
Abstract
G protein-coupled receptors (GPCRs) are a group of membrane proteins that mediate most of the physiological responses to various signaling molecules such as hormones, neurotransmitters, and environmental stimulants. Inflammatory bowel disease (IBD) is a chronic relapsing disorder of the gastrointestinal tract and presents a spectrum of heterogeneous disorders falling under two main clinical subtypes including Crohn’s disease (CD) and ulcerative colitis (UC). The pathogenesis of IBD is multifactorial and is related to a genetically dysregulated mucosal immune response to environmental drivers, mainly microbiotas. Although many drugs, such as 5-aminosalicylic acid, glucocorticoids, immunosuppressants, and biological agents, have been approved for IBD treatment, none can cure IBD permanently. Emerging evidence indicates significant associations between GPCRs and the pathogenesis of IBD. Here, we provide an overview of the essential physiological functions and signaling pathways of GPCRs and their roles in mucosal immunity and IBD regulation.
Similar content being viewed by others
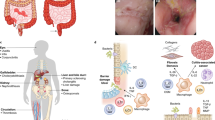

Introduction
G protein-coupled receptors (GPCRs) constitute the largest and most functionally diverse membrane protein superfamilies in eukaryotes1. More than 800 individual genes encode GPCRs in human2, which are widely distributed in various tissues and control an incredible array of cellular functions and physiological processes. All GPCRs are characterized by common seven-transmembrane α-helical segments separated by variable intracellular and extracellular loop regions. Based on the sequences and structures, GPCRs in vertebrates are divided into six subfamilies: class A (rhodopsin-like), class B1 (secretin receptor-like), class B2 (adhesion receptors), class C (metabotropic glutamate receptor-like), class F (frizzled-like), and class T2 (taste 2 sensory receptor)2. GPCRs recognize and respond to multitudinous ligands that range from light photons, odor molecules, ions, and small neurotransmitters to large peptide hormones, glycoprotein hormones, and chemokines3. These ligands interact with the extracellular domains of GPCRs, causing conformational changes in the transmembrane domains and intracellular sequence to transmit signals.
The intestinal mucosal immune system is considered the largest immunological organ in the human body4. A network of immune cells distributes in the intestinal mucosa and constitutes an essential barrier against food antigens, gut microbiota, and foreign pathogens. A growing body of evidence reveals that GPCRs are critical signaling elements involved in regulating intestinal mucosal immunity and maintaining intestinal barrier5. Disorder of intestinal mucosal immunity is an important event in the pathogenesis of inflammatory bowel disease (IBD)6. Further elucidation of the roles of GPCRs in intestinal mucosal immunity is essential to provide novel targets and strategies for the management of IBD. Here, we summarize the complex interplay between GPCRs and mucosal cells in humans and animal models and the dysregulation of GPCR signaling in IBD.
Overview of fundamental biology of GPCRs
To elicit signaling, GPCRs need to couple with intracellular heterotrimeric G proteins. The heterotrimeric complexes are, in turn, dissociated into Gα and Gβγ subunits and interact with different downstream effectors7. In humans, 16 Gα, 5 Gβ, and 13 Gγ subunits have been found2. Each Gα subunit transmits signal independently, whereas Gβ and Gγ subunits are obligate heterodimers that function as an integral (Gβγ). The 16 Gα subunits are classified into 4 major subtypes (including Gs, Gi/o, Gq/11, and G12/13), which trigger distinct signaling cascades (Fig. 1)8. Two representative pathways referring to GPCRs and Gα subunits have been widely studied during the past decades. One is the adenylyl cyclase (AC)/cyclic adenosine monophosphate (cAMP) signaling pathway7,9. Gs proteins stimulated by GPCRs signaling activate AC, resulting in an accumulation of intracellular cAMP and activation of protein kinase A (PKA). In contrast, activation of Gi inhibits AC, causing a decrease in cAMP and a reduced activity of PKA. Pathophysiological processes regulated by AC/cAMP pathway include immune and inflammatory responses, cell growth, and tumorigenesis10,11. Activated Gq protein triggers another fundamental signaling cascade mediated by phospholipase C (PLC)7,9. PLC can hydrolyze phosphatidylinositol 4,5-bisphosphate (PIP2) to generate 2 second messengers, inositol triphosphate (IP3) and diglyceride (DAG), which participate in the regulation of calcium- and calmodulin-dependent protein kinases, and activation of PKC. The PLC signaling pathway mediates the modulation of substance metabolism, neurotransmitter synthesis, and cellular growth and proliferation12. In addition to coupling with G proteins that serve as canonical transducer proteins, GPCRs also interact with regulatory and scaffolding proteins, such as arrestins13. Arrestins can prevent the activated receptors from binding to G proteins and target ligand-occupied GPCRs for endocytosis, therefore exerting a negative moderating effect. However, recent studies have shown that arrestins also serve as scaffolds for initiating additional signaling, such as activation of various mitogen-activated protein kinases (MAPKs)14. Given the universal roles of GPCRs in normal physiological processes, it is not surprising that disturbance in GPCRs and/or their transducers participates in the initiation and progression of various diseases, including IBD.

Canonical GPCR signaling occurs by dissociating heterotrimeric G proteins into Gα and Gβγ subunits. Gα proteins are subdivided into four major subtypes interacting with different downstream effectors. Gs proteins activate AC, resulting in an accumulation of intracellular cAMP and activation of PKA. In contrast, the activated Gi inhibits AC. Gi proteins also activate both PI3K/AKT and MAPK pathways. Activated Gq proteins stimulate PLC, eventually regulating Ca2+ signal and PKC activity, and G12/13 participates in the regulation of small GTPase RhoA-related signaling pathways. GPCRs also interact with other regulatory and scaffolding proteins, such as arrestins. Arrestins mediate the internalization of ligand-occupied GPCRs to exert a negative moderating effect. However, in some contexts, arrestins can also serve as scaffolds for the activation of MAPK.
GPCRs in intestinal mucosal homeostasis and immunity
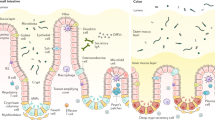
The intestinal mucosal immune system is composed of the epithelium layer, the lamina propria layer, and the gut-associated lymphoid tissues (GALTs). Intestinal immune cells express a variety of GPCRs (Table 1). These GPCRs mediate the complex interactions between immune cells and different environmental factors (Fig. 2).

Intestinal epithelial barrier constitutes the first line of defense against pathogen invasion. Activation of ADRA2, CysLTR1, BLT2, PAR2, and FPR2 promotes the survival and proliferation of IECs. LPAR1, GPR39, and T1R3 participate in the regulation of tight junctions. During an immune response, UDP and SCFAs promote the production of cytokines and chemokines in IECs via recognition of P2Y6 and FFAR2/3, respectively. On the contrary, GPR120 signaling suppresses NF-κB activation in IECs. Multiple GPCR-mediated signalings (such as FPR1/2, C5AR1, BLT1) induce neutrophil migration to inflammatory sites, whereas CB1/2 limits the recruitment of neutrophils. The SCFA-FFAR2 interaction in neutrophils restrains MPO and ROS production, although FFAR2 is also a chemoattractant receptor for neutrophils. GPCRs (e.g., CHRM3, GPBAR1, GPR40, and A2A) are involved in the polarization of macrophages. Moreover, several GPCRs also control the production of distinct cytokines to regulate the function of lymphocytes indirectly. GPCRs such as GPR174, CB2, A2A, FFAR2, and GPR65 directly modulate the differentiation and function of different T cell subsets. In ILC2, activation of NMUR1 and CRTH2 induces the secretion of type 2 cytokines (e.g., IL-4, 5, and 13), whereas the CGRP and ADRB2 signaling play an opposite role. In ILC3, both FFAR2 and GPR34 promote the production of IL-22, which is critical to intestinal mucosal homeostasis. In Peyer’s patches (PPs), B cells depend on CCR6 to migrate to the sup-epithelial dome (SED) and interact with DCs, which is essential for B cells to accomplish antibody class switch and produce IgA. BLT1 signaling enhances the proliferation of IgA+ plasma cells in the intestinal lamina propria.
Intestinal epithelial cells
Intestinal epithelial cells (IECs) participate in the innate immune response of the intestinal mucosa in various ways, including constructing a physical barrier between the exterior environment and the host, as well as actively secreting and responding to various cytokines and other immune active molecules15. Multiple GPCRs are involved in regulating the proliferation and regeneration of IECs. In human IECs, α2A adrenergic receptors are coupled with Gi2/Gi3 and accelerate cell proliferation16. Mechanistically, Gβγ subunit of Gi2/Gi3 proteins facilitates the formation of SHC-transforming protein 1 (SHC1)-growth factor receptor-bound protein 2 (GRB2)-son of sevenless homolog (SOS) complex, which subsequently mediates the activation of MAPK/ERK kinase (MEK) 1 and MAPK to promote cell proliferation16,17. Besides, endogenous production of leukotriene D4 promotes the survival and proliferation of IECs via cysteinyl leukotriene receptor 1 (CysLTR1)18. Another leukotriene receptor, leukotriene B4 receptor type 2 (BLT2), also promotes epithelial cell proliferation and intestinal wound repair through Gi/o protein- and PLC/PKC-dependent signaling pathways19. N-formyl peptide receptors comprise a group of Gi-coupled receptors that participate in the regulation of innate immune responses. Interestingly, both formylpeptide receptor-1 (FPR1) and formylpeptide receptor-2 (FPR2) are expressed in IECs20,21, and enhance epithelial cell proliferation and renewal upon activation, leading to epithelial wound healing. The proteinase-activated receptors (PARs), a unique subset of GPCRs, are activated through proteolytic cleavage of the N terminus of the receptor, instead of traditional ligand binding22. PAR2 stimulation activates MAPK/ERK kinase (MEK) 1/2 and phosphoinositide-3-kinase (PI3K) to protect colonic epithelial cells from apoptosis induced by proinflammatory cytokines such as TNF-α and IFN-γ23. GPR81 (also known as hydroxycarboxylic acid receptor 1, HCA1) is an endogenous receptor of lactate24. Interestingly, it has been found that microbiota-derived lactate activates GPR81 signaling to induce Wnt3 secretion by Paneth cells and stromal cells and eventually promotes intestinal stem cell proliferation25. Furthermore, some GPCRs are capable of regulating epithelial barrier function. For example, lysophosphatidic acid (LPA) receptor 1 participates in maintaining apical junction integrity26. Consistently, deletion of Lpar1 in mice results in the decrease of claudin-4, claudin-7, and E-cadherin and the augment of epithelial permeability. Additionally, activation of GPR39, a zinc sensor, promotes an assembly of tight junctions in IECs via the PLC-calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2)-AMP-activated protein kinase (AMPK) pathways27. In contrast, artificial sweeteners can activate the sweet taste receptor (T1R3) to induce claudin-3 internalization and promote cell apoptosis, thereby significantly impairing the intestinal barrier function28. The small intestinal tuft cells, which play critical roles in immunity against parasite infection29, use succinate receptor (SUCNR1, also known as GPR91) to monitor helminth Nippostrongylus brasiliensis and protist Tritrichomonas rainier, and initiate type 2 immunity30,31. Tuft cells also express another GPCR, the bitter taste receptor (type 2 taste receptor, T2R) whose signaling via G protein cascade activates tuft cells to protect the intestine against Trichinella spiralis infection32. Recent work has shown that tuft cells express an olfactory-related GPCR Vmn2r2633. By sensing the bacterial metabolite N-undecanoylglycine through Vmn2r26, tuft cells quickly expand in response to bacterial infection and release prostaglandin D2 (PGD2) to enhance the mucus secretion of goblet cells. These findings illustrate the additional effects of tuft cells on intestinal mucosal immunity against bacterial infection through various GPCRs.
The secretion of cytokines by IECs, such as IL-6, TNF-α, MCP-1, CCL10, GM-CSF, and CXCL8 (IL-8), is tightly regulated by GPCRs. P2Y receptors are a group of GPCRs that sense extracellular nucleotides34. Intestinal inflammation concurrently upregulates P2Y6 receptor expression and promotes the release of extracellular UDP in the colon mucosa of colitic mice35. The activation of P2Y6 by UDP in turn promotes the transcription and release of CXCL8, which is critical for recruiting neutrophils. Activation of either PAR1 or PAR2 also induces the transcriptional upregulation of CXCL8 in IECs via a pathway that involves ERK/RSK p90, NF-κB, and histone acetyltransferase (HAT) activity36. Colon epithelial cells highly express free fatty acid receptor 3 (FFAR3, also known as GPR41) and free fatty acid receptor 2 (FFAR2, also known as GPR43)37, which are able to sense short chain fatty acids (SCFAs) and activate the MAPK signaling pathway to promote the production of various cytokines and chemokines. Therefore, at the initial stages of intestinal bacterial infection, recognition of SCFAs by IECs promotes an acute inflammatory response, which is crucial for the clearance of pathogens. Furthermore, SCFAs act through FFAR2 to induce IECs to express antimicrobial peptides such as RegIIIγ and β-defensins 1/3/4 in a mTOR- and STAT3-dependent manner38. In contrast to FFAR2 and FFAR3, hydroxycarboxylic acid receptor 2 (HCAR2, also known as GPR109A, a receptor for niacin and butyrate) signaling profoundly suppresses lipopolysaccharide (LPS)-induced NF-κB activation in normal and colon cancer cell lines as well as in normal mouse colon39. Another GPCR calcium-sensing receptor (CaSR) plays an important role in sensing nutrients and monitoring ion balance in the intestine. A recent report has demonstrated that activation of CaSR by L-tryptophan or L-valine could block TNF-α-induced inflammatory response in IECs40. Free fatty acid receptor 4 (FFAR4, also known as GPR120), which senses medium- and long-chain unsaturated fatty acids, also exerts an anti-inflammatory effect by binding to β-arrestin2 and attenuating NF-κB activation in IECs41.
Neutrophils
Neutrophils are the first leukocytes mobilized and recruited into the sites of infection and inflammation42. They are responsive to a broad array of chemically diverse chemoattractants, many of which are sensed by GPCRs, including FPR1/2 (sensing bacteria-derived N-formyl peptides), C5AR1 (sensing C5a), BLT1 (sensing LTB4) and multiple chemokines receptors43. FFAR2 is also a chemoattractant receptor highly expressed in neutrophils44,45. During Clostridium difficile infection in the intestine, acetate enhances neutrophil accumulation in the inflamed sites and prevents bacterial translocation. This effect is partly mediated by FFAR246. Moreover, it has also been demonstrated that the SCFA-FFAR2 interaction in neutrophils could induce apoptosis, reduce MPO and ROS production and inhibit the chemotaxis to N-formyl peptides and C5a44. Collectively, the current evidence indicates that the SCFA-FFAR2 signaling fine-tunes neutrophils by concurrently guiding the recruitment of neutrophils at the early stage of infection and preventing the overactivation during the inflammatory response, which is crucial for host defense against pathogens and resolution of inflammation. Recent data have also demonstrated that FPR1 is not just a chemoattractant receptor that modulates the recruitment of neutrophils, but also promotes local immune cell activation and survival, thereby contributing to the inflammatory process47. Consistently, Fpr1 knockout mice have shown an increased resistance to 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced experimental colitis. The endocannabinoid system consists of cannabinoid receptors, the endogenous lipid ligands (known as the endocannabinoids), and the enzymes that catalyze the metabolism of these lipids48. Both cannabinoid receptor 1 (CNR1) and cannabinoid receptor 2 (CNR2) are GPCRs mainly coupling with Gs. Accumulating evidence indicates that the endocannabinoid system profoundly inhibits inflammatory response through multiple mechanisms, including direct regulation of neutrophils49,50,51,52. Indeed, neutrophils isolated from Cnr2−/− mice exhibit an upregulated migration-related transcriptional profile and enhanced adhesion and transmigration capacity toward activated endothelial cells, suggesting that CNR2 restrains the recruitment of neutrophils to local inflammatory sites52.
Mononuclear phagocytes
The intestinal lamina propria contains a variety of mononuclear phagocyte subsets, including monocytes, intestinal resident macrophages, and dendritic cells (DCs), which are critical for triggering proper active immune responses against various commensal microorganisms and pathogens53. Intestinal mononuclear phagocytes express a series of GPCRs, which exert a broad regulatory effect in response to various tissue microenvironment-related stimuli. Adenosine accumulates extracellularly in states of stress54,55, such as hypoxia and cell damage. Extracellular adenosine acts as an endogenous regulator of immune response by binding to and activating four kinds of GPCRs, designated A1, A2A, A2B, and A356. A2A receptor signaling has been shown to augment IL-4- or IL-13-induced M2-like macrophage polarization in vitro57, whereas adenosine acts via the A2B receptor to induce small intestinal DCs to express IL-6, which further promotes the development of Th17 cells58. In chemically induced murine models of colitis, activation of GPBAR1, a GPCR for secondary bile acids, has been found to shift colonic lamina propria macrophages to a M2-like phenotype to alleviate the intestinal inflammation59. Moreover, the effect of GPBAR1 on macrophage polarization is dependent on IL-10. Similarly, activation of both D prostanoid receptor 1 by prostaglandin D2 and GPR40 by α-linolenic acid (ALA) facilitates intestinal macrophage polarization toward M2-like cells60,61. Colonic macrophages and DCs also express GPR109A62,63, and the butyrate- or niacin-GPR109A signaling endows macrophages and DCs with an anti-inflammatory phenotype characterized by upregulated expression of IL-10 and aldehyde dehydrogenase 1A1 and enables them to induce differentiation of regulatory T cells (Tregs)64. Furthermore, it has been shown recently that GPR109A signaling could limit microbiota-induced production of IL-23 by colonic DCs to restrain the overactivation of ILC3s65. GPR81, a metabolite-sensing receptor for lactate, also imparts a regulatory phenotype on colonic macrophages and DCs to maintain the balance of Treg and Th1/Th17 effector populations, which is critical to maintaining intestinal homeostasis66. In the lamina propria of the small intestine, CX3CR1+ macrophages, a well-known immune regulatory subtype, express the highest level of CNR2, which can be activated by endogenous cannabinoid anandamide (AEA). Therefore, the AEA-CNR2 axis regulates immune tolerance in the intestine through maintaining CX3CR1+ macrophages and enhancing the competence of macrophages to induce a subset of regulatory T cells (namely Tr1 cells) in an IL-27-dependent manner67. In addition, CX3CR1+ macrophages can protrude their dendrites into the lumen to access luminal antigens, which is indispensable for intestinal immune surveillance68,69. Recent study has also reported that intestinal bacterium-derived pyruvic acid and lactic acid induce dendrite extension of CX3CR1+ macrophages to enhance immune responses via GPR31 signaling70.
The enteric nervous system conveys important information about the environment to the immune system. β2-adrenergic receptor (β2-AR) mediates the catecholamine-induced activation of AC through the action of G proteins. Activation of β2-adrenergic receptor in macrophages converts a general proinflammatory signal in response to TLRs into an anti-inflammatory pathway and induces the rapid transcription and secretion of IL-1071,72. In responding to luminal infection, intestinal muscularis macrophages orchestrate a neuroprotective program via a β2-adrenergic receptor signaling pathway and prevent neurons from death post-infection through an arginase 1-polyamine axis73. In contrast, muscarinic stimulation through type 3 muscarinic receptors acts synergistically with interferon-γ to promote the development of classically activated macrophages, which contributes to host defense against the enteric bacterium Citrobacter rodentium74. GPR55 responds to endogenous L-α-lysophosphatidylinositol (LPI)75,76 and signals through G12/13 and Gq proteins, leading to release of Ca2+ and activation of NF-κB and nuclear factor of activated T-cells (NFAT)77. Consistently, pharmacological blockade of GPR55 could alleviate experimental colitis by inhibiting macrophage accumulation and activation in the intestine78. BLT1 can act on DCs to upregulate the expression of proinflammatory cytokines such as IL-6, IL-12, and TNF-α, further promoting Th1/Th17 cell differentiation79. DC-specific knockout of Blt1 significantly ameliorates TNBS-induced colitis in mice. GPCR signaling also indirectly affects the function of B cells through DCs. For example, acetate activation of FFAR2 induces the intestinal DCs to express aldehyde dehydrogenase 1A2, which converts vitamin A into its metabolite retinoic acid (RA) to promote B cell IgA class switch and increase IgA concentration in the intestinal lumen80.
Innate lymphoid cells
Innate lymphoid cells (ILCs) comprise a special population of hematopoietic effector cells that mainly reside in mucosa-associated tissues and play a crucial role in immune defense and tissue remodeling81,82. They share numerous developmental and functional characteristics with CD4+ T cells and are divided into three subtypes based on the signature transcription factors and effector cytokines: (1) ILC1s require the transcription factor T-BET and express interferon-γ; (2) ILC2s depend on the transcription factor GATA3 and mainly express the type 2 cytokines IL-5 and IL-13; and (3) ILC3s are dependent on the transcription factor RORγt and produce IL-17 and IL-22.
ILC2s have emerged as critical regulators of defense against parasites and protozoa and type 2 inflammation in intestinal mucosa83,84,85. It has been shown that neuronal signals orchestrate intestinal ILC2 activity through multiple GPCRs. Cholinergic neurons-derived neuropeptide neuromedin U (NMU) acts through NMUR1, which is coupled with Gαq, to promote ILC2 activation, proliferation, and secretion of IL-5, IL-9, and IL-1386,87,88. In vivo administration of NMU to mice triggers potent type 2 cytokine responses and accelerates the expulsion of the gastrointestinal nematode Nippostrongylus brasiliensis. Interestingly, cholinergic neurons in the intestine also release another neuropeptide named alpha-calcitonin gene-related peptide (α-CGRP), which inhibits ILC2 proliferation and restrains type 2 immunity through a Gs-AC-cAMP pathway89,90. Moreover, murine ILC2s express β2-adrenergic receptor and colocalize with adrenergic neurons in the intestine91. The β2-adrenergic receptor pathway functions as a cell-intrinsic negative regulator of ILC2 responses by restraining cell proliferation and effector function, and its deficiency results in exaggerated ILC2 responses and type 2 inflammation in the intestine. Chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2), a receptor for prostaglandin D2 (PGD2), is found to be expressed in human ILC2s92. PGD2 activates ILC2s through CRTH2 to induce cell migration, production of type 2 cytokines, and upregulation of IL-33 and IL-25 receptor subunits (ST2 and IL-17RA). Furthermore, during helminth infection in the small intestine, leukotrienes released by tuft cells cooperate with IL-25 to activate ILC2s and drive rapid anti-helminth immunity. The effect of leukotrienes is mediated through CysLTR1 and CysLTR2, both of which belong to the GPCR family93.
ILC3s are enriched in the intestine, where they maintain intestinal homeostasis by inducing lymphoid-tissue development, containment of commensal bacteria, and regulation of host defense and adaptive immunity94,95. Distinct signals control the spatial and functional compartmentalization of ILC3s in the intestine. Migration of CCR6−NKp46+ ILC3 to the lamina propria occurs in a CXCL16- and CXCR6-dependent manner96, while 7α,25-hydroxycholesterol (7α,25-OHC) acts through GPR183 to direct CCR6+ ILC3s to colonic cryptopatches (CPs) and isolated lymphoid follicles (ILFs), where they induce normal lymphoid tissue development97,98. Consistently, Gpr183 deficiency in mice results in a disorganized distribution of ILC3 in mesenteric lymph nodes (MLNs), decreased ILC3 accumulation in the intestine, and increased susceptibility to enteric bacterial infections. Colonic ILC3 also expresses FFAR2, and activation of FFAR2 promotes cell proliferation and IL-22 production in an AKT- and STAT3-dependent manner99. Accordingly, deletion of Ffar2 in mice leads to impaired intestinal barrier function characterized by decreased production of mucus-associated proteins and antimicrobial peptides and increased susceptibility to colonic injury and bacterial infection. When intestinal tissue is damaged, lysophosphatidylserine (LysoPS) released by apoptotic neutrophils activates GPR34 in ILC3s to enhance IL-22 production and tissue repair in a PI3K-AKT- or ERK-dependent manner100. Collectively, this uncovers a novel mechanism whereby GPCRs coordinate different immune cells in the intestinal mucosa.
T lymphocytes
The establishment and maintenance of adaptive immune responses, immunological memory, and self-tolerance largely depend on T cells. In the intestine, diverse effector T cells are responsible for defense against the invasion of various pathogens, while Tregs play a critical role in maintaining immune tolerance to gut commensal microbiotas and innocent food antigens. As discussed above, GPCRs orchestrate innate immune, especially macrophages and DCs, to regulate T cells indirectly. Moreover, T cells also express various GPCRs and are robustly regulated by GPCR signaling.
The small intestine epithelium releases the chemokine CCL25, an important ligand for the chemokine receptor CCR9101. Memory and effector T cells activated in GALTs are recruited to the small intestine in a CCR9-dependent manner, and Tregs homing to small intestine lamina propria also express CCR9102,103. Another chemokine receptor, GPR15, is important for the localization of both regulatory and effector/memory T cells in the colon104,105. It has been shown that environmental sensor aryl hydrocarbon receptor directly binds to open chromatin regions of the GPR15 locus to enhance its expression in CD4+ T cells but not CD8+ T cells in the intestines106. However, GPR55, whose endogenous ligand is lysophosphatidylinositol, has been demonstrated to function as a negative regulator of T cell homing to the small intestine via a Gα13/Rho/ROCK-mediated pathway107. Another important mediator that regulates T cell trafficking is sphingosine-1-phosphate (S1P). There are five GPCRs (namely S1PR1 to S1PR5) widely expressed in vertebrates that can respond to extracellular S1P108,109. S1PR1 signaling is crucial for the egress of lymphocytes from lymphoid organs110,111. Its agonists induce internalization and degradation of S1PR1, thus maintaining T cell retention in secondary lymphoid organs110,112. This results in a reversible decline of circulating lymphocytes in the periphery blood.
Distinct GPCRs also regulate specific subsets of T cells. GPR174, which senses lysophosphatidylserine, is abundantly expressed in developing and mature Tregs113. In vitro study has shown that lysophosphatidylserine could act via activating GPR174 to inhibit T cell proliferation and Treg differentiation. CNR2 is preferentially expressed on Tregs, and selective agonists of CNR2 have been shown to exert an anti-inflammatory effect in different settings of murine colitis models114,115. Mechanistically, CNR2 activation induces Treg differentiation and enhances their suppressive capacity via the p38-STAT5A pathway115. In contrast, the CCL20-CCR6 signaling suppresses the TGF-β1-induced Treg (iTreg) differentiation and dictates them towards the pathogenic Th17 phenotype116. Interestingly, such iTregs that differentiate in the presence of CCL20 also show impaired suppressive functions characterized by decreased expression of suppressor molecules such as CD39, CD73, and FasL. A microbiota-modulated metabolite, inosine, significantly activates adenosine A2A receptor to inhibit the differentiation of Th1 and Th2 cells and associated proinflammatory cytokine production117. Furthermore, SCFAs have been found to directly act via FFAR2 to induce IL-10 expression in Th1 cells118. Mechanistically, this effect is mediated through activating STAT3 and mTOR, consequently upregulating the transcription factor B lymphocyte induced maturation protein 1 (Blimp-1), which directly targets IL-10. Recently, it has been reported that activation of GPR120 promotes the production of IL-10 in CD4+ T cells by inducing Blimp1 and enhancing glycolysis119.
B lymphocytes
B lymphocytes participate in the first line of defense against gut antigens by providing neutralizing immunoglobulins (mainly IgA) directed against pathogens or toxins. Therefore, the effect of GPCRs on B cells mainly involves the modulation of migration and IgA production. Similar to T cells, IgA+ B cells in Peyer’s patches (PPs) express high levels of CCR9120, which is required for migration of IgA+ B cells via the draining MLN and thoracic duct into the small intestinal lamina propria. During the migration, IgA+ B cells gradually mature into plasma cells with local IgA secretion121,122. Accordingly, knockout of Ccr9 in mice results in a loss of IgA+ plasma cells in the lamina propria of the small intestine and impaired IgA response to an orally administered antigen, ovalbumin (OVA). The CCL20-CCR6 and CXCL13-CXCR5 signals are essential for B cell recruitment and isolated lymphoid follicle formation in the small intestine123,124. The microbiota has been shown to promote the expression of CCL20 and CXCL13 in the small intestine, but not in the colon97. Interestingly, activated B cells also depend on CCR6 to access the sup-epithelial dome (SED) of PPs and interact with SED DCs, which is critical for B cells to accomplish IgA class switch125. In PPs, B cells gradually upregulate BLT1 expression during their differentiation into IgA+ B cells and retain BLT1 expression after migrating to lamina propria126. BLT1 signaling enhances the commensal bacteria-dependent proliferation of IgA+ plasma cells in the intestinal lamina propria by inducing MyD88. Therefore, BLT1 plays a critical role in the production of antigen-specific intestinal mucosal IgA.
GPCRs and IBD
It is well established that GPCRs play important roles in the pathogenesis of various autoimmune diseases, including IBD. Recent research advances have shown that GPCRs have enormous potentials in the diagnosis, treatment, and monitoring of IBD.
An acidic microenvironment with a decreased local tissue pH is a hallmark of chronic intestinal inflammation, including IBD127,128,129. Therefore, it is essential to determine the effect of the acidic microenvironment on intestinal cells and the underlying signaling pathways. Up to date, a pH-sensing GPCR family including GPR4, GPR68 (also known as OGR1), GPR65 (also known as TDAG8), and GPR132 (also known as G2A) has been found130,131,132. GPR4 is highly expressed on vascular endothelial cells133,134. Activation of GPR4 in response to acidosis exerts a proinflammatory effect, including induction of adhesion molecules and chemokines, which collectively enhance leukocyte adhesion to endothelial cells and subsequent extravasation into inflamed tissues135,136. GPR4 is found to be increased in mouse and human IBD intestinal mucosal tissues and highly enriched in microvessels adjacent to ILFs and the specialized high endothelial venules (HEVs) in MLNs137. GPR68 has been reported to act through a Gq-coupled signaling pathway to induce proinflammatory cytokine production. Concurrent knockout of Gpr68 prevents Il10−/− mice from spontaneous colitis138. Moreover, GPR68 is also increased in fibrosis-affected human terminal ileum compared to that in the non-fibrotic resection margin and positively correlated with pro-fibrotic cytokines (such as TGF-β1 and CTGF) and pro-collagens139. Consistently, Gpr68 deletion alleviates the intestinal fibrosis formation in a heterotopic mouse intestinal transplant model, further indicating a critical role in the development of intestinal fibrosis. GPR65 is critical to maintaining lysosomal function and efficient pathogen defense140. Loss of Gpr65 increases mouse susceptibility to Citrobacter rodentium infection-associated colitis. Mechanistically, increased intracellular cAMP by GPR65 signaling induces V-ATPase trafficking to support a lysosomal acidic state and normal protein-degrading function. In patients with an IBD-associated GPR65 missense variant, I231L, lymphoblasts also display disrupted lysosomal function and intracellular bacterial clearance. Recently, we have found that GPR65 is significantly upregulated in the inflamed mucosa of IBD patients and promotes Th1 and Th17 cell differentiation and immune response by suppressing the expression of the NUAK family SNF1-like kinase 2 (NUAK2)141. Consistently, selective knockout of Gpr65 in CD4+ T cells markedly alleviates acute and chronic murine colitis. Our findings further reveal the important role of GPR65 in regulating intestinal homeostasis and inflammatory response.
GPR84 is a putative medium-chain free fatty acid receptor that is mainly expressed in myeloid cells142. Macrophages expressing GPR84 are increased in the colonic mucosa of active UC patients143. Consistently, deletion of Gpr84 reduces the susceptibility of mice to dextran sulfate sodium (DSS)-induced colitis. CLH536, a novel GPR84 antagonist, is observed to alleviate acute colitis in mice by inhibiting the polarization and function of proinflammatory macrophages. In contrast, GPR132 signaling functions to dampen intestinal inflammation via the production of IFN-γ, promoting monocyte maturation with a less proinflammatory program144.
The endocannabinoid system exerts an immunosuppressive effect, partly mediated by CNR2, which is decreased in the ileum of CD patients114. In vitro treatment with CNR2 agonist promotes epithelial cell proliferation and reduces MMP9 and IL-8 levels in inflamed biopsies from IBD patients145, indicating that CNR2 contributes to mucosal healing in IBD. However, a common CNR2 functional variant, Q63R, is associated with a more severe phenotype in both UC and CD146. Therefore, the critical role of endocannabinoid system in the pathogenesis of IBD needs to be further investigated.
It has been shown that succinate participates in the pathogenesis of IBD through SUCNR1 (GPR91), which is highly expressed in fibroblasts from CD patients compared to healthy controls147. Stimulation with succinate induces fibrotic markers (such as COL1A1, α-SMA) and proinflammatory cytokines through SUCNR1. Deletion of Sucnr1 protects mice against intestinal fibrosis induced by the heterotopic transplant of colonic tissue. Interestingly, in penetrating CD patients, succinate and SUCNR1 are upregulated in intestinal tissue that surrounds the fistula tract148. In vitro assay has shown that in HT29 cell lines, SUCNR1 activation induces the expression of Wnt ligands and a Wnt-mediated epithelial-to-mesenchymal transition (EMT) process, which is associated with fistula formation. Collectively, these data reveal that SUCNR1 may serve as a novel target for the prevention and alleviation of IBD complications, including intestinal fibrosis and fistula formation.
Mast cells have been shown to involve in IBD pathogenesis, although the precise roles remain unclear. Mast cells are highly infiltrated in the ileal tissues of CD patients and the colonic tissues of UC patients, and highly express vasoactive intestinal polypeptide receptor 1 (VIPR1), a GPCR coupled with Gs for vasoactive intestinal polypeptide (VIP)149. Other studies have also demonstrated that colonic mast cell-derived histamine promotes granulocyte infiltration into the colonic mucosa through the histamine H4 receptor (GPR105). Specific blockade of histamine production in mast cells reduces mucosal neutrophil infiltration and intestinal inflammation in both oxazolone- and DSS-induced experimental colitis models in mice. Another GPCR, MRGPRX2, has been recently identified as a novel mast cell-specific receptor to facilitate IgE-independent mast cell activation150,151. A unique variant of MRGPRX2, D62S, appears protective in UC through enhancing β-arrestin recruitment, decreasing IP-1, and increasing phosphorylated ERK152.
GPCRs as therapeutic targets in IBD
GPCRs constitute the largest membrane protein family and participate in various cellular responses and pathophysiological processes, which makes GPCRs among the most concerned drug targets. Statistics show that more than 700 drugs targeting GPCRs have been approved by the FDA, which target 134 unique GPCRs and occupy almost 35% of all FDA-approved drugs153. Central nervous system diseases (e.g., Alzheimer’s disease, Huntington’s disease, and multiple sclerosis) remain the most widely used field for GPCR-targeted drugs. In recent years some digestive diseases have also been added to the expanding list of indications. Naloxegol, a purely peripherally acting μ-opioid receptor antagonist, has been used to manage opioid-induced constipation and other gastrointestinal symptoms of opioid-induced bowel dysfunction154. Pentoxifylline, an A2B antagonist, is the first GPCR-targeted agent for irritable bowel syndrome, which was approved by FDA in 1984. Eluxadoline is a mixed µ-opioid receptor agonist and δ-opioid receptor antagonist that exerts effect locally in the gastrointestinal tract, and has been approved by FDA to treat adults who have irritable bowel syndrome with diarrhea (IBS-D) since 2015155. Concerning IBD, although GPCRs play critical roles in the regulation of inflammatory responses, intestinal barrier and intestinal fibrosis, and even some agonists and antagonists have been proven to be effective in animal models of IBD, translation from basic research to clinical application is just in its infancy. Only a limited number of drugs have entered clinical trials, including prostaglandin E receptor 4 (EP4) agonist, CCR9 antagonist, GPR84 antagonist, FFAR2 antagonist, and some S1P modulators (Table 2)156,157,158,159,160. More research efforts are warranted to expedite GPCR-targeted drug discovery further.
Conclusions
GPCRs are receiving a great deal of attention as critical intestinal mucosal immune system regulators. IBD is a highly complex disease, and its pathogenesis remains fully undefined. With an in-depth understanding of the biological role of GPCRs in different intestinal immune cells, increasing evidence has supported the involvement of GPCR signaling pathways in IBD-associated pathophysiological processes, including leukocyte adhesion and accumulation, inflammatory mediator production, intestinal barrier maintenance, and defense against pathogens. Although many agents targeting GPCRs have shown to be effective in the preclinical stage, including in vitro cell assays and animal models, translation to clinical and pharmaceutical practice is still a long way off. New experimental technologies and drug biotechnologies are urgently needed to promote GPCR-targeted drug discovery and disease treatment further.
References
Rosenbaum, D. M., Rasmussen, S. G. & Kobilka, B. K. The structure and function of G-protein-coupled receptors. Nature 459, 356–363 (2009).
Wootten, D., Christopoulos, A., Marti-Solano, M., Babu, M. M. & Sexton, P. M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell. Biol. 19, 638–653 (2018).
Katritch, V., Cherezov, V. & Stevens, R. C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2013).
Yap, Y. A. & Mariño, E. An insight into the intestinal web of mucosal immunity, microbiota, and diet in inflammation. Front. Immunol. 9, 2617 (2018).
Sun, M., Wu, W., Liu, Z. & Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 52, 1–8 (2017).
Graham, D. B. & Xavier, R. J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 578, 527–539 (2020).
Cabrera-Vera, T. M. et al. Insights into G protein structure, function, and regulation. Endocr. Rev. 24, 765–781 (2003).
Duc, N. M., Kim, H. R. & Chung, K. Y. Structural mechanism of G protein activation by G protein-coupled receptor. Eur. J. Pharmacol. 763, 214–222 (2015).
Ulloa-Aguirre, A., Stanislaus, D., Janovick, J. A. & Conn, P. M. Structure-activity relationships of G protein-coupled receptors. Arch. Med. Res. 30, 420–435 (1999).
Arumugham, V. B. & Baldari, C. T. cAMP: a multifaceted modulator of immune synapse assembly and T cell activation. J. Leukoc. Biol. 101, 1301–1316 (2017).
Zhang, H., Kong, Q., Wang, J., Jiang, Y. & Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 9, 32 (2020).
Bill, C. A. & Vines, C. M. Phospholipase C. Adv. Exp. Med. Biol. 1131, 215–242 (2020).
Peterson, Y. K. & Luttrell, L. M. The diverse roles of arrestin scaffolds in G protein-coupled receptor signaling. Pharmacol. Rev. 69, 256–297 (2017).
Smith, J. S., Lefkowitz, R. J. & Rajagopal, S. Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 17, 243–260 (2018).
Müller, C. A., Autenrieth, I. B. & Peschel, A. Innate defenses of the intestinal epithelial barrier. Cell. Mol. Life Sci. 62, 1297–1307 (2005).
Schaak, S. et al. Alpha(2) adrenoceptors regulate proliferation of human intestinal epithelial cells. Gut 47, 242–250 (2000).
van Biesen, T. et al. Receptor-tyrosine-kinase- and G beta gamma-mediated MAP kinase activation by a common signalling pathway. Nature 376, 781–784 (1995).
Paruchuri, S., Mezhybovska, M., Juhas, M. & Sjölander, A. Endogenous production of leukotriene D4 mediates autocrine survival and proliferation via CysLT1 receptor signalling in intestinal epithelial cells. Oncogene 25, 6660–6665 (2006).
Matsumoto, Y. et al. Leukotriene B(4) receptor Type 2 accelerates the healing of intestinal lesions by promoting epithelial cell proliferation. J. Pharmacol. Exp. Ther. 373, 1–9 (2020).
Chen, K. et al. Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J. Clin. Invest. 123, 1694–1704 (2013).
Babbin, B. A. et al. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J. Immunol. 179, 8112–8121 (2007).
Macfarlane, S. R., Seatter, M. J., Kanke, T., Hunter, G. D. & Plevin, R. Proteinase-activated receptors. Pharmacol. Rev. 53, 245–282 (2001).
Iablokov, V. et al. Proteinase-activated receptor 2 (PAR2) decreases apoptosis in colonic epithelial cells. J. Biol. Chem. 289, 34366–34377 (2014).
Hu, J. et al. The roles of GRP81 as a metabolic sensor and inflammatory mediator. J. Cell. Physiol. 235, 8938–8950 (2020).
Lee, Y. S. et al. Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe 24, 833–846.e836 (2018).
Lin, S. et al. Lysophosphatidic acid receptor 1 is important for intestinal epithelial barrier function and susceptibility to colitis. Am. J. Pathol. 188, 353–366 (2018).
Pongkorpsakol, P., Buasakdi, C., Chantivas, T., Chatsudthipong, V. & Muanprasat, C. An agonist of a zinc-sensing receptor GPR39 enhances tight junction assembly in intestinal epithelial cells via an AMPK-dependent mechanism. Eur. J. Pharmacol. 842, 306–313 (2019).
Shil, A. et al. Artificial sweeteners disrupt tight junctions and barrier function in the intestinal epithelium through activation of the sweet taste receptor, T1R3. Nutrients 12, 1862 (2020).
Schneider, C., O’Leary, C. E. & Locksley, R. M. Regulation of immune responses by tuft cells. Nat. Rev. Immunol. 19, 584–593 (2019).
Nadjsombati, M. S. et al. Detection of succinate by intestinal tuft cells triggers a Type 2 innate immune circuit. Immunity 49, 33–41.e37 (2018).
Schneider, C. et al. A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 174, 271–284.e214 (2018).
Luo, X. C. et al. Infection by the parasitic helminth Trichinella spiralis activates a Tas2r-mediated signaling pathway in intestinal tuft cells. Proc. Natl Acad. Sci. USA 116, 5564–5569 (2019).
Xiong, Z. et al. Intestinal Tuft-2 cells exert antimicrobial immunity via sensing bacterial metabolite N-undecanoylglycine. Immunity 55, 686–700 (2022).
von Kügelgen, I. & Hoffmann, K. Pharmacology and structure of P2Y receptors. Neuropharmacology 104, 50–61 (2016).
Grbic, D. M., Degagné, E., Langlois, C., Dupuis, A. A. & Gendron, F. P. Intestinal inflammation increases the expression of the P2Y6 receptor on epithelial cells and the release of CXC chemokine ligand 8 by UDP. J. Immunol. 180, 2659–2668 (2008).
Wang, H., Moreau, F., Hirota, C. L. & MacNaughton, W. K. Proteinase-activated receptors induce interleukin-8 expression by intestinal epithelial cells through ERK/RSK90 activation and histone acetylation. FASEB J. 24, 1971–1980 (2010).
Kim, M. H., Kang, S. G., Park, J. H., Yanagisawa, M. & Kim, C. H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 145, 396–406.e391-310 (2013).
Zhao, Y. et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol. 11, 752–762 (2018).
Thangaraju, M. et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 69, 2826–2832 (2009).
Mine, Y. & Zhang, H. Calcium-sensing receptor (CaSR)-mediated anti-inflammatory effects of L-amino acids in intestinal epithelial cells. J. Agric. Food Chem. 63, 9987–9995 (2015).
Anbazhagan, A. N. et al. A novel anti-inflammatory role of GPR120 in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 310, C612–C621 (2016).
Kolaczkowska, E. & Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 (2013).
Sadik, C. D., Kim, N. D. & Luster, A. D. Neutrophils cascading their way to inflammation. Trends Immunol. 32, 452–460 (2011).
Maslowski, K. M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009).
Sina, C. et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J. Immunol. 183, 7514–7522 (2009).
Fachi, J. L. et al. Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J. Exp. Med. 217, 1–18 (2020).
Di Paola, R. et al. Formyl peptide receptor 1 signalling promotes experimental colitis in mice. Pharmacol. Res. 141, 591–601 (2019).
Lu, H. C. & Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 79, 516–525 (2016).
Engel, M. A. et al. Mice lacking cannabinoid CB1-, CB2-receptors or both receptors show increased susceptibility to trinitrobenzene sulfonic acid (TNBS)-induced colitis. J. Physiol. Pharmacol. 61, 89–97 (2010).
Maresz, K. et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat. Med. 13, 492–497 (2007).
Wen, J., Ribeiro, R., Tanaka, M. & Zhang, Y. Activation of CB2 receptor is required for the therapeutic effect of ABHD6 inhibition in experimental autoimmune encephalomyelitis. Neuropharmacology 99, 196–209 (2015).
Kapellos, T. S. et al. Cannabinoid receptor 2 deficiency exacerbates inflammation and neutrophil recruitment. FASEB J. 33, 6154–6167 (2019).
Sanders, T. J., Yrlid, U. & Maloy, K. J. Intestinal mononuclear phagocytes in health and disease. Microbiol. Spectr. 5, 1–13 (2017).
Haskó, G. & Cronstein, B. N. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 25, 33–39 (2004).
Linden, J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 41, 775–787 (2001).
Jacobson, K. A. & Gao, Z. G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 5, 247–264 (2006).
Csóka, B. et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 26, 376–386 (2012).
Wilson, J. M. et al. The A2B adenosine receptor promotes Th17 differentiation via stimulation of dendritic cell IL-6. J. Immunol. 186, 6746–6752 (2011).
Biagioli, M. et al. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J. Immunol. 199, 718–733 (2017).
Li, J. et al. Niacin ameliorates ulcerative colitis via prostaglandin D(2)-mediated D prostanoid receptor 1 activation. EMBO Mol. Med. 9, 571–588 (2017).
Ohue-Kitano, R. et al. α-Linolenic acid-derived metabolites from gut lactic acid bacteria induce differentiation of anti-inflammatory M2 macrophages through G protein-coupled receptor 40. FASEB J. 32, 304–318 (2018).
Blad, C. C., Tang, C. & Offermanns, S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat. Rev. Drug Discov. 11, 603–619 (2012).
Ganapathy, V., Thangaraju, M., Prasad, P. D., Martin, P. M. & Singh, N. Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr. Opin. Pharmacol. 13, 869–874 (2013).
Singh, N. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40, 128–139 (2014).
Bhatt, B. et al. Gpr109a limits microbiota-induced IL-23 production to constrain ILC3-mediated colonic inflammation. J. Immunol. 200, 2905–2914 (2018).
Ranganathan, P. et al. GPR81, a cell-surface receptor for lactate, regulates intestinal homeostasis and protects mice from experimental colitis. J. Immunol. 200, 1781–1789 (2018).
Acharya, N. et al. Endocannabinoid system acts as a regulator of immune homeostasis in the gut. Proc. Natl Acad. Sci. USA 114, 5005–5010 (2017).
Niess, J. H. et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258 (2005).
Chieppa, M., Rescigno, M., Huang, A. Y. & Germain, R. N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 203, 2841–2852 (2006).
Morita, N. et al. GPR31-dependent dendrite protrusion of intestinal CX3CR1(+) cells by bacterial metabolites. Nature 566, 110–114 (2019).
Nijhuis, L. E. et al. Adrenergic β2 receptor activation stimulates anti-inflammatory properties of dendritic cells in vitro. PLoS One 9, e85086 (2014).
Ağaç, D., Estrada, L. D., Maples, R., Hooper, L. V. & Farrar, J. D. The β2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain. Behav. Immun. 74, 176–185 (2018).
Matheis, F. et al. Adrenergic signaling in muscularis macrophages limits infection-induced neuronal loss. Cell 180, 64–78.e16 (2020).
McLean, L. P. et al. Type 3 muscarinic receptors contribute to clearance of citrobacter rodentium. Inflamm. Bowel Dis. 21, 1860–1871 (2015).
Oka, S. et al. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: a possible natural ligand for GPR55. J. Biochem. 145, 13–20 (2009).
Ross, R. A. The enigmatic pharmacology of GPR55. Trends Pharmacol. Sci. 30, 156–163 (2009).
Kargl, J. et al. A selective antagonist reveals a potential role of G protein-coupled receptor 55 in platelet and endothelial cell function. J. Pharmacol. Exp. Ther. 346, 54–66 (2013).
Stančić, A. et al. The GPR55 antagonist CID16020046 protects against intestinal inflammation. Neurogastroenterol. Motil. 27, 1432–1445 (2015).
Zhou, J. et al. BLT1 in dendritic cells promotes Th1/Th17 differentiation and its deficiency ameliorates TNBS-induced colitis. Cell. Mol. Immunol. 15, 1047–1056 (2018).
Wu, W. et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 10, 946–956 (2017).
Sonnenberg, G. F. & Artis, D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37, 601–610 (2012).
Bostick, J. W. & Zhou, L. Innate lymphoid cells in intestinal immunity and inflammation. Cell. Mol. Life Sci. 73, 237–252 (2016).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Molofsky, A. B., Savage, A. K. & Locksley, R. M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 42, 1005–1019 (2015).
Cardoso, V. et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281 (2017).
Klose, C. S. N. et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 (2017).
Wallrapp, A. et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 (2017).
Wallrapp, A. et al. Calcitonin gene-related peptide negatively regulates alarmin-driven Type 2 innate lymphoid cell responses. Immunity 51, 709–723.e706 (2019).
Xu, H. et al. Transcriptional atlas of intestinal immune cells reveals that neuropeptide α-CGRP modulates group 2 innate lymphoid cell responses. Immunity 51, 696–708.e699 (2019).
Moriyama, S. et al. β(2)-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061 (2018).
Xue, L. et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 133, 1184–1194 (2014).
McGinty, J. W. et al. Tuft-cell-derived leukotrienes drive rapid anti-helminth immunity in the small intestine but are dispensable for anti-protist immunity. Immunity 52, 528–541.e527 (2020).
Hepworth, M. R. et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113–117 (2013).
Hepworth, M. R. et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 348, 1031–1035 (2015).
Satoh-Takayama, N. et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970 (2008).
Emgård, J. et al. Oxysterol sensing through the receptor GPR183 promotes the lymphoid-tissue-inducing function of innate lymphoid cells and colonic inflammation. Immunity 48, 120–132.e128 (2018).
Chu, C. et al. Anti-microbial functions of group 3 innate lymphoid cells in gut-associated lymphoid tissues are regulated by g-protein-coupled receptor 183. Cell Rep. 23, 3750–3758 (2018).
Chun, E. et al. Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity 51, 871–884.e876 (2019).
Wang, X. et al. GPR34-mediated sensing of lysophosphatidylserine released by apoptotic neutrophils activates type 3 innate lymphoid cells to mediate tissue repair. Immunity 54, 1123–1136.e1128 (2021).
Kunkel, E. J. et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: Epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J. Exp. Med. 192, 761–768 (2000).
Siewert, C. et al. Induction of organ-selective CD4+ regulatory T cell homing. Eur. J. Immunol. 37, 978–989 (2007).
Lee, J. H., Kang, S. G. & Kim, C. H. FoxP3+ T cells undergo conventional first switch to lymphoid tissue homing receptors in thymus but accelerated second switch to nonlymphoid tissue homing receptors in secondary lymphoid tissues. J. Immunol. 178, 301–311 (2007).
Kim, S. V. et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 340, 1456–1459 (2013).
Nguyen, L. P. et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat. Immunol. 16, 207–213 (2015).
Xiong, L. et al. Ahr-Foxp3-RORγt axis controls gut homing of CD4(+) T cells by regulating GPR15. Sci. Immunol. 5, 1–14 (2020).
Sumida, H. et al. GPR55 regulates intraepithelial lymphocyte migration dynamics and susceptibility to intestinal damage. Sci. Immunol. 2, 1–15 (2017).
Proia, R. L. & Hla, T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J. Clin. Invest. 125, 1379–1387 (2015).
Cartier, A. & Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 366, (2019).
Mandala, S. et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296, 346–349 (2002).
Schwab, S. R. & Cyster, J. G. Finding a way out: lymphocyte egress from lymphoid organs. Nat. Immunol. 8, 1295–1301 (2007).
Gräler, M. H. & Goetzl, E. J. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 18, 551–553 (2004).
Barnes, M. J. et al. The lysophosphatidylserine receptor GPR174 constrains regulatory T cell development and function. J. Exp. Med. 212, 1011–1020 (2015).
Leinwand, K. L. et al. Cannabinoid receptor-2 ameliorates inflammation in murine model of crohn’s disease. J. Crohn’s colitis 11, 1369–1380 (2017).
Gentili, M. et al. Selective CB2 inverse agonist JTE907 drives T cell differentiation towards a Treg cell phenotype and ameliorates inflammation in a mouse model of inflammatory bowel disease. Pharmacol. Res. 141, 21–31 (2019).
Kulkarni, N. et al. CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. J. Autoimmun. 88, 121–130 (2018).
He, B. et al. Resetting microbiota by Lactobacillus reuteri inhibits T reg deficiency-induced autoimmunity via adenosine A2A receptors. J. Exp. Med. 214, 107–123 (2017).
Sun, M. et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 9, 3555 (2018).
Yang, W. et al. GPR120 inhibits colitis through regulation of CD4(+) T cell interleukin 10 production. Gastroenterology 162, 150–165 (2021).
Pabst, O. et al. Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. J. Exp. Med. 199, 411–416 (2004).
Macpherson, A. J., McCoy, K. D., Johansen, F. E. & Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal Immunol. 1, 11–22 (2008).
Lamm, M. E. & Phillips-Quagliata, J. M. Origin and homing of intestinal IgA antibody-secreting cells. J. Exp. Med. 195, F5–F8 (2002).
McDonald, K. G. et al. CC chemokine receptor 6 expression by B lymphocytes is essential for the development of isolated lymphoid follicles. Am. J. Pathol. 170, 1229–1240 (2007).
Buettner, M. & Lochner, M. Development and function of secondary and tertiary lymphoid organs in the small intestine and the colon. Front. Immunol. 7, 342 (2016).
Reboldi, A. et al. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 352, aaf4822 (2016).
Nagatake, T. et al. BLT1 mediates commensal bacteria-dependent innate immune signals to enhance antigen-specific intestinal IgA responses. Mucosal Immunol. 12, 1082–1091 (2019).
Fallingborg, J., Christensen, L. A., Jacobsen, B. A. & Rasmussen, S. N. Very low intraluminal colonic pH in patients with active ulcerative colitis. Dig. Dis. Sci. 38, 1989–1993 (1993).
Sasaki, Y., Hada, R., Nakajima, H., Fukuda, S. & Munakata, A. Improved localizing method of radiopill in measurement of entire gastrointestinal pH profiles: colonic luminal pH in normal subjects and patients with Crohn’s disease. Am. J. Gastroenterol. 92, 114–118 (1997).
Nugent, S. G., Kumar, D., Rampton, D. S. & Evans, D. F. Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 48, 571–577 (2001).
Ludwig, M. G. et al. Proton-sensing G-protein-coupled receptors. Nature 425, 93–98 (2003).
Seuwen, K., Ludwig, M. G. & Wolf, R. M. Receptors for protons or lipid messengers or both? J. Recept. Signal Transduct. Res. 26, 599–610 (2006).
Justus, C. R., Dong, L. & Yang, L. V. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 4, 354 (2013).
Yang, L. V. et al. Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol. Cell. Biol. 27, 1334–1347 (2007).
Mahadevan, M. S. et al. Isolation of a novel G protein-coupled receptor (GPR4) localized to chromosome 19q13.3. Genomics 30, 84–88 (1995).
Dong, L. et al. Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS One 8, e61991 (2013).
Chen, A. et al. Activation of GPR4 by acidosis increases endothelial cell adhesion through the cAMP/Epac pathway. PLoS One 6, e27586 (2011).
Sanderlin, E. J. et al. GPR4 deficiency alleviates intestinal inflammation in a mouse model of acute experimental colitis. Biochim Biophys. Acta Mol. Basis Dis. 1863, 569–584 (2017).
de Vallière, C. et al. G Protein-coupled pH-sensing receptor OGR1 is a regulator of intestinal inflammation. Inflamm. Bowel Dis. 21, 1269–1281 (2015).
Hutter, S. et al. Intestinal activation of pH-sensing receptor OGR1 [GPR68] contributes to fibrogenesis. J. Crohns Colitis 12, 1348–1358 (2018).
Lassen, K. G. et al. Genetic coding variant in GPR65 alters lysosomal ph and links lysosomal dysfunction with colitis risk. Immunity 44, 1392–1405 (2016).
Lin, R. et al. GPR65 promotes intestinal mucosal Th1 and Th17 cell differentiation and gut inflammation through downregulating NUAK2. Clin. Transl. Med. 12, e771 (2022).
Wang, J., Wu, X., Simonavicius, N., Tian, H. & Ling, L. Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J. Biol. Chem. 281, 34457–34464 (2006).
Zhang, Q. et al. GPR84 signaling promotes intestinal mucosal inflammation via enhancing NLRP3 inflammasome activation in macrophages. Acta Pharmacol. Sin., online ahead of print (2021).
Frasch, S. C. et al. G2A signaling dampens colitic inflammation via production of IFN-γ. J. Immunol. 197, 1425–1434 (2016).
Tartakover Matalon, S. et al. Cannabinoid receptor 2 agonist promotes parameters implicated in mucosal healing in patients with inflammatory bowel disease. U. Eur. Gastroenterol. J. 8, 271–283 (2020).
Strisciuglio, C. et al. Cannabinoid receptor 2 functional variant contributes to the risk for pediatric inflammatory bowel disease. J. Clin. Gastroenterol. 52, e37–e43 (2018).
Macias-Ceja, D. C. et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. 12, 178–187 (2019).
Ortiz-Masiá, D. et al. Succinate activates EMT in intestinal epithelial cells through SUCNR1: a novel protagonist in fistula development. Cells 9, 1–17 (2020).
Casado-Bedmar, M., Heil, S. D. S., Myrelid, P., Söderholm, J. D. & Keita, Å. V. Upregulation of intestinal mucosal mast cells expressing VPAC1 in close proximity to vasoactive intestinal polypeptide in inflammatory bowel disease and murine colitis. Neurogastroenterol. Motil. 31, e13503 (2019).
McNeil, B. D. et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519, 237–241 (2015).
Pundir, P. et al. A connective tissue mast-cell-specific receptor detects bacterial quorum-sensing molecules and mediates antibacterial immunity. Cell Host Microbe 26, 114–122.e118 (2019).
Chen, E. et al. Inflamed ulcerative colitis regions associated With MRGPRX2-mediated mast cell degranulation and cell activation modules, defining a new therapeutic target. Gastroenterology 160, 1709–1724 (2021).
Sriram, K. & Insel, P. A. G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 (2018).
Leppert, W. & Woron, J. The role of naloxegol in the management of opioid-induced bowel dysfunction. Ther. Adv. Gastroenterol. 9, 736–746 (2016).
Keating, G. M. Eluxadoline: a review in diarrhoea-predominant irritable bowel syndrome. Drugs 77, 1009–1016 (2017).
Nakase, H. et al. Effect of EP4 agonist (ONO-4819CD) for patients with mild to moderate ulcerative colitis refractory to 5-aminosalicylates: a randomized phase II, placebo-controlled trial. Inflamm. Bowel Dis. 16, 731–733 (2010).
Feagan, B. G. et al. Randomised clinical trial: vercirnon, an oral CCR9 antagonist, vs. placebo as induction therapy in active Crohn’s disease. Aliment. Pharmacol. Ther. 42, 1170–1181 (2015).
Sandborn, W. J. et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N. Engl. J. Med. 374, 1754–1762 (2016).
Sandborn, W. J. et al. Efficacy and safety of etrasimod in a phase 2 randomized trial of patients with ulcerative colitis. Gastroenterology 158, 550–561 (2020).
Sandborn, W. J. et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 385, 1280–1291 (2021).
Acknowledgements
This work was funded by grants from the National Natural Science Foundation of China (91942312, 81630017). Figures were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Z.L. and Y.C. were responsible for the conception, literature review, and revising of the manuscript. Z.F. and R.S. drafted the manuscript and interpreted the results. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Feng, Z., Sun, R., Cong, Y. et al. Critical roles of G protein-coupled receptors in regulating intestinal homeostasis and inflammatory bowel disease. Mucosal Immunol 15, 819–828 (2022). https://doi.org/10.1038/s41385-022-00538-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00538-3
