicotine day. Cotinine, the primary metabolite of nicotine, was measured after scanning as were the side effects (see Supplement).
Study drug
Nicotine and placebo were administered on separate study days ~1 week apart. On the study day, drugs were administered 1 h prior to fMRI scanning to attain peak plasma nicotine levels during scanning. Peak plasma levels for the 2-mg lozenge used in this study are reached at 1 h and have a half-life of 2.3 h [24]. On both nicotine and placebo study days, participants were asked to place the lozenge inside their mouth, next to their cheek and let it dissolve without chewing. This took ~15 min. A 2-mg dose of nicotine was administered in the form of a lozenge (Nicorette Lozenge, GlaxoSmithKline, Brentford London). Placebo was a Tums antacid (GlaxoSmithKline, Brentford London). Both the nicotine and placebo lozenges were mint flavored, roughly the same size, shape, and color and could only be identified through careful side-by-side inspection, which was not possible as these drugs were administered ~1 week apart (see Supplementary Figure 1). At the beginning of the study, the PI (AJ) created the blind so drug could be administered in a randomized, counterbalanced, double-blind manner. This schema resulted in no group difference in drug-administration order (χ2 = 0.77). At the onset of the study, alphabetized subject-specific containers were created that held study drug 1 and 2 (the order of which was randomize/counterbalanced). These containers were kept in a locked drug cabinet and were accessed only when administering drug. When a subject entered the study, she/he was assigned the next available drug code letter such that they received both study drug 1 and 2 from the same container. Nicotine administration order had no impact on brain coupling as demonstrated in the supplement. To maintain the blind, the lozenges were delivered to the participant on a plate with any identifying marks placed face down and the participant was asked to place the lozenge directly in their mouth without any inspection. A 2-mg dose of nicotine was chosen to reduce potential side effects of nicotine at higher doses; in addition, a 2-mg nicotine lozenge was chosen as this yields 1-mg of systemic nicotine [24], which is comparable to smoking a cigarette [25]. Thus, we expected that this dose would induce brain changes comparable to those following exposure to nicotine during more naturalistic tobacco smoking.
Functional neuroimaging
Scans were conducted on a Siemens Trio 3T scanner (Erlangen, Germany) with a 32-channel head coil. Multiecho multi-planar rapidly acquired gradient echo-structural images and multi-band fMRI were collected using standard parameters used previously [26]. Multiecho multi-planar rapidly acquired gradient echo-structural images were acquired with the following parameters (TR = 2.1 s, TE = 3.3 ms, slices = 128, matrix = 256, 256, flip angle 7°, resolution 1.0×1.0×1.33 mm). Gradient echo-planar images were collected during a 6-min resting state. Slices were acquired aligned to the anterior and posterior commissures and the phase encode direction was set from the posterior to anterior direction to prevent prefrontal signal loss. A multi-band acquisition was conducted using the following parameters (TR = 0.72 s, TE = 0.32 s, multi-band acceleration factor = 8, flip angle 66°, slices = 64, voxel size = 2.5×2.5×2.5 mm). During the resting state, participants were asked to keep their eyes open.
fMRI pre-processing
Briefly, tools from the fMRI of the Brain (FMRIB) Software Library (FSL; www.fmri-b.ox.ac.uk/fsl) were used and standard pre-processing was applied (motion correction with MCFLIRT, brain extraction using BET, slice time correction, spatial smoothing with a Gaussian kernel of full-width half-maximum 6 mm, and high-pass temporal filtering). To prevent motion and other noise sources, each participant’s data were denoised using FSL’s multivariate exploratory linear decomposition into ICs (MELODIC). First, MELODIC was used to identify all ICs on an individual basis. This individual spatial and temporal information was then visually inspected and components representing noise, including motion, were regressed out using FSL’s fsl_regfilt command. Even prior to this denoising step motion (the absolute mean displacement) was minimal across the nicotine (0.28 mm ± 0.04) and placebo visits (0.26 mm ± 0.06). When considering absolute mean displacement, there was no effect of Group (MDD, HC), Drug (nicotine, placebo) or an interaction (F1,33 = 0.54, P = .47) of physiological signals such as heart rate and respiration was possible given the fast TR (0.72 s).
A seed-based analysis was used to evaluate cortico-striatal connectivity. The striatal subregions were defined using the striatal parcellations by Choi et al. [27] (Supplementary Figure 2). The right and left NAc were defined as well as the dorsal striatal subregion that shows coupling with the frontoparietal network, a common resting-state network that encompasses the DLPFC. The rACC was comprised of a 5 mm sphere located at MNI coordinates x = 0, y = 38, z = −4, which is just anterior of the genu of the corpus callosum and overlaps with the portion of the rACC emerging as being influenced by nicotine in a recent meta-analysis [28]. The right and left DLPFC seeds were located at MNI coordinates ±42, 38, 28, which falls within Brodmann’s area 9/46 [29]. Specific coordinates were highlighted in the review by Curtis and D’Esposito [30] (Supplementary Figure 3). Using methods described in our prior work [26], the average time courses for these regions of interest were demeaned, detrended, Hamming windowed, and correlated. A single correlation value (r) was identified for each network (right NAc–rACC, left NAc–rACC, right DS—right DLPFC, left DS—left DLPFC) and each participant under both drug conditions and z-transformed.
Statistics
The interaction of study drug and group on cortico-striatal connectivity was evaluated using repeated measure analysis of variance (ANOVAs) in SPSS (IBM SPSS Statistics Version 24). Questionnaires and peripheral measures noted above also were evaluated by repeated measure ANOVAs. Significant ANOVA findings were followed up using post hoc t-tests.
Graph theory
The brain was parcellated into 129 nodes using the Yeo atlas for the cortex [31], the Choi atlas for the striatum [27] and the Automated Anatomical Labeling atlas for the amygdala (http://www.gin.cnrs.fr/en/tools/aal-aal2/). Graph theory metrics were carried out as defined in the Supplement. We evaluated the global functional network organization focusing on the efficiency of the network using global efficiency [32, 33], local efficiency, and vulnerability. Global efficiency is calculated by considering the efficiency of all nodes concurrently. In contrast, local efficiency evaluates only each node’s neighbors and they are averaged over the network to provide a measure of the whole network. Vulnerability is a proportional drop in global efficiency when a node is removed from the graph. Global vulnerability was calculated, which is the maximum vulnerability for all of its nodes. All network metrics were calculated using R packages (igraph and brainGraph).
Results
Cotinine
An ANOVA revealed a significant main effect of Drug (nicotine, placebo; F1,30 = 154.8,0 P < .001) but no Group (MDD, HC) × Drug interaction (F1,30 = 0.18, P = .67) as cotinine rose significantly in both groups following nicotine administration. A lack of cotinine was confirmed on the placebo day in both groups. Three of the HCs were unable to provide a blood sample, but cotinine levels were evaluated using urinalysis. While urine samples confirmed the presence of cotinine on the nicotine day, these values could not be combined with the serum data.
PANAS
An ANOVA on positive affect was run entering Group, Drug, and Time (pre, post drug administration). A main effect of Group emerged (F1,32 = 39.66, P < .001) as the HC reported overall higher positive affect relative to MDD (averaged across study days and time points). The Group × Time interaction was significant (F1,32 = 9.18, P = .005) due to the fact that HCs had a reduction in positive affect from before to after scanning (t16 = 2.50, P = .023), while this effect was not significant for the MDD group (t16 = 1.81, P = .090). This effect was not qualified by a Drug × Group × Time interaction (F1,32 = 1.37, P > .25).
To explore whether nicotine had an effect in those with MDD, an exploratory ANOVA was run within the MDD group only. Of note, a significant Drug × Time interaction emerged (F1,16 = 8.63, P = .010), which was due to nicotine significantly increasing positive affect (t16 = 3.08, P = .007) while placebo had no impact (t17 = 0.04, P > .96). Evaluating individuals, 8/18 had enhanced positive affect after placebo (binomial P(8/18) = .17), while 14 of 17 showed increased positive affect following nicotine (binomial P(14/17) = .005; Fishers exact test P = .02)
For negative affect, there was a significant Group × Time interaction (F1,32 = 6.09, P = .019) as well as main effects of Time (F1,32 = 9.57, P = .004) and Group (F1,32 = 19.2, P < .001), due to significantly greater overall negative affect in the MDD group relative to HCs. Across drug visits, individuals with MDD had a significant reduction in negative affect from before to after the scan (t16 = 2.83, P = .012) while HCs showed no change (t16 = 1.07, P = .29). For those with MDD, on the placebo day 14/18 showed a reduction in negative affect post-scan (binomial P(14/18) = .01) and 14/17 showed a reduction on the nicotine day (binomial P(14/17) < .01; Fisher’s exact test P = .31).
NAc–rACC coupling
A repeated measures ANOVA was conducted on NAc–rACC coupling entering Laterality (right, left), Drug, and Group factors. In addition to a main effect of Group (F1,33 = 5.76, P = .023), there was a significant Group × Drug interaction (F1,33 = 5.76, P = .03, Fig. 1), whereas the three-way interaction failed to reach significance (F1,33 = 1.6, P = .21). Follow-up analyses for the Group × Drug interaction indicated that, as expected, on the placebo day MDD had significantly lower NAc–rACC coupling than the HC group (t33 = −3.35, P = .002; Cohen’s value: −1.13), replicating prior findings [4, 5]. Critically, no group difference was observed after nicotine administration (t33 = 0.42, P > .67; Cohen’s value: −0.14). Moreover, while nicotine had no impact on NAc–rACC coupling in HCs (t16 = 0.86, P > .40), nicotine significantly enhanced NAc–rACC coupling in the MDD group (t17 = 2.69, P = .016; Cohen’s d: 0.63). On an individual level, 13 of the 18 MDD participants (binomial P(13/18) = .033) but only 8 of 17 HC participants (binomial P(8/17) > .18) showed greater NAc–rACC coupling in the nicotine vs. placebo condition (Fisher’s exact test P = .09).

Coupling between the nucleus accumbens (NAc) and rostral anterior cingulate cortex (rACC): an ANOVA revealed a significant Group × Drug interaction (F1,33 = 5.76, P = .03). Relative to healthy controls, individuals with MDD had significantly reduced NAc–rACC coupling on the placebo day. Acute nicotine administration significantly increased NAc–rACC coupling in the MDD group but not healthy control group. Green overlay on brain images represent NAc and rACC ROIs. *P < .05, **P < .001
DLPFC–DS coupling
An analogous ANOVA revealed a significant Group × Drug interaction (F1,33 = 4.94, P = .033; Fig. 2), as well main effects of Laterality (F1,33 = 5.30, P = .028) and Drug (F1,33 = 4.75, P = .036). The three-way interaction was not significant (F1,33 = 0.91, P > .34). Relative to HCs, individuals with MDD showed a trend for greater DS–DLPFC coupling on the placebo day (t33 = 1.72, P = .095; Cohen’s d: 0.58). Groups did not differ on the nicotine day (t33 = 0.83, P > .40; Cohen’s d: −0.28). Critically, individuals with MDD showed a significant reduction in DS–DLPFC coupling after nicotine relative to placebo administration (t17 = −2.84, P = .011; Cohen’s d: −0.43), while HCs showed no differences between nicotine and placebo (t16 = 0.09, P > .92). On an individual level, 16 of the 18 MDD participants (binomial P(16/18) < .001) but only 9 of 17 HC participants (binomial P(9/17) > .18) showed lower DS–DLPFC coupling in the nicotine vs. placebo condition (Fisher’s exact test P = .020).

Coupling between the dorsal striatum (DS) and dorsolateral prefrontal cortex (DLPFC): an ANOVA revealed a significant Group × Drug interaction ((F1,33 = 4.94, P = .03). Relative to healthy controls, individuals with MDD showed a trend for greater DS–DLPFC coupling on the placebo day. Acute nicotine administration significantly reduced DS–DLPFC coupling in the MDD group but not heathy control group. Green overlay on brain images represent DS and DLPFC ROIs. *P < .05
Anhedonia and cortico-striatal coupling
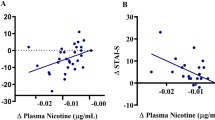
Separate ANCOVAs were run to evaluate the influence of anhedonia on nicotine-induced changes in cortico-striatal connectivity in the MDD group. For NAc–rACC coupling, the main effect of Drug (F1,17 = 7.23, P = .016) was abolished when including SHAPS as a covariate (F1,15 = 0.62, P > .44). Moreover, a significant Drug by SHAPS (measured on the nicotine day) emerged (F1,15 = 5.61, P = .032). To further investigate this effect, a NAc–rACC difference score was computed by subtracting coupling on the placebo day from coupling on the nicotine day. These difference scores were then correlated with the SHAPS scores acquired on the nicotine day. A Pearson’s correlation showed that MDD individuals with the greatest level of anhedonia had the largest increase in nicotine-induced NAc–rACC coupling (r = 0.45, P = .029, one-tailed; Fig. 3).

Association between anhedonia and nicotine-induced change in NAc–rACC Coupling. The y-axis represents the SHAPS scores measured on the nicotine day. The x-axis shows the change in NAc–rACC coupling (nicotine day–placebo day). Individuals with MDD reporting more anhedonia showed the greatest increase in NAc–rACC coupling on the nicotine relative to the placebo day (r = 0.45, P = .029, one-tailed)
In contrast, when evaluating DS–DLPFC connectivity, the main effect of Drug (F1,17 = 8.06, P = .011) remained at a trend level (F1,15 = 4.18, P = .059) when entering SHAPS scores as covariates, and no Drug × SHAPS interaction emerged (F1,15 = 2.16, P > .16).
Graph theory
When considering global efficiency, a significant Group × Drug interaction emerged (F1,33 = 4.66, P = .038; Fig. 4a). Individuals with MDD showed significantly greater global efficiency after receiving nicotine compared to placebo (t17 = −2.41, P = .028, Cohen’s d = 0.41), whereas global efficiency of HCs remained the same (t16 = 0.48, P = .63). Moreover, individuals with MDD had significantly smaller global efficiency compared to HCs on placebo (t33 = −2.63, P = .013, Cohen’s d = 0.81). Conversely, there was no difference in global efficiency between groups on nicotine (t33 = −1.11, P = .27). When considering local efficiency, no significant effects were found (Fig. 4b). When assessing vulnerability, there was a significant main effect of Group (F1,33 = 5.02, P = .032) and Drug (F1,33 = 6.95, P < .013) but no interaction (F1,33 = 0.29, P = .59; Fig. 4c). When evaluating individuals 14/18 of those with MDD showed nicotine-induced enhancement of global efficiency (binomial P(14/18) = .0117) compared with 9/17 HCs showing a similar increase HC (P(9/17) = .19) Fisher’s exact test P = .09)

Graph theory metrics. a Global efficiency, highlighting that individuals with MDD had significantly lower global efficiency on placebo relative to healthy controls. Global efficiency was enhanced in the MDD group after receiving nicotine. b Local efficiency, which yielded no effects of group or drug. c Vulnerability, which revealed main effects of Group and Drug. *P < .05
Discussion
The current findings indicate that nicotine acutely normalizes cortico-striatal connectivity in unmedicated, non-smokers with MDD. Replicating prior findings [5, 6, 17], individuals with MDD had reduced NAc–rACC coupling and a trend for greater DS–DLPFC coupling following placebo administration. While nicotine had no impact on HCs, it enhanced NAc–rACC coupling and reduced DS–DLPFC coupling in those with MDD to the levels of HCs. Anhedonia modulated these effects, as individuals with MDD reporting the greatest level of anhedonia had the largest nicotine-induced increase in NAc–rACC coupling relative to the placebo day. The link between anhedonia and NAc–rACC coupling was expected given this pathway’s role in affect, reward, and positive mood [13, 34, 35]. These findings raise the possibility that nicotine’s ability to reduce anhedonia [16] may be due to normalized NAc–rACC communication. These findings are intriguing, particularly in light of independent evidence indicating that functional connectivity between the NAc and mPFC regions encompassing the rACC is implicated in anhedonia [7].
The fact that nicotine normalized both pathways is noteworthy as nicotine enhanced and decreased connectivity in a network-specific manner. The importance of opposing effects in these pathways is supported by prior work showing that recovery from depression is associated with reduction of rACC but potentiation of DLPFC metabolism [36]. Collectively, individuals with MDD appear to have an overactive yet weakly connected rACC, and an underactive yet hyperconnected DLPFC [5, 6, 17, 36]. While activity of these regions was not assessed, the current findings indicate that nicotine re-balances these systems in relation to cortico-striatal communication. Such differential effects may be due to distinct nicotinic acetylcholine receptor (nAchR) expression patterns in each pathway. Additionally, acute nicotine has been found to enhance dopamine (DA) in a region-specific manner. Specifically, in nicotine-naive rats, an acute dose of nicotine resulted in significantly greater DA release in the NAc compared to the DS [37, 38]. Thus, enhanced DA release in the NAc may increase the otherwise deficient NAc–rACC communication in those with MDD.
While our interpretation centers on subcortical influences of nicotine, it is plausible that nicotine impacted connectivity via cortical targets. Cortical modulation fits with TMS findings showing that targeting medial parts of the PFC induces DA release in the NAc [39], while DLPFC stimulation induces DA release in the DS [40]. These findings confirm the pathway-specific nature of cortico-striatal communication, and suggest that nicotine may impact communication via the PFC. Unlike the focal nature of TMS, nicotine acts systemically, making it difficult to determine the specific site of action or how the directional flow of information within the cortico-striatal path is influenced. Regardless, the current findings highlight that cortico-striatal coupling was impacted by nicotine among non-smokers with MDD.
When evaluating acute changes in mood, an ANOVA did not reveal an interaction between Group and Drug. However, nicotine significantly increased positive affect when exploring the MDD group alone. It is plausible that the large overall group difference in mood obscured the subtle influence of nicotine in those with MDD. A weak effect of nicotine on mood was expected as others have shown that a longer duration of nicotine administration is needed to reduce depressive symptoms [41]. The current findings suggest circuit normalization (and associated subtle changes in affect) may precede overt changes in depressive symptoms.
Finally, we examined nicotine’s broader impact on brain connectivity using graph theory. Vulnerability findings suggest that in those with MDD, each nodal element is more critical to maintaining efficient communication relative to HCs. However, nicotine reduced global vulnerability in both groups. Further, nicotine reduced the deficit in global efficiency noted in those with MDD on placebo. This finding mirrored nicotine’s influence on cortico-striatal connectivity suggesting that nicotine had more overt effects on network connectivity when dysfunction is present at baseline. Prior work shows that nicotine acutely increases global efficiency during nicotine withdrawal in chronic smokers and such enhancement is associated with a reduction in errors on a go/no-go task [42]. This implies that enhancing global efficiency may have a cognitive benefit, yet the impact of such nicotine-induced changes in those with MDD was not evaluated. However, these results should be considered preliminary as it was difficult to define a clear a priori hypothesis based on existing literature. Despite this limitation, the current findings indicate that nicotine is having a more global influence on brain connectivity in those with MDD.
Several considerations must be taken into account when interpreting the current findings. First, the sample size was limited, preventing us from including other biological variables in the model. The use of a within-subject design somewhat counteracted this limitation, and our a priori effects were confirmed. Additionally, mechanisms through which nicotine mediates change within these circuits require further testing. Despite these limitations the current work shows, we believe for the first time, that acute nicotine enhances reward circuitry among unmedicated and non-smoking individuals with MDD. These findings fit with preclinical findings [12] and suggest several important paths forward. In particular, the findings support the idea that cholinergic agents might have utility in treating depression. However, as the current work focused only on the impact of acute nicotine administration in non-smokers, it is unclear how these connectivity patterns change following chronic use. Additionally, both nicotinic agonists and antagonists have anti-depressant properties (for review see ref. [43]), mirroring the mixed literature on the causal link between depression and nicotine dependence [3]. As discussed in the Introduction section, this inconsistent literature implies that nicotine’s impact on causing or mitigating symptoms of depression may be network-specific [4]. The current work indicates that acute nicotine administration has a beneficial impact at least in terms of normalizing the cortico-striatal circuits evaluated, which may predispose individuals with MDD to develop dependence. These effects might be particularly pronounced for individuals reporting elevated anhedonia, since nicotine-induced change in NAc–rACC connectivity correlated with anhedonia. Finally, the current findings show that differences in baseline neurobiology determine nicotine’s impact on brain circuitry, strengthening the idea that pre-existing neurobiological vulnerabilities may predispose individuals for addiction.
References
Diwan A, Castine M, Pomerleau CS, Meador-Woodruff JH, Dalack GW. Differential prevalence of cigarette smoking in patients with schizophrenic vs mood disorders. Schizophr Res. 1998;33:113–8.
Glassman AH, Helxer JE, Covey LS, Cottler LB, Stetner F, Tipp JE, et al. Smoking, smoking cessation, and major depression. JAMA. 1990;264:1546–9.
Fluharty M, Taylor AE, Grabski M, Munafò MR. The association of cigarette smoking with depression and anxiety: a systematic review. Nicotine Tob Res. 2017;19(1):3–13.
Picciotto MR, Lewis AS, van Schalkwyk GI, Mineur YS. Mood and anxiety regulation by nicotinic acetylcholine receptors: A potential pathway to modulate aggression and related behavioral states. Neuropharmacol. 2015;96(B):235–43.
Furman DJ, Hamilton JP, Gotlib IH. Frontostriatal functional connectivity in major depressive disorder. Biol Mood Anxiety Disord. 2011;1(1):11.
Kaiser RH, Andrews-Hanna JR, Wager TD, Pizzagalli DA. Large-scale network dysfunction in major depressive disorder: a meta-analysis of resting-state functional connectivity. JAMA Psychiatry. 2015;72(6):603–11.
Kaiser RH, Treadway MT, Wooten DW, Kumar P, Goer F, Murray L, et al. Frontostriatal and dopamine markers of individual differences in reinforcement learning: a multi-modal investigation. Cereb Cortex. 2017. https://doi.org/10.1093/cercor/bhx281
Admon R, Pizzagalli DA. Corticostriatal pathways contribute to the natural time course of positive mood. Nat Commun. 2015;6:10065.
Leventhal A, Ramsey S, Brown R, LaChance H, Kahler C. Dimensions of depressive symptoms and smoking cessation. Nicotine Tob Res. 2008;10(3):507–17.
Leventhal AM, Waters AJ, Kahler CW, Ray LA, Sussman S. Relations between anhedonia and smoking motivation. Nicotine Tob Res. 2009;11(9):1047–54.
Carton S, Le Houezec J, Lagrue G, Jouvent R. Early emotional disturbances during nicotine patch therapy in subjects with and without a history of depression. J Affect Disord. 2002;72:195.
Ferrari R, Le Novère N, Picciotto MR, Changeux JP, Zoli M. Acute and long-term changes in the mesolimbic dopamine pathway after systemic or local single nicotine injections. Eur J Neurosci. 2002;15(11):1810–8.
Kenny PJ, Markou A. Nicotine self-administration acutely activates brain reward systems and induces a long-lasting increase in reward sensitivity. Neuropsychopharmacology. 2006;31(6):1203–11.
Barr RS, Pizzagalli DA, Culhane MA, Goff DC, Evins AE. A single dose of nicotine enhances reward responsiveness in nonsmokers: implications for development of dependence. Biol Psychiatry. 2008;63(11):1061–5.
Janes AC, Pedrelli P, Whitton AE, Pechtel P, Douglas S, Martinson MA, et al. Reward responsiveness varies by smoking status in women with a history of major depressive disorder. Neuropsychopharmacology. 2015;40(8):1940–6.
Pergadia ML, Der-Avakian A, D’Souza MS, Madden PAF, Heath AC, Shiffman S, et al. Association between nicotine withdrawal and reward responsiveness in humans and rats. JAMA Psychiatry. 2014;71(11):1238–45.
Kerestes R, Harrison BJ, Dandash O, Stephanou K, Whittle S, Pujol J, et al. Specific functional connectivity alterations of the dorsal striatum in young people with depression. NeuroImage Clin. 2015;7(C):266–72.
Avissar M, Powell F, Ilieva I, Respino M, Gunning FM, Liston C, et al. Functional connectivity of the left DLPFC to striatum predicts treatment response of depression to TMS. Brain Stimul. 2017;10(5):919–25.
First MB, Spitzer RL, Gibbon M, Williams JB. Structured clinical interview for DSM-IV-TR axis I disorders, research version, patient edition. SCID-I/P. New York: Biometrics Research, New York State Psychiatric Institute, 2002.
Hamilton M. A rating scale for depression. J Neurol Neurosurg PS. 1960;23:56–62.
Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32:50–55.
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiat. 1995;167(1):99–103.
Watson D, Clark LA, Tellegen A. Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol. 1988;54:1063–70.
Choi J, Dresler C, Norton M, Strahs K. Pharmacokinetics of a nicotine polacrilex lozenge. Nicotine Tob Res. 2003;5(5):635–44.
Benowitz NL, Jacob P. Daily intake of nicotine during cigarette smoking. Clin Pharmacol Ther. 1984;35(4):499–504.
McCarthy JM, Zuo CS, Shepherd JM, Dias N, Lukas SE, Janes AC. Reduced interhemispheric executive control network coupling in men during early cocaine abstinence: a pilot study. Drug Alcohol Depend. 2017;181:1–4.
Choi EY, Yeo BT, Buckner RL. The organization of the human striatum estimated by intrinsic functional connectivity. J Neurophysiol. 2012;108(8):2242–63.
Sutherland MT, Ray KL, Riedel MC, Yanes JA, Stein EA, Laird AR. Neurobiological impact of nicotinic acetylcholine receptor agonists: an activation likelihood estimation meta-analysis of pharmacologic neuroimaging studies. Biol Psychiatry. 2015;78(10):711–20.
Petrides M. Lateral prefrontal cortex: architectonic and functional organization. Philos Trans R Soc Lond B Biol Sci. 2005;360(1456):781–95.
Curtis CE, D’Esposito M. Persistent activity in the prefrontal cortex during working memory. Trends Cogn Sci. 2003;7(9):415–23.
Yeo BT, Krienen FM, Sepulcre J, Sabuncu MR, Lashkari D, Hollinshead M, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106(3):1125–65.
Latora V, Marchiori M. Efficient behavior of small-world networks. Phys Rev Lett. 2001;87(19):440–4.
Bullmore E, Sporns O. Complex brain networks: graph theoretical analysis of structural and functional systems. Nat Rev Neurosci. 2009;10(3):186–98.
Haber S. Parallel and integrative processing through the Basal Ganglia reward circuit: lessons from addiction. J Neurosci. 2008;64(3):173–4.
Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2009;35(1):4–26.
Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiat. 1999;156(5):675–82.
Adermark L, Morud J, Lotfi A. Temporal rewiring of striatal circuits initiated by nicotine. Neuropsychopharmacology. 2016;41:1–33.
Brazell MP, Mitchell SN, Joseph MH, Gray JA. Acute administration of nicotine increases the invivo extracellular levels of dopamine, 3,4-dihydrozyphenylacetic acid and ascorbic acid preferentially in the nucleus accumbens of the rat: comparison with caudate-putamen. Neuropharmacology. 1990;29:1177–85.
Hill, DF, Parent, KL, Atcherley, CW, Cowen, SL, Heien, ML. Differential release of dopamine in the nucleus accumbens evoked by low-versus high-frequency medial prefrontal cortex stimulation. Brain Stimul. 2017;11:426–34. https://doi.org/10.1016/j.brs.2017.11.010.
Strafella AP, Paus T, Barrett J, Dagher A. Repetitive transcranial magnetic stimulation of the human prefrontal cortex induces dopamine release in the caudate nucleus. J Neurosci. 2001;21(15):RC157.
McClernon FJ, Hiott FB, Westman EC, Rose JE, Levin ED. Transdermal nicotine attenuates depression symptoms in nonsmokers: a double-blind, placebo-controlled trial. Psychopharmacol. 2006;189(1):125–33.
Giessing C, Thiel CM, Alexander-Bloch AF, Patel AX, Bullmore ET. Human brain functional network changes associated with enhanced and impaired attentional task performance. J Neurosci. 2013;33(14):5903–14.
Rahman S. Targeting brain nicotinic acetylcholine receptors to treat major depression and co-morbid alcohol or nicotine addiction. CNS Neurol Disord Drug Targets. 2015;14(5):647–53.
Acknowledgements
This project was supported by the National Institute on Drug Abuse grants K10 DA029645 and K02 DA042987 (ACJ). DAP was partially supported by National Institute of Mental Health grant R37 MH068376. Over the past 3 years, DAP has received consulting fees from Akili Interactive Labs, BlackThorn Therapeutics, Boehringer Ingelheim, Pfizer and Posit Science, for activities unrelated to the current research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Janes, A.C., Zegel, M., Ohashi, K. et al. Nicotine normalizes cortico-striatal connectivity in non-smoking individuals with major depressive disorder. Neuropsychopharmacol 43, 2445–2451 (2018). https://doi.org/10.1038/s41386-018-0069-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0069-x
This article is cited by
-
Hydroxynorketamine, but not ketamine, acts via α7 nicotinic acetylcholine receptor to control presynaptic function and gene expression
Translational Psychiatry (2024)
-
rTMS for Co-occurring Psychiatric and Substance Use Disorders: Narrative Review and Future Directions
Current Addiction Reports (2024)
-
Mechanisms affecting brain remodeling in depression: do all roads lead to impaired fibrinolysis?
Molecular Psychiatry (2022)
-
Safety and target engagement of an oral small-molecule sequestrant in adolescents with autism spectrum disorder: an open-label phase 1b/2a trial
Nature Medicine (2022)
-
Reduced nucleus accumbens functional connectivity in reward network and default mode network in patients with recurrent major depressive disorder
Translational Psychiatry (2022)
