
Abstract
Despite compartmentalization within the lumen of the gastrointestinal tract, the gut microbiota has a far-reaching influence on immune cell development and function throughout the body. This long-distance relationship is crucial for immune homeostasis, including effective host defense against invading pathogens that cause systemic infections. Herein, we review new insights into how commensal microbes that are spatially restricted to the gut lumen can engage in long-distance relationships with innate and adaptive immune cells at systemic sites to fortify host defenses against infections. In addition, we explore the consequences of intestinal dysbiosis on impaired host defense and immune-mediated pathology during infections, including emerging evidence linking dysbiosis with aberrant systemic inflammation and immune-mediated organ damage in sepsis. As such, therapeutic modification of the gut microbiota is an emerging target for interventions to prevent and/or treat systemic infections and sepsis by harnessing the long-distance relationships between gut microbes and systemic immunity.
Similar content being viewed by others

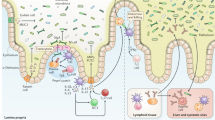

The intestinal microbiota and host defense against infections
From birth, the intestinal microbiota educates and regulates the development and function of our immune system1. The crosstalk between commensal microbes and the immune system is crucial in establishing and maintaining immune homeostasis, and preventing immune-mediated disorders such as autoimmunity, allergies, and chronic inflammatory diseases2,3. However, in addition to maintaining immune homeostasis and self-tolerance, the microbiota and its shaping of immunity are also critical for protection of the host against infections.
The most direct example of host protection conferred by the gut microbiota is through colonization resistance against mucosal pathogens like Clostridium difficile. Extensive mechanistic and clinical evidence has demonstrated that disruption of the intestinal microbiota by antibiotics plays a causal role in C. difficile infection of the gut, whereas restoration of a diverse intestinal microbiota through fecal transplant or microbial consortia therapy can be curative4,5,6. A diverse gut microbiota aides the host in resisting pathogen colonization through various mechanisms including inter-microbial competition for nutrients, metabolic competition, and direct antagonism/killing strategies7, as well as fortification of mucosal barrier integrity8,9 and local mucosal immune defenses10. In this way, gut commensals protect the host against mucosal infection, as well as secondary systemic infections that can occur as a result of overgrowth and translocation of intestinal pathogens into the circulation and distal organs7,11,12,13,14,15.
However, in addition to its locally protective role against mucosal pathogens, the gut microbiota also exerts a crucial influence on the shaping of systemic host defense in the bloodstream and extra-intestinal organs. Germ-free (GF) and antibiotic-conditioned SPF mice display increased susceptibility to a wide variety of infection models including bloodstream infections, bacterial pneumonia, intra-abdominal sepsis, endotoxemia, as well as viral infections like influenza A, Lymphocytic Choriomeningitis Virus (LCMV), and others16,17,18,19,20,21,22,23,24. The observed defects in systemic host defense in GF and antibiotic-conditioned mice can be remedied by repopulating the gut with commensal microbes. Herein, we will review how commensal microbes that are spatially restricted to the gut lumen can engage in long-distance relationships with extra-intestinal immune cells to fortify host defense against systemic infections. We will also explore the consequences of intestinal dysbiosis on extra-intestinal host defense and susceptibility to systemic infections, and opportunities for mechanism-guided microbial-immunotherapy to bolster immune defense against severe infections and sepsis.
Mechanisms of long-distance communication between gut microbes and extra-intestinal immune cells
The vast spatial separation between immune cells in extra-intestinal sites and commensal microbes in the lumen of the gut necessitates the use of long-distance mechanisms of communication to enact functional regulation of systemic host defense. Various organ-specific axes of communication have been well described (e.g., gut-brain axis, gut-liver axis, gut-lung axis) and have been the subject of recent focused reviews25,26,27,28,29. Here, we aim to provide a summary of the general mechanisms by which bacteria compartmentalized in the gut lumen can communicate with, and functionally regulate, immune cells in the bloodstream and distant organs (Fig. 1).

Intestinal microbes communicate with immune cells in extra-intestinal organs through multiple mechanisms. Gut microbes produce small molecule metabolites, PRR ligands (MAMPs), extra-cellular vesicles, neurotransmitters, and hormones that can enter the lymph and circulation, travelling to distant organs where they impact immune cell development and function. In addition to soluble signals that act at distant sites, the gut microbiota also locally educates immune cells in the mucosa, including dendritic cells, ILCs, T cells, and others, that then egress from the gut and migrate to systemic sites to contribute to host defense. Lastly, long-distance communication by the gut microbiota can also be mediated via neuronal communication between the gut and distant organs including the brain. Microbiota-derived compounds are sensed by the enteric nervous system, and afferent signalling through the vagus nerve can enable coordinated systemic responses via the central nervous system.
Perhaps the most well studied mechanism involves microbe-associated molecular pattern (MAMP) components, like LPS and peptidoglycan from bacteria or β-glucan from fungal colonizers, that access the circulation and bind to pattern recognition receptors (PRRs) on immune cells throughout the body (Fig. 1). Even in the absence of gut barrier pathology, low levels of MAMPs can be detected in the circulation of mice and humans that regulate myeloid and lymphoid cell production in the bone marrow and thymus, and impart regulatory influences on immune cell functions throughout the body, as discussed further below30,31,32. Although commensal-derived MAMPs and pathogen-derived PAMPs can act on the same PRRs, the immunological outputs may differ to prevent commensal-derived MAMPs from inducing pathological inflammation. For example, LPS from Bacteroidales colonizers has a hypoacetylated structure that supresses TLR4-mediated cytokine release from human mononuclear cells, in contrast to LPS from pathogenic E. coli that induced robust cytokine production33.
Similar to long-distance MAMP signals, gut bacteria also produce a variety of immune-active small molecule metabolites that gain access to the bloodstream and communicate with systemic immune cells (Fig. 1). Short-chain fatty acids (SCFAs) have emerged as pleiotropic immunoregulatory mediators produced by intestinal microbes through fermentation of dietary fibre. SCFAs provide a crucial energy source for intestinal epithelial cells, and can modulate immune cell function epigenetically through inhibition of histone deacetylases (HDACs), as well as through activation of G-protein coupled receptors (GPR41, GPR43, GPR109a) expressed on immune cells34. As such, the transit of SCFAs to extra-intestinal tissues contributes to the regulation of systemic inflammation, directing leukocyte recruitment, anti-pathogen cell programming, and cytokine release34. In addition to SCFAs, gut microbes produce a range of other immune-regulatory metabolites from modification of dietary molecules (tryptophan metabolites, TMAO), host bile acids (secondary bile acid metabolites), as well as de novo bacterial metabolites (polyamines, vitamins, branched chain amino acids) that can facilitate long-distance regulation of systemic immune responses34.
Signalling between the gut and extra-intestinal organs can also be accomplished through neuro-immune mechanisms that are tuned by gut microbes (Fig. 1). Intestinal bacteria can synthesize neurotransmitters like γ-aminobutyric acid (GABA), as well as modulate host production of a variety of neurotransmitters that can impact immune cell functions25. In addition, gut bacteria stimulate the release of neuroendocrine peptide hormones by enteroendocrine cells that can access the circulation and act at systemic sites35,36. Interestingly, gut bacteria can also communicate with distal organs via neuronal pathways. For example, autonomic nerve terminals in the intestinal mucosa have been found to sense bacterial metabolites and relay signals to the central nervous system37,38. Additional research is needed to understand how these neuronal mechanisms of long-distance microbiota-immune communication may impact host defense, but one would hypothesize an important contribution given the role of neural reflexes in the regulation of immune cell functions during infections and sepsis39,40.
More recently, bacterial extracellular vesicles (EVs) were identified that instruct differential host responses dependent on the source microbe (Fig. 1)41. As with LPS, the immunogenicity of microbiota-derived EVs differs between microbial taxa, with EVs from pathogenic bacteria associated with pro-inflammatory responses compared to anti-inflammatory responses initiated by commensal EVs41. For example, EVs from a commensal strain of E. coli enhanced production of mucosal cytokines important for host defense42. While much of what we know about microbiota-derived EVs is based on investigations of their local effects in the gut, recent findings showed that there is also systemic regulation of host immunity, with microbiota-derived EVs priming neutrophils for inflammatory responses to secondary stimuli43.
Lastly, gut microbes preside over the local education of immune cells in the mucosa that then recirculate to other body compartments to participate in systemic host defense (Fig. 1). Notably, it is well established that mucosal antigen-presenting cells (APCs) can carry microbiota antigens to distant organs to direct peripheral T cell function44. Dendritic cells (DC) migrate from the gut to the thymus to help shape the T cell receptor repertoire of microbiota-reactive T-cells, and to peripheral lymph nodes to shape the effector, regulatory, and memory T cell pools45,46,47. As described further below, intestinal microbiota-imprinting of peripheral CD4+ T cells with reactivity to intestinal microbiota may support a systemic monitoring system for a rapid memory response to disseminated gut pathobionts48.
Collectively, these mechanisms help expand the host-microbe interface beyond the local environment of the gut to enable a long-distance relationship between gut commensal and systemic immunity. In the next section, we will explore how these communication mechanisms are utilized to modulate the development, polarization, and functions of various immune cell populations involved in systemic host defense against infections.
Shaping systemic host defense through microbiota-immune interactions
The contribution of gut microbes towards shaping extra-intestinal host defense encompasses all major immune effector cells of the innate and adaptive immune systems. Below, we will focus our discussion on the primary effector cell populations involved in anti-pathogen host defense within the bloodstream as the key compartment for invasion and dissemination of systemic infections (Fig. 2). Organ-specific microbiota-immune axes between the gut and brain, lung, liver, and other key organs are beyond the scope of the current review but have been recently summarized25,26,27,28.

a During eubiosis, homeostatic communication between the gut microbiota and systemic immune cell populations maintains key mechanisms of systemic host defense against infections. b In contrast, intestinal dysbiosis results in a breakdown of microbiota-dependent mechanisms of systemic host defense and impaired clearance of pathogens, as well as aberrant immune cell functions that contribute to immune-mediated organ damage in sepsis.
Circulating Neutrophils
Neutrophils in the bloodstream are rapidly deployed to sites of infection where they contribute a powerful payload of anti-pathogen effector mechanisms that are essential to the containment and eradication of pathogens. As such, effective neutrophil responses are dependent on their continuous supply in the bloodstream (primarily through granulopoiesis in the bone marrow), recruitment to sites of infection, and regulation of their arsenal of pathogen killing mechanisms. Emerging evidence has revealed that signals emanating from the gut microbiota are amongst the most influential modulators of neutrophil production and function during infection (Fig. 2).
For many years, it has been appreciated that germ-free and antibiotic-conditioned animals displayed suppressed myelopoiesis in the bone marrow, resulting in relative neutropenia and monocyte defects that contribute to impaired pathogen clearance and death in experimental models of infections21,30,31,49. Bone marrow myelopoiesis is rescued in GF and antibiotic-conditioned mice by reconstituting the gut with microbes30 and can be partially reversed by gastrointestinal administration of non-viable microbes (heat killed E. coli or autoclaved cecal contents) in a manner that is dependent on PRR signaling via MyD8821. Furthermore, experiments using transient (~48–72 h) gut colonization with auxotrophic E. coli demonstrated that myelopoiesis is dynamically regulated via continuous input from gut-derived signals, rather than long-term imprinting30. Specifically, granulopoiesis at steady-state is controlled through a feedback circuit involving TLR-mediated recognition of microbiota products by innate lymphoid cells (ILCs) in the gut mucosa, which produce IL-17 that stimulates production of G-CSF, culminating in augmented granulopoiesis in the bone marrow21,50,51. This microbiota-dependent mechanism of granulopoiesis is negatively regulated by homeostatic migration of neutrophils back to tissues (possibly the gut) which inhibits the IL-17/G-CSF axis52. Collectively, these findings implicate the gut microbiota as a key controller of the circulating pool of phagocytes available to respond to invading pathogens.
In addition to their supply in the bloodstream, the gut microbiota also fine-tunes the anti-pathogen functionality of neutrophils. Neutrophils from germ-free or microbiota-depleted mice display marked functional impairment, including defects in migration, phagocytosis, and production of anti-microbial molecules like myeloperoxidase and ROS, culminating in impaired capacity to kill pathogens53,54. PRR-dependent priming of neutrophils by gut microbiota-derived peptidoglycan was found to enhance phagocytosis and killing of S. pneumoniae and S. aureus ex vivo and improve survival during in vivo infection22. Similar functional priming has been observed in response to gut-microbe derived SCFA55. Zhang et al. reported that the gut microbiota induces a specific functional program of neutrophil aging in vivo, whereby neutrophils undergo phenotypic and functional maturation characterized by upregulation of CXCR4 and downregulation of L-selectin, enhanced phagocytic capacity, inflammatory cytokine production, and neutrophil extracellular trap formation56. Microbiota-induced neutrophil aging was dependent on gut-derived TLR ligands in the circulation that directly stimulated neutrophils in a MyD88-dependent manner56. Lastly, there is emerging evidence that the gut microbiota may contribute to the orchestration of neutrophil-mediated host defense thought the regulation of cell trafficking to sites of infection. In germ-free zebrafish, the absence of intestinal microbiota was associated with aberrant neutrophil trafficking towards sites of injury57. In mice, germ-free status was associated with marked impairment of neutrophil recruitment to the inflamed peritoneum54, as well as defective neutrophil trafficking to the lungs in a model of bacterial pneumonia58. Taken together, the cumulative evidence demonstrates that long-distance communication between intestinal commensals and neutrophils in the bone marrow and circulation is critical for the orchestration of effective host defense against systemic infections.
Macrophages and monocytes
Well-defined axes of communication exist between the gut microbiota and tissue resident macrophages in the gut59,60, brain61,62, lung63, spleen64, liver16,65,66, and other organs. Among these, the impact of the gut microbiota on systemic host defense against infection is perhaps best exemplified by the unique intravascular macrophages of the liver (Kupffer cells, KC) (Fig. 2). Their unique residence within the vascular lumen of liver sinusoids positions these cells as defenders of the bloodstream, displaying a remarkable capacity to rapidly detect, capture, and engulf pathogens circulating in the blood as they pass through the liver. KC are continuously bathed in gut-derived signals reaching the liver via the portal veinous drainage of the GI tract. Studies in KC-depleted mice reveal the crucial role of these intravascular macrophages in preventing dissemination of microbes in the systemic circulation during infection, as well as preventing steady-state dissemination of gut commensals that translocate into the portal blood16,67,68.
Recent evidence has uncovered important contributions of the gut microbiota to the anatomical localization and antimicrobial actions of KC. Recently, Gola et al. reported that the spatial distribution of KC to the periportal regions of the liver is dependent on tonic input from gut microbial signals, rather than developmental imprinting in early life65. MyD88-dependent signaling from gut microbial MAMPs to liver sinusoidal endothelial cells induced the establishment of chemokine gradients in the liver that guided KC to reside in periportal regions. This spatial zonation in colonized mice was critical for effective clearance of intravascular pathogens in a model of Listeria monocytogenes infection, compared to the uniform distribution of cells that occurs in the absence of gut microbial input65. Beyond spatial regulation, the gut microbiota also provides crucial input to KC to enhance their ability to phagocytize circulating pathogens and perform intracellular killing. Using intravital imaging of the liver, it was observed that KC in GF mice displayed defects in the capture, phagocytosis, and killing of circulating S. aureus and E. coli, contributing to an enhanced susceptibility of GF mice to disseminated bloodstream infections16. The compartmentalized shuttling of high concentrations of D-lactate from the gut to the liver via the portal vein was found to control pathogen uptake and killing by KC, and pathogen clearance in GF mice was restored by repopulating the gut with D-lactate producing bacteria16. In addition to pathogen killing, others have reported that microbial signals may also regulate KC density within the liver66. The exact mechanisms by which gut-derived mediators regulate KC anti-pathogen activities remain to be elucidated, but these findings uncover an important mechanism of long-distance communication mediated by bacterial metabolites that are crucial for macrophage host defense against systemic infections that enter the bloodstream.
Circulating monocytes contribute to the acute response against pathogens through recruitment to sites of infection, where they phagocytose and kill pathogens, regulate local inflammation, and also participate in shaping tissue immunity through their differentiation into resident macrophages69. In a ground-breaking study of 500 healthy humans, gut microbiota composition and function were shown to modulate cytokine production by peripheral blood mononuclear cells in responses to ex vivo stimulation with microbial products70. In mice, the absence of an intestinal microbiota results in aberrant programming of inflammatory monocytes, as shown recently that splenic monocyte homeostasis was dependent on MAMPs produced by an intact gut microbiota64. Kolypetri et al. reported that antibiotic-treated mice harbour an altered splenic monocyte pool with impaired inflammatory cytokine production following LPS challenge which could be rescued through treatment with PRR ligands such as muramyl-dipeptide (MDP)64. Differentiation of monocytes into tissue macrophages is also regulated by the intestinal microbiota and may contribute to anti-pathogen defense. For example, the SCFA butyrate directs enhanced and long-lived anti-bacterial activity in human macrophages that differentiate from CD14 + monocytes71. Interestingly, microbiota-programming of monocytes likely differs between mice and humans. Ang et al., show that human monocytes stimulated with SCFA acetate experienced inflammatory re-programming, resulting in reduced expression of inflammatory cytokines such as GM-CSF, IL-1β and ICAM-1, whereas mouse monocytes responded to acetate with enhanced inflammatory cytokine production via distinct signalling pathways72. In mice fed high fiber diets, microbiota-derived SCFA promoted generation of non-classical Ly6C− monocytes that differentiated into alternatively activated macrophages and helped blunt excessive inflammation in response to influenza A infection73. Overall, pleiotropic modulation of monocytes is emerging as an important mechanism by which the gut microbiota shapes both acute anti-pathogen responses as well as the landscape of macrophage-mediated tissue immunity.
Innate lymphoid cells (ILCs)
NK cells and other innate lymphoid cells (ILCs) are intimately tied to the gut microbiota through its role in directing the development and programming of ILCs in vivo74,75. ILCs primarily reside at mucosal sites including the gastrointestinal tract, and make well described contributions to host defenses in the gut. However, microbiota-educated ILCs also participate in systemic host defense against infections both directly and indirectly. Mucosal ILC3 in the intestine produce IL-17 in response to gut microbes, which regulates G-CSF production and granulopoiesis in the bone marrow21. This ILC3-mediated response to gut commensals was critical for effective neutrophil-mediated host defense against pneumonia in neonatal mice21. Their powerful ability to modulate the function of other immune cells represents an important indirect contribution of microbiota-responsive ILCs to systemic host defense against infections.
More directly, microbiota-ILC interactions can impact pathogen clearance at extra-intestinal sites of infection. NK cell-mediated protection against viral infections is markedly impaired in germ free mice75. The gut microbiota was shown to be critical for effective NK cell priming and antiviral activity, however this was not an NK cell-intrinsic effect but was instead mediated by microbiota-educated mononuclear cells75. In addition to antiviral immunity, collaborations between the microbiota and ILCs can impact host defense against bacterial infections. Gray et al. reported that exposure of neonatal mice to intestinal commensals induced trafficking of ILC3 to the lungs, where they were key to effectively combatting pneumonia pathogens76. Through an intestinal dendritic cell intermediary, gut microbes induced upregulation of the lung homing chemokine receptor CCR4 on ILCs, leading to their selective recruitment to the lungs of colonized neonatal mice. Microbiota-directed homing of ILC3s to the lungs and production of IL-22 was crucial for survival in a neonatal model of S. pneumonia infection76. Collectively, the influence of intestinal microbes on ILC biology provides the host with multiple important direct and indirect benefits for enhanced defense against bacterial and viral infections at systemic sites.
T cell mediated host defense
The shaping of T cell immunity by gut microbes has been identified as an important aspect of host defense against a wide range of bacterial, fungal, and viral infections. Multiple mechanisms have been identified that mediate microbiota-T cell cross-talk, including direct and indirect modulation of T cell development, polarization, memory formation/maintenance, and effector regulation (Fig. 2)77.
The shaping of T cell-mediated host defense by the microbiota begins during thymic development, where commensal microbes help shape diversity and specificities of T cell receptors in a manner that may protect against future infection by related pathogens. The impact of intestinal microbes on thymic T cell development is illustrated by the stark reduction in thymic cellularity and T cell output in germ-free and antibiotic-conditioned SPF mice47,78,79. Gnotobiotic mouse models have revealed that gut colonization leads to the production of microbiota-specific T cells in the thymus that provide protection against future infections by pathobionts and commensal-related pathogens47. In these mice, gut bacterial antigens were delivered to the thymus by CX3CR1+ dendritic cells where they induced positive selection of CD4+ T cells that then trafficked to peripheral tissues47. Additional mechanisms of microbiota-thymic communication have been identified that help shape the developing T cell repertoire, including regulation of autoimmune regulator (AIRE) expression in medullary thymic epithelial cells (mTEC), thus influencing the deletion of auto-reactive thymocytes and induction of thymic regulatory T cells (Treg)80,81,82. Further work is needed to better understand how this long-distance communication between gut microbes and the thymus impacts host defense, and the implications of intestinal dysbiosis during early life development on susceptibility to infections in later life.
Gut microbes also help shape systemic effector and memory T cell responses. Shaping of T cell polarization and effector responses by gut microbes is exemplified by the influence of segmented filamentous bacteria (SFB) on CD4+ T cell polarization towards Th17 phenotype, which was originally shown to improve host resistance against C. rodentium infection in the murine gut83. More recently, intestinal SFB has also been found to promote systemic Th17 mediated responses against extraintestinal pathogens. Mice harbouring SFB in the gut had augmented Th17 responses in extraintestinal organs including the lungs, and were more resistant to pneumonia caused by diverse pathogens including MRSA and Aspergillus fumigatus84,85. In addition to SFB, intestinal fungi including C. albicans can influence T cell polarization towards Th17 phenotype in the spleen and peripheral lymph nodes86. Priming of systemic Th17 effector T cells by C. albicans yielded improved host defense against systemic candidemia, as well as augmented host defense against a heterologous pathogen S. aureus, indicating that T cell activation and polarization by intestinal Candida is not an antigen-specific response86. In addition to Th17-mediated immunity, specific mechanisms linking gut microbes to the polarization and effector functions of other CD4+ T cell subsets have also been established87,88,89,90,91. Together, these studies support a key role for the gut microbiota in fortifying host defense by promoting effector CD4+ T cell polarization in the systemic compartment.
Akin to CD4+ T cells, CD8+ T cells rely on signals and metabolites from the microbiota for optimal function in response to intracellular infections92. Germ free and antibiotic-conditioned SPF mice display reduced CD8+ numbers, cytotoxicity, cytokine production, and enhanced susceptibility to infection by a variety of different viral pathogens including Influenza A virus, LCMV, flavivirus infections, and others17,93,94. In mice infected with Influenza A virus, defects in virus-specific CD8+ T cell responses could be restored by intra-rectal administration of TLR ligands, suggesting PRR-dependent communication from gut microbes contributes to the regulation of effector CD8+ responses17. In addition to TLR ligands, evidence from high-fat diet fed mice revealed that gut derived SCFA played a critical role in effective expansion and function of CD8+ T cells during influenza through selective regulation of cellular metabolism73. Others have shown that SCFA like butyrate promote transition of extraintestinal CD8+ T cells into memory cells and the maintenance of these specific memory cells in response to viral infection95,96. Taken together, the cumulative evidence supports the existence of multiple direct and indirect mechanisms of communication between gut microbes and systemic T cells that is critical for effective CD4+ and CD8+ T cell-mediated host defense against extracellular and intracellular pathogens.
Microbiota regulation of antibody-mediated host defense
At mucosal surfaces, the interplay between gut microbes and secretory immunoglobulins is paramount in maintaining host-microbial homeostasis97. Within the systemic circulation, antibody responses are also heavily modulated by gut commensals in a manner that bolsters humoral immunity against invading pathogens. Germ-free and antibiotic-treated mice show marked reductions of serum IgA98,99 and IgG100 and an increase in IgE which is associated with predisposition to allergies101. Effects of antibiotic-driven IgA depletion are likewise observed in humans18. Correspondingly, probiotics and high dietary fiber increase IgG and IgA levels and suppress IgE87,102. Bacteria-produced SCFAs promote B cell maturation into germinal center B cells and plasma cells in secondary lymphoid tissues102. In addition to regulating Ig production, the gut microbiota also shapes the repertoire of protective antibody specificities in the systemic circulation. Commensal-specific IgG, IgA, and IgM can be detected in the circulation of microbiota colonized hosts100,103,104. These anti-commensal antibodies contribute to protection against cross-reactive invading pathogens, as well as against infection caused by translocated gut pathobionts. Transfer of commensal specific Ig from naïve SPF mice to GF animals improves their resistance against systemic bacterial infections105. Gut microbiota-induced systemic IgG specific for bacterial lipoprotein renders colonized mice more resistant to bloodstream infections by E. coli and Salmonella enterica100. In addition, microbiota-specific IgA in the blood has also been shown to improve survival in models of Pseudomonas pneumonia and polymicrobial sepsis18,103. Like bacterial colonizers, gut fungi also elicit a systemic protective anti-fungal IgG response. Intestinal Candida drive the production of anti-fungal IgG that protects against systemic infection by C. albicans and the emerging pathogen C. auris106.
Although it was originally believed that systemic anti-commensal Ig responses were due solely to “leaking” of commensals and their antigens out of the gut during intestinal pathology107, it is now established that mucosal colonization alone is sufficient to drive a systemic Ig response108. In fact, mucosal versus translocation exposure to commensals direct unique programs of Ig responses in the circulation, with systemic exposure eliciting a more broad IgG repertoire of anti-commensal antibodies in the blood109. Lastly, in addition to the specific repertoire of conventional B cell and Ig responses, the intestinal microbiota may also contribute to the generation of polyreactive natural antibodies (NAbs) by innate-like B-1 cells. Although debate exists as to whether NAbs are generated in response to microbiota antigens directly110 or independently of exogenous antigen exposure in utero111, polyreactive NAbs do bind to commensal microbes112 and the repertoire of certain NAbs may be influenced by gut microbial composition113. These findings suggest at least a partial influence of select microbial species on NAb levels and repertoires, but many questions remain as to the exact mechanism and scope of this influence. These questions notwithstanding, research supporting the action of microbiota-induced NAbs in systemic host defense has been observed in experiments where natural antibodies transferred from conventional mice to germ free mice restored protection against Klebsiella pneumoniae lung infection105.
Intestinal dysbiosis and pathological systemic host response to infection
Many of the microbiota-immune connections noted above were discovered, in part, through the demonstration that these mechanisms of systemic host defense fail when the microbiota is disrupted by antibiotics or absent in GF mice16,17,18,19,20,21,22,23,24. Intestinal dysbiosis in both animals and humans has been linked to increased susceptibility to bacterial, viral, and fungal pathogens20,21,114,115,116,117. However, failure of microbiota-dependent host defense mechanisms is not the only consequence of intestinal dysbiosis during infection, as it is now appreciated that dysbiosis can also actively induce pathological systemic inflammation and immune-mediated organ damage in response to infection (ie. sepsis). For example, mouse models of pneumonia induced by a variety of pathogens have demonstrated that intestinal dysbiosis caused by pre-conditioning with antibiotics led to impaired pathogen clearance, but also exacerbated inflammation, multi-organ damage, and increased mortality19,20,117,118. Additionally, dysbiosis-induced defects in host defense in a model of post-surgical infection could be ameliorated with live (but not autoclaved) fecal microbiota transplantation from healthy littermates, mediated by restoration of butyrate levels and interferon regulator factor-3 signaling119. Further stark examples of how gut microbiota composition and diversity impact sepsis severity can be found in studies using “dirty” mice derived from pet shops or mice colonized with a wild/natural microbiome120. Mice harbouring a pet shop microbiome subjected to cecal ligation and puncture or endotoxin shock exhibited dramatically increased systemic inflammatory organ injury and mortality compared to conventional SPF mice121. In these animals, pathogen burden in the bloodstream was equivalent, suggesting that the exacerbated sepsis in dirty mice may be driven by a state of impaired disease tolerance mediated by pathologically altered systemic inflammation. Indeed, a role for the gut microbiota in mediating disease tolerance to infection has previously been demonstrated in gnotobiotic mice harbouring E. coli O21:H + that induced inflammasome-dependent resistance to muscle wasting during infection122. Overall, pathological alterations of gut microbiota composition appear to drive not only defects in host defense against pathogens, but may also actively participate in promoting a septic response to inflection mediated by dysregulated systemic inflammation and immune-mediated organ damage.
Observational studies of humans with sepsis and other hospitalized and critically ill patients have also reported extreme shifts in microbiota composition and diversity that have been linked with adverse outcomes, including increased infections, organ dysfunction, prolonged requirement for life support and hospitalization, and even mortality15,123,124,125,126,127,128,129,130,131,132. In particular, multiple studies have reported that the gut microbiota of patients with sepsis is depleted of anaerobic fermenters, with corresponding reductions in fecal SCFA levels130,133,134,135. Furthermore, the dysbiotic microbiome observed in septic patients is notably enriched with pathobionts of the Proteobacteria phylum as well as Enterococcus and Staphylococcus123,124,125,126,132. In addition to intestinal bacterial communities, Haak et al. recently reported the presence of multi-kingdom dysbiosis in critically ill septic patients including altered gut fungal ecology together with shifts in endogenous viruses126. This marked dysbiosis suffered by patients with sepsis and critical illness has been associated with worsening multi-organ dysfunction, as well as an increased risk of secondary (hospital-acquired) infections123,125,127,128,131,132,136. Furthermore, epidemiologic data generated from hospitalized patients revealed that individuals who sustained the greatest insult to their microbiota (ie. highest degree of dysbiosis) were at increased risk of being re-hospitalized with a recurrent bout of sepsis in the subsequent 90 days137. Together, these interesting hypothesis-generating observational data suggest that a pathologically altered gut microbiota may predispose to both a defective host response to infection, as well as exacerbation of disease severity through detrimental immune-mediated organ damage. This presents an exciting potential for therapeutic interventions targeting the microbiota for the prevention and treatment of systemic infections and sepsis.
Therapeutic implications of microbiota-immune interactions in systemic infections and sepsis
Mouse models have shown that microbiota modulation can augment host defense and improve outcomes in response to a wide range of severe infections by therapeutic supplementation with immune-modulatory commensals16, FMT21,119, dietary modulation of microbial SCFA production73, and administration of bacterial products and metabolites16,56. In humans, microbiota modifying therapeutics have been used very successfully to treat mucosal infections like C. difficile colitis138, but the application of microbiota-based treatments for systemic infection and sepsis are in their infancy. As described above, widespread multi-taxa dysbiosis has been observed in septic humans, and the optimal approach to microbiota modulation has not been defined. Preliminary clinical trials of probiotics in adult patients with critical illness showed promising signals towards a reduction in systemic hospital-acquired infections139, but a recent large randomized controlled trial of a Lactobacillus rhamnosus GG probiotic in this patient population failed to impact rates of secondary (hospital-acquired) infections or survival140. Case reports of FMT therapy in septic patients have been published, but limited data is available on the safety and efficacy of this approach141. Aside from restorative approaches like probiotics and FMT, other investigators have tested an opposing strategy of gut microbe depletion with antibiotics, a strategy called “selective digestive decontamination” (SDD) that is hypothetically combatting disease-associated pathobiont overgrowth in the gut142. This polarization of approaches to therapeutic microbiota modification (supplementation versus depletion) in sepsis and critical illness is perhaps the clearest indication that further research is needed to understand mechanisms of microbiota-host interactions to guide rationalized and targeted approaches to microbiota therapies that improve host defense and modulate pathological inflammation in sepsis.
Lastly, the use of microbial therapeutics in this sick and vulnerable patient population is not without risks. Recent high-profile publications of serious and deadly infections caused by FMT in vulnerable patients, as well as bloodstream dissemination of enterally administered probiotics in critically ill patients, reinforce the need to comprehensively understand the efficacy and safety of microbial therapeutics in these populations143,144.
Conclusions
The cumulative evidence from mice and emerging evidence from humans supports an intimate and essential long-distance relationship between gut microbes and systemic immune responses that mediate host defense against infection. Breakdown of this relationship caused by intestinal dysbiosis has important consequences for the impairment of pathogen clearance as well as the induction of pathological immune dysregulation and sepsis. Furthermore, the therapeutic malleability of the gut microbiota makes this relationship an attractive target for intervention with microbiota-based immunotherapy to prevent and/or treat systemic infections and sepsis.
Although tremendous strides have been made in our understanding of the mechanisms underlying microbiota-immune communication in systemic host defense, much remains to be learned. Most notably, much of our understanding of these mechanisms is derived from inbred mouse models, whereas less is known about systemic microbiome-immune communication in humans. Emerging evidence from humans in both health and disease support an important connection between the gut microbiota and systemic immune function70,145,146,147. However, heterogeneity contributed by genetic diversity, environmental and dietary factors as well as co-morbidities and treatments are likely to make the impact of microbiota more nuanced in humans compared to highly controlled laboratory mouse models. Improvements in mouse models, including wild-microbiota and human-microbiota associated mice, may help by more effectively phenocopying human microbiota-immune responses including during infections and sepsis148,149,150. Nevertheless, further human translational research is crucial to understand how the microbiota, as well as microbiota-modulating therapeutics, impact immunity and host defense. Developing a comprehensive understanding of long-distance interactions between gut microbes and systemic immunity in humans will require integrating multi-omics profiling using systems-level analyses of immunity together with microbiota composition and function, and will also benefit from longitudinal study designs that sample the changing landscape of the microbiome and immune system throughout the course of infection and the impact of anti-microbial (and other) treatments. Defining these mechanisms in humans, and how they change in response to microbiome perturbations, will be key to developing effective microbiota-based therapies to treat infection and sepsis.
In addition, much remains to be learned about how other important biological inputs affect microbiome-immune communication that may also have important therapeutic implications. For example, sex-based differences in gut microbiota composition have been shown to impact systemic immunity in diseases like diabetes151,152. In mice, sexual dimorphism is associated with differences in protection against sepsis, yet the mechanisms underlying this remain unexplored including a potential role for sex-based differences in the microbiota153. Lastly, our understanding of microbiota-immune interactions to date has been very bacteria-centric, and yet there is emerging data that other kingdoms of commensal microbes have a powerful impact on systemic immunity and host defense. Gut fungi have been implicated in the regulation of host defense against infections caused by both fungal and bacterial pathogens, and it has been suggested that gut fungi may be important mediators of systemic trained immunity and resistance to heterologous infections154. Much remains to be learned about how commensal fungi, viruses, and protozoans, as well as cross-kingdoms interactions amongst gut microbes, contribute to the regulation of systemic host defense. Gaining a deeper understand of the breadth of communication between intestinal microorganisms and systemic immune mechanisms will undoubtedly pay dividends for the development of rationalized, precision-based therapies that harness long-distance microbiota-immune communication to prevent and treat infections and sepsis.
References
Zheng, D., Liwinski, T. & Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 30, 492–506 (2020).
Pascal, M. et al. Microbiome and allergic diseases. Front. Immunol. 9, 1584 (2018).
De Luca, F. & Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 195, 74–85 (2019).
Kao, D. et al. Effect of Oral Capsule– vs Colonoscopy-delivered fecal microbiota transplantation on recurrent clostridium difficile Infection: A randomized clinical trial. JAMA 318, 1985–1993 (2017).
Feuerstadt, P. et al. SER-109, an Oral microbiome therapy for recurrent clostridioides difficile infection. N. Engl. J. Med. 386, 220–229 (2022).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208 (2015).
Sorbara, M. T. & Pamer, E. G. Interbacterial mechanisms of colonization resistance and the strategies pathogens use to overcome them. Mucosal Immunol. 12, 1–9 (2019).
Paone, P. & Cani, P. D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 69, 2232–2243 (2020).
Leonardi, I. et al. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 185, 831–846.e814 (2022).
Kayama, H., Okumura, R. & Takeda, K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu. Rev. Immunol. 38, 23–48 (2020).
Ubeda, C. et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Invest 120, 4332–4341 (2010).
Tamburini, F. B. et al. Precision identification of diverse bloodstream pathogens in the gut microbiome. Nat. Med. 24, 1809–1814 (2018).
Spadoni, I. et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350, 830–834 (2015).
Taur, Y. et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 55, 905–914 (2012).
Dickson, R. P. et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 1, 16113 (2016).
McDonald, B. et al. Programing of an intravascular immune firewall by the gut microbiota protects against pathogen dissemination during infection. Cell Host Microbe 28, 660–668.e664 (2020).
Ichinohe, T. et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl Acad. Sci. 108, 5354–5359 (2011).
Robak, O. H. et al. Antibiotic treatment–induced secondary IgA deficiency enhances susceptibility to Pseudomonas aeruginosa pneumonia. J. Clin. Investig. 128, 3535–3545 (2018).
Clarke, T. B. Early innate immunity to bacterial infection in the lung is regulated systemically by the commensal microbiota via nod-like receptor ligands. Infect. Immun. 82, 4596–4606 (2014).
Schuijt, T. J. et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 65, 575–583 (2016).
Deshmukh, H. S. et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 20, 524–530 (2014).
Clarke, T. B. et al. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 16, 228–231 (2010).
Jacobs, M. C. et al. Effect of antibiotic gut microbiota disruption on LPS-induced acute lung inflammation. PLoS One 15, e0241748 (2020).
Souza, D. G. et al. The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J. Immunol. 173, 4137–4146 (2004).
Morais, L. H., Schreiber, H. L. T. & Mazmanian, S. K. The gut microbiota-brain axis in behaviour and brain disorders. Nat. Rev. Microbiol 19, 241–255 (2021).
Cryan, J. F. et al. The microbiota-gut-brain axis. Physiol. Rev. 99, 1877–2013 (2019).
Dumas, A., Bernard, L., Poquet, Y., Lugo-Villarino, G. & Neyrolles, O. The role of the lung microbiota and the gut-lung axis in respiratory infectious diseases. Cell Microbiol. 20, e12966 (2018).
Tripathi, A. et al. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 15, 397–411 (2018).
Yang, T., Richards, E. M., Pepine, C. J. & Raizada, M. K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 14, 442–456 (2018).
Balmer, M. L. et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J. Immunol. 193, 5273–5283 (2014).
Khosravi, A. et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 15, 374–381 (2014).
Hergott, C. B. et al. Peptidoglycan from the gut microbiota governs the lifespan of circulating phagocytes at homeostasis. Blood 127, 2460–2471 (2016).
d’Hennezel, E., Abubucker, S., Murphy, L. O., Cullen, T. W. & Lozupone, C. Total lipopolysaccharide from the human gut microbiome silences toll-like receptor signaling. mSystems 2, e00046–00017. (2017).
Yang, W. & Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol. Immunol. 18, 866–877 (2021).
Tolhurst, G. et al. Short-chain fatty acids stimulate glucagon-like Peptide-1 secretion via the G-Protein–coupled receptor FFAR2. Diabetes 61, 364–371 (2012).
Lebrun, L. J. et al. Enteroendocrine L cells sense LPS after gut barrier injury to enhance GLP-1 secretion. Cell Rep. 21, 1160–1168 (2017).
Muller, P. A. et al. Microbiota modulate sympathetic neurons via a gut-brain circuit. Nature 583, 441–446 (2020).
Bravo, J. A. et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 108, 16050–16055 (2011).
Baral, P. et al. Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat. Med 24, 417–426 (2018).
Borovikova, L. V. et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462 (2000).
Macia, L., Nanan, R., Hosseini-Beheshti, E. & Grau, G. E. Host- and Microbiota-Derived Extracellular Vesicles, Immune Function, and Disease Development. Int. J. Mol. Sci. 21, 107 (2019).
Fabrega, M. J. et al. Activation of immune and defense responses in the intestinal mucosa by outer membrane vesicles of commensal and probiotic escherichia coli strains. Front Microbiol 7, 705 (2016).
Lajqi, T. et al. Gut Microbiota-Derived Small Extracellular Vesicles Endorse Memory-like Inflammatory Responses in Murine Neutrophils. Biomedicines. 10, 442 (2022).
Cerovic, V. et al. Intestinal CD103(-) dendritic cells migrate in lymph and prime effector T cells. Mucosal Immunol. 6, 104–113 (2013).
Fleming, C. et al. Microbiota-activated CD103(+) DCs stemming from microbiota adaptation specifically drive gammadeltaT17 proliferation and activation. Microbiome 5, 46 (2017).
Coombes, J. L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med 204, 1757–1764 (2007).
Zegarra-Ruiz, D. F. et al. Thymic development of gut-microbiota-specific T cells. Nature 594, 413–417 (2021).
Hegazy, A. N. et al. Circulating and Tissue-Resident CD4(+) T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology 153, 1320–1337.e1316 (2017).
Goris, H., de Boer, F. & van der Waaij, D. Myelopoiesis in experimentally contaminated specific-pathogen-free and germfree mice during oral administration of polymyxin. Infect. Immun. 50, 437–441 (1985).
Bugl, S. et al. Steady-state neutrophil homeostasis is dependent on TLR4/TRIF signaling. Blood 121, 723–733 (2013).
Hidalgo, A. & Casanova-Acebes, M. Dimensions of neutrophil life and fate. Semin Immunol. 57, 101506 (2021).
Mei, J. et al. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J. Clin. Invest 122, 974–986 (2012).
Ohkubo, T., Tsuda, M., Tamura, M. & Yamamura, M. Impaired superoxide production in peripheral blood neutrophils of germ-free rats. Scand. J. Immunol. 32, 727–729 (1990).
Karmarkar, D. & Rock, K. L. Microbiota signalling through MyD88 is necessary for a systemic neutrophilic inflammatory response. Immunology 140, 483–492 (2013).
Maslowski, K. M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009).
Zhang, D. et al. Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532 (2015).
Kanther, M. et al. Commensal microbiota stimulate systemic neutrophil migration through induction of serum amyloid A. Cell Microbiol 16, 1053–1067 (2014).
Fagundes, C. T. et al. Transient TLR activation restores inflammatory response and ability to control pulmonary bacterial infection in germfree mice. J. Immunol. 188, 1411–1420 (2012).
Kang, B. et al. Commensal microbiota drive the functional diversification of colon macrophages. Mucosal Immunol. 13, 216–229 (2020).
Honda, M. et al. Perivascular localization of macrophages in the intestinal mucosa is regulated by Nr4a1 and the microbiome. Nat. Commun. 11, 1329 (2020).
Abdel-Haq, R., Schlachetzki, J. C. M., Glass, C. K. & Mazmanian, S. K. Microbiome-microglia connections via the gut-brain axis. J. Exp. Med 216, 41–59 (2019).
Erny, D. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977 (2015).
Khan, N. et al. Intestinal dysbiosis compromises alveolar macrophage immunity to Mycobacterium tuberculosis. Mucosal Immunol. 12, 772–783 (2019).
Kolypetri, P. et al. Regulation of splenic monocyte homeostasis and function by gut microbial products. iScience 24, 102356 (2021).
Gola, A. et al. Commensal-driven immune zonation of the liver promotes host defence. Nature 589, 131–136 (2021).
Corbitt, N. et al. Gut bacteria drive Kupffer cell expansion via MAMP-mediated ICAM-1 induction on sinusoidal endothelium and influence preservation-reperfusion injury after orthotopic liver transplantation. Am. J. Pathol. 182, 180–191 (2013).
Balmer, M. L. et al. The Liver May Act as a Firewall Mediating Mutualism Between the Host and Its Gut Commensal Microbiota. Sci. Transl. Med. 6, 237ra266–237 (2014).
Zeng, Z. et al. CRIg Functions as a Macrophage Pattern Recognition Receptor to Directly Bind and Capture Blood-Borne Gram-Positive Bacteria. Cell Host Microbe 20, 99–106 (2016).
Serbina, N. V., Jia, T., Hohl, T. M. & Pamer, E. G. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 26, 421–452 (2008).
Schirmer, M. et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167, 1125–1136 (2016).
Schulthess, J. et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 50, 432–445 (2019).
Ang, Z. et al. Human and mouse monocytes display distinct signalling and cytokine profiles upon stimulation with FFAR2/FFAR3 short-chain fatty acid receptor agonists. Sci. Rep. 6, 34145 (2016).
Trompette, A. et al. Dietary Fiber Confers Protection against Flu by Shaping Ly6c− Patrolling Monocyte Hematopoiesis and CD8+ T Cell Metabolism. Immunity 48, 992–1005 (2018).
Gury-BenAri, M. et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid. Cells Are Shaped Microbiome. Cell 166, 1231–1246.e1213 (2016).
Ganal, S. C. et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37, 171–186 (2012).
Gray, J. et al. Intestinal commensal bacteria mediate lung mucosal immunity and promote resistance of newborn mice to infection. Sci. Transl. Med. 9, eaaf9412 (2017).
Brown, E. M., Kenny, D. J. & Xavier, R. J. Gut Microbiota Regulation of T Cells During Inflammation and Autoimmunity. Annu. Rev. Immunol. 37, 599–624 (2019).
Ennamorati, M. et al. Intestinal microbes influence development of thymic lymphocytes in early life. Proc. Natl Acad. Sci. USA 117, 2570–2578 (2020).
Varian, B. J. et al. Beneficial bacteria inhibit cachexia. Oncotarget 7, 11803–11816 (2016).
Nakajima, A. et al. Commensal Bacteria Regulate Thymic Aire Expression. PLoS ONE 9, e105904 (2014).
Hu, M. et al. Decreased maternal serum acetate and impaired fetal thymic and regulatory T cell development in preeclampsia. Nat. Commun. 10, 3031 (2019).
Nakajima, A. et al. Maternal High Fiber Diet during Pregnancy and Lactation Influences Regulatory T Cell Differentiation in Offspring in Mice. J. Immunol. 199, 3516–3524 (2017).
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Gauguet, S. et al. Intestinal Microbiota of Mice Influences Resistance to Staphylococcus aureus Pneumonia. Infect. Immun. 83, 4003–4014 (2015).
McAleer, J. P. et al. Pulmonary Th17 anti-fungal immunity is regulated by the gut microbiome. J. Immunol. (Baltim., Md: 1950) 197, 97–107 (2016).
Shao, T. Y. et al. Commensal Candida albicans Positively Calibrates Systemic Th17 Immunological Responses. Cell Host Microbe 25, 404–417.e406 (2019).
Kim, C. H. Control of lymphocyte functions by gut microbiota-derived short-chain fatty acids. Cell Mol. Immunol. 18, 1161–1171 (2021).
Atarashi, K. et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236 (2013).
Sun, M. et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 9, 3555 (2018).
Tan, J. et al. Dietary Fiber and Bacterial SCFA Enhance Oral Tolerance and Protect against Food Allergy through Diverse Cellular Pathways. Cell Rep. 15, 2809–2824 (2016).
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O. & Kasper, D. L. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 122, 107–118 (2005).
Yang, M. et al. Intestinal Microbiota—A Promising Target for Antiviral Therapy? Front. Immunol. 12, 676232 (2021).
Abt, M. C. et al. Commensal Bacteria Calibrate the Activation Threshold of Innate Antiviral Immunity. Immunity 37, 158–170 (2012).
Thackray, L. B. et al. Oral antibiotic treatment of mice exacerbates the disease severity of multiple flavivirus infections. Cell Rep. 22, 3440–3453.e3446 (2018).
Bachem, A. et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T Cells. Immunity 51, 285–297.e285 (2019).
Tanaka, K., Sawamura, S., Satoh, T., Kobayashi, K. & Noda, S. Role of the Indigenous Microbiota in Maintaining the Virus-Specific CD8 Memory T Cells in the Lung of Mice Infected with Murine Cytomegalovirus. J. Immunol. 178, 5209–5216 (2007).
Pabst, O. & Slack, E. IgA and the intestinal microbiota: the importance of being specific. Mucosal Immunol. 13, 12–21 (2020).
Benveniste, J., Lespinats, G. & Salomon, J.-C. Serum and Secretory IgA in Axenic and Holoxenic Mice. J. Immunol. 107, 1656–1662 (1971).
Crabbe, P., Bazin, H., Eyssen, H. & Heremans, J. The normal microbial flora as a major stimulus for proliferation of plasma cells synthesizing IgA in the gut. Int. Arch. allergy Immunol. 34, 362–375 (1968).
Zeng, M. Y. et al. Gut Microbiota-Induced Immunoglobulin G Controls Systemic Infection by Symbiotic Bacteria and Pathogens. Immunity 44, 647–658 (2016).
Cahenzli, J., Köller, Y., Wyss, M., Geuking Markus, B. & McCoy Kathy, D. Intestinal Microbial Diversity during Early-Life Colonization Shapes Long-Term IgE Levels. Cell Host Microbe 14, 559–570 (2013).
Kim, M., Qie, Y., Park, J. & Kim, C. H. Gut microbial metabolites fuel host antibody responses. Cell host microbe 20, 202–214 (2016).
Wilmore, J. R. et al. Commensal microbes induce serum IgA responses that protect against polymicrobial sepsis. Cell Host Microbe 23, 302–311.e303 (2018).
Rollenske, T. et al. Cross-specificity of protective human antibodies against Klebsiella pneumoniae LPS O-antigen. Nat. Immunol. 19, 617–624 (2018).
Cisalpino, D. et al. Microbiota-induced antibodies are essential for host inflammatory responsiveness to sterile and infectious stimuli. J. Immunol. 198, 4096–4106 (2017).
Doron, I. et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell 184, 1017–1031.e1014 (2021).
Landers, C. J. et al. Selected loss of tolerance evidenced by Crohn’s disease–associated immune responses to auto- and microbial antigens. Gastroenterology 123, 689–699 (2002).
Christmann, B. S. et al. Human seroreactivity to gut microbiota antigens. J. Allergy Clin. Immunol. 136, 1378-1386.e1371-1375 (2015).
Li, H. et al. Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature 584, 274–278 (2020).
Khasbiullina, N. R. & Bovin, N. V. Hypotheses of the origin of natural antibodies: a glycobiologist’s opinion. Biochem. (Mosc.) 80, 820–835 (2015).
Savage, H. P. & Baumgarth, N. Characteristics of natural antibody-secreting cells. Ann. N. Y. Acad. Sci. 1362, 132–142 (2015).
Bunker, J. J. et al. Natural polyreactive IgA antibodies coat the intestinal microbiota. Sci. (NY.) 358, eaan6619 (2017).
Bello-Gil, D. et al. The formation of Glycan-specific natural antibodies repertoire in GalT-KO mice is determined by gut microbiota. Front. Immunol. 10, 342 (2019).
Dessein, R. et al. Antibiotic-related gut dysbiosis induces lung immunodepression and worsens lung infection in mice. Crit. Care 24, 611 (2020).
Li, M. et al. Microbiota-driven interleukin-17 production provides immune protection against invasive candidiasis. Crit. Care 24, 268 (2020).
Sencio, V., Machado, M. G. & Trottein, F. The lung-gut axis during viral respiratory infections: the impact of gut dysbiosis on secondary disease outcomes. Mucosal Immunol. 14, 296–304 (2021).
Lankelma, J. M. et al. The gut microbiota as a modulator of innate immunity during melioidosis. PLoS Negl. Trop. Dis. 11, e0005548 (2017).
Dumas, A. et al. The Host Microbiota Contributes to Early Protection Against Lung Colonization by Mycobacterium tuberculosis. Front Immunol. 9, 2656 (2018).
Kim, S. M. et al. Fecal microbiota transplant rescues mice from human pathogen mediated sepsis by restoring systemic immunity. Nat. Commun. 11, 2354 (2020).
Hamilton, S. E. et al. New Insights into the Immune System Using Dirty Mice. J. Immunol. 205, 3–11 (2020).
Huggins, M. A. et al. Microbial Exposure Enhances Immunity to Pathogens Recognized by TLR2 but Increases Susceptibility to Cytokine Storm through TLR4 Sensitization. Cell Rep. 28, 1729–1743.e1725 (2019).
Schieber, A. M. et al. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science 350, 558–563 (2015).
Lamarche, D. et al. Microbial dysbiosis and mortality during mechanical ventilation: a prospective observational study. Respir. Res. 19, 245 (2018).
McDonald, D. et al. Extreme dysbiosis of the microbiome in critical illness. mSphere. 1, e00199 (2016).
Freedberg, D. E. et al. Pathogen colonization of the gastrointestinal microbiome at intensive care unit admission and risk for subsequent death or infection. Intensive Care Med 44, 1203–1211 (2018).
Haak, B. W. et al. Integrative transkingdom analysis of the gut microbiome in antibiotic perturbation and critical illness. mSystems. 6, 01148-20 (2021).
Shimizu, K. et al. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig. Dis. Sci. 56, 1171–1177 (2011).
Taur, Y. et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124, 1174–1182 (2014).
Peled, J. U. et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N. Engl. J. Med. 382, 822–834 (2020).
Haak, B. W. et al. Impact of gut colonization with butyrate-producing microbiota on respiratory viral infection following allo-HCT. Blood 131, 2978–2986 (2018).
Dickson, R. P. et al. Lung Microbiota Predict Clinical Outcomes in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 201, 555–563 (2020).
Kullberg, R. F. J. et al. Rectal bacteriome and virome signatures and clinical outcomes in community-acquired pneumonia: An exploratory study. eClinicalMed. 39, 101074 (2021).
Valdes-Duque, B. E. et al. Stool Short-Chain Fatty Acids in Critically Ill Patients with Sepsis. J. Am. Coll. Nutr. 39, 706–712 (2020).
Yamada, T. et al. Rapid and Sustained Long-Term Decrease of Fecal Short-Chain Fatty Acids in Critically Ill Patients With Systemic Inflammatory Response Syndrome. J. Parenter. Enter. Nutr. 39, 569–577 (2015).
Lee, J. R. et al. Butyrate-producing gut bacteria and viral infections in kidney transplant recipients: A pilot study. Transpl. Infect. Dis. 21, e13180 (2019).
Klingensmith, N. J. & Coopersmith, C. M. The gut as the motor of multiple organ dysfunction in critical illness. Crit. Care Clin. 32, 203–212 (2016).
Prescott, H. C., Dickson, R. P., Rogers, M. A., Langa, K. M. & Iwashyna, T. J. Hospitalization Type and Subsequent Severe Sepsis. Am. J. Respir. Crit. Care Med 192, 581–588 (2015).
Sorbara, M. T. & Pamer, E. G. Microbiome-based therapeutics. Nat. Rev. Microbiol. 20, 365–380 (2022).
Manzanares, W., Lemieux, M., Langlois, P. L. & Wischmeyer, P. E. Probiotic and synbiotic therapy in critical illness: a systematic review and meta-analysis. Crit. Care 19, 262 (2016).
Johnstone, J. et al. Effect of Probiotics on Incident Ventilator-Associated Pneumonia in Critically Ill Patients: A Randomized Clinical Trial. JAMA 326, 1024–1033 (2021).
Limketkai, B. N., Hendler, S., Ting, P. S. & Parian, A. M. Fecal Microbiota Transplantation for the Critically Ill Patient. Nutr. Clin. Pr. 34, 73–79 (2019).
Wittekamp, B. H. J., Oostdijk, E. A. N., Cuthbertson, B. H., Brun-Buisson, C. & Bonten, M. J. M. Selective decontamination of the digestive tract (SDD) in critically ill patients: a narrative review. Intensive Care Med 46, 343–349 (2020).
DeFilipp, Z. et al. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med 381, 2043–2050 (2019).
Yelin, I. et al. Genomic and epidemiological evidence of bacterial transmission from probiotic capsule to blood in ICU patients. Nat. Med 25, 1728–1732 (2019).
Schluter, J. et al. The gut microbiota is associated with immune cell dynamics in humans. Nature 588, 303–307 (2020).
Henrick, B. M. et al. Bifidobacteria-mediated immune system imprinting early in life. Cell 184, 3884–3898 (2021).
Wipperman, M. F. et al. Gastrointestinal microbiota composition predicts peripheral inflammatory state during treatment of human tuberculosis. Nat. Commun. 12, 1141 (2021).
Rosshart, S. P. et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 171, 1015–1028 e1013 (2017).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019).
Walter, J., Armet, A. M., Finlay, B. B. & Shanahan, F. Establishing or exaggerating causality for the gut microbiome: Lessons from human microbiota-associated rodents. Cell 180, 221–232 (2020).
Yurkovetskiy, L. et al. Gender bias in autoimmunity is influenced by microbiota. Immunity 39, 400–412 (2013).
Markle, J. G. M. et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339, 1084–1088 (2013).
Zhang, M. et al. The effects of biological sex on sepsis treatments in animal models: A systematic review and a narrative elaboration on sex- and gender-dependent differences in sepsis. Crit. Care Explorations 3, e0433 (2021).
Tso, G. H. W. et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science 362, 589–595 (2018).
Acknowledgements
J.S. is funded by an Alberta Graduate Education Scholarship (AGES). B.M. receives funding from the Canadian Institutes of Health Research, Alberta Health Services, the Cumming School of Medicine, and the Canadian Foundation for Innovation. MBG receives funding from the Canadian Institutes of Health Research. Figures were created using BioRender.com (http://biorender.com/). Figure 2 was adapted from “Vitamin D Deficiency Impacts Immunity in the Gut”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
Author information
Authors and Affiliations
Contributions
J.S., I.S., M.B.G., and B.M. conceptualized, wrote, and edited the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Schlechte, J., Skalosky, I., Geuking, M.B. et al. Long-distance relationships - regulation of systemic host defense against infections by the gut microbiota. Mucosal Immunol 15, 809–818 (2022). https://doi.org/10.1038/s41385-022-00539-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00539-2
This article is cited by
-
Altered gut microbiota composition in children and their caregivers infected with the SARS-CoV-2 Omicron variant
World Journal of Pediatrics (2023)
