
Abstract
Crohn’s disease (CD) and ulcerative colitis (UC) are both characterized by chronic inflammation and severe dysfunction of the gastrointestinal tract. These two forms of inflammatory bowel disease (IBD) represent distinct clinical disorders with diverse driving mechanisms; however, this divergence is not reflected in currently approved therapeutics that commonly target general proinflammatory pathways. A compelling need therefore remains to understand factors that differentiate the topology and the distinct clinical manifestations of CD versus UC, in order to develop more effective and specialized therapies. Animal models provide valuable platforms for studying IBD heterogeneity and deciphering disease-specific mechanisms. Both the established and the newly developed ileitis mouse models are characterized by various disease initiating mechanisms and diverse phenotypic outcomes that reflect the complexity of human CD-ileitis. Microbial dysbiosis, destruction of epithelial barrier integrity, immune cell deregulation, as well as the recently described genome instability and stromal cell activation have all been proposed as the triggering factors for the development of ileitis-associated pathology. In this review, we aim to critically evaluate the mechanistic underpinnings of murine models of CD-ileitis, discuss their phenotypic similarities to human disease, and envisage their further exploitation for the development of novel targeted and personalized therapeutics.
Similar content being viewed by others

Introduction
Inflammatory bowel disease (IBD) is a severe chronic inflammatory gastrointestinal (GI) disorder encompassing two clinical subtypes, Crohn’s disease (CD) and ulcerative colitis (UC), which exhibit different pathophysiology, disease location, and clinical manifestations.1,2 CD can affect any part of the GI tract with the majority of patients having ileal involvement, while UC is restricted to the colon.1 CD is characterized by inflammation that penetrates the deeper intestinal layers, the submucosa, and the muscularis propria, as well as by granuloma formation. The presence of skip lesions (patchy inflammatory lesions surrounded by healthy areas) is evident among CD patients contrary to UC patients, where inflammation is seen in a more continuous pattern.2 CD mostly presents with diarrhea, abdominal pain, weight loss, and perianal disease, while over time it can lead to complications such as fistulae, fissures, and bowel obstruction.2 Conversely, chronic inflammation in UC affects primarily the innermost lining layer of the intestinal wall, the lamina propria (LP), often forming sores, termed ulcers.3 The hallmark feature of UC is bloody diarrhea associated with lower abdominal cramping, but it can also present with various symptoms common in CD patients.2 Moreover, both subtypes of IBD may exhibit extraintestinal manifestations involving almost every system, such as the musculoskeletal, the dermatologic, and the metabolic ones.4
During the past few decades, the use of biologic agents, such as anti-tumor necrosis factor (TNF) and anti-integrin blockers, improved dramatically the clinical management of IBD patients.5 Despite these great advances, there are still many IBD patients who either do not respond at all or eventually lose response to the clinically approved drugs.6 It is notable that most of the approved treatments are common for UC and CD, despite the heterogeneity of the two clinical subtypes. This discrepancy is due to the fact that the recently approved biologic agents, as well as the more conventional therapeutic approaches, are targeting the chronic inflammatory mediators that are shared between the two diseases. As the incidence of IBD is rising worldwide placing an increasing economic burden on the health care system and the economy as a whole,7 there is an urgent need for the development of new therapeutic concepts leading to individualized treatments that could more accurately reflect the substantial heterogeneity underlying the IBD clinical manifestations and disease course.
A great number of IBD mouse models has been extensively used in the last few decades for preclinical research and aided the discovery of new therapeutic targets.8 Given the broad variety of inducible and spontaneous colitis models and the limited number of ileitis models, the majority of the studies have been conducted in mouse models with colonic inflammation. The results from these studies are often generalized and, at times, mistakenly attributed to CD-ileitis pathophysiology underestimating the topological and molecular differences of the IBD subtypes. Although it is important to highlight the common basis of IBD, it is equally important to discern the underlying differences of its subtypes to allow a deeper understanding of their distinct pathophysiologies for better patient stratification and development of personalized therapeutic approaches. For example, a unique gene expression profile and a distinct microbial community swift was shown in the ileum of CD patients with either exclusive ileum or colon involvement, which was not identified in UC patients underlining the ileum as a core location for the development of CD.9 In this review we focus on describing the spontaneous ileitis mouse models and discuss how these inform our understanding of CD pathophysiology, as well as their relevance to preclinical and clinical research (summarized in Fig. 1 and Table 1).

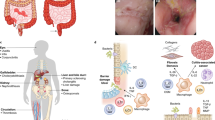
Inflammation in the ileum can be induced by different genetic perturbations reflecting the complexity of the disease triggering mechanisms occurring in CD-ileitis. Inflammation in NCADΔ (a), Xbp1IECKO (b), FaddIECKO (c), Casp8IECKO (d), and Setdb1IECKO(e) is caused by the disruption of intestinal epithelial cell (IEC) homeostasis through diverse mechanisms, including impaired epithelial cell adhesion, IEC death pathway activation and defective ER-stress responses. f ShipKO model shows deregulated hematopoiesis and a granulomatous inflammation, while the infiltration of immune cells in the ileum of TnfΔΑRE mice (g) is mediated by the TNF-dependent activation of intestinal fibroblasts. SAMP1/Yit model (H) is the only congenic ileitis model, which pathology is mediated by genetic defects in the stromal cell compartment. Ecad E-cadherin, ER endoplasmic reticulum, RIPK3 receptor interacting serine/threonine kinase 3, IECs intestinal epithelial cells, ISCs intestinal stem cells, MLKL mixed lineage kinase domain-like, GSDMD gasdermin D, SAMP1 senescence accelerated mouse P1. Created with BioRender.com.
Ileitis mouse models
The congenic SAMP1/Yit(Fc) model
The earliest ileitis mouse model recapitulating some of the CD features in the small intestine (SI) was described in 1995 by Hermiston et al., who generated chimeric mice expressing a dominant negative N-cadherin mutant (NCADΔ); however, no further studies were conducted using these mice.10 Later in 1998, Matsumoto et al. generated the inbred SAMP1/Yit mouse strain through continuous and selective brother–sister matings of SAMP1 mice, a senescence-accelerated prone (SAMP) strain derived from continuous mating of parental AKR/J mice from Jackson.11 The spontaneous intestinal inflammation developed by these mice is segmented throughout the distal SI with the more severe pathology detected in the terminal ileum. The first histological signs of inflammation appear at the age of 20 weeks and involve lymphocytic and myeloid cell infiltration across the whole intestinal wall. As the disease progresses, inflammation mainly penetrates the muscle layer, which becomes hypertrophic, while villous atrophy and crypt distortion are observed. Defective stromal, and not immunological responses have been identified as the initiating etiologic factor of ileitis development in this model.12 A few years after the first description of the SAMP1/Yit mouse model, the SAMP1/YitFc substrain was generated by Rivera-Nieves et al., which exhibited some additional macroscopic and histological features closely resembling the characteristics present in CD patients.13 Besides the accelerated inflammatory phenotype, this new substrain developed perianal lesions, fistula formation, muscular hypertrophy, as well as collagen deposition in the muscularis propria. In an effort to define the genetic causality of ileitis, four susceptibility loci were identified in the SAMP1/YitFc model (chromosomes 9, 6, 8, and X) including genes associated with epithelial changes (Nod2, Pparγ) and immune response (Tnfrsf1a, Il10ra, il18).14 Interestingly, the SAMP1/YitFc mouse model develops periodontitis, an inflammatory disorder of the periodontium with high incidence among CD patients, providing a preclinical platform to study the association between the two disorders.15
The TNF-driven inflammatory ileitis of Tnf ΔΑRE mice
Genetic overexpression of TNF in TnfΔΑRE mice leads to spontaneous, patchy intestinal inflammation, located mainly in the terminal ileum and occasionally in the proximal colon. The generation of these mice, in the late 90s, was a result of a targeted deletion of the AU-rich elements (ARE) located in the 3’untranslated region of Tnf, which resulted in stabilization of TNF mRNA and enhanced translational efficiency.16 Despite some indications from previously reported IBD animal models,17,18 TnfΔΑRE model was the first mouse model that unequivocally established the causal role of TNF in the inflammatory response of Crohn’s-like ileitis; a discovery that paralleled the first approval of a monoclonal antibody targeting TNF for CD patients.19,20 Intestinal epithelial cells (IEC), macrophages, and, to a lesser extent, T cells were shown to be major cellular sources of TNF in the LP of TnfΔΑRE mice, as restricted genetic overexpression of TNF by each of these cell types induced chronic ileitis in mice with different disease onset.16,21 Studies in TnfΔΑRE mice propose that TNF initiates a proinflammatory cascade by stimulating the LP mesenchymal cells to elicit innate and adaptive immune responses.22 As a result, heterozygous TnfΔΑRE mice develop mucosal inflammation at 6–8 weeks of age, and with disease progression the inflammation extents to the submucosa and muscularis propria and becomes chronic. Histopathological characteristics recapitulating human CD include villous blunting, development of tertiary lymphoid structures (TLOs) with active germinal centers in the inflamed LP, and formation of granulomas in the deeper intestinal layers.16 Over the years, many studies in this model have revealed the complexity of the immune responses generated either topically in the intestine or in lymph nodes, and proposed numerous promising therapeutic targets23 (Table 2), some of which were later developed into approved drugs, such as Ustekinumab, vedolizumab, and natalizumab. It is noteworthy that apart from ileitis, TnfΔΑRE mice develop also inflammatory polyarthritis16 and cardiovascular disease,24 two of the most common concomitant diseases among CD patients, rendering these mice a useful tool to study CD-ileitis comorbid conditions.
The ER stress-dependent Xbp1 IECKO model
X-box binding protein 1 (XBP1) is a transcription factor involved in endoplasmic reticulum (ER) stress resolution through regulation of the unfolded protein response (UPR).25,26 Single nucleotide polymorphisms (SNPs) in the XBP1 gene causing defective transcriptional activity have been identified as genetic risk factors for the development of IBD.27,28 IEC-specific ablation of Xbp1 in mice, using the Villin-cre line (Xbp1IECKO), results in increased ER stress and spontaneous enteritis.27 The intestinal pathology, which is not fully penetrant, is not topologically limited in the ileum, but rather extents to whole SI forming skip lesions.27 Epithelial XBP1 deficiency results in absent Paneth cells (PCs) due to programmed cell death and reduced number of goblet cells, which coincide with LP inflammation, crypt distortion, and development of granulomas. This model highlights the importance of a balanced ER stress response in PCs for the maintenance of intestinal homeostasis and reveals a causative link between defective UPR and intestinal inflammation. Moreover, IEC-specific deletion of both Xbp1 and the autophagy mediating gene Atg16l1 (autophagy related 16 like 1) causes an exacerbated phenotype with excessive submucosal and transmural inflammation revealing that concurrent defects in ER stress response and autophagy can act synergistically to induce intestinal inflammation.29
The ShipKO model of granulocytic ileitis
ShipKO model develops granulocytic ileitis. Another reported ileitis model with distinct pathophysiological features is generated by the Src homology 2 domain-containing inositol 5 phosphatase (SHIP) deficiency, due to deregulation of immune cell homeostasis.30 SHIP acts as a negative regulator of phosphatidylinositol-3-kinase (PI3K) by converting the lipid second messenger phosphatidylinositol‐3,4,5‐trisphosphate [PI(3,4,5)P3] to phosphatidylinositol‐3,4‐bisphosphate [PI(3,4)P2] and participates in a variety of regulatory and inflammatory signals in hematopoietic cells.31 Reduced SHIP activity and protein levels have been reported in inflamed ileal tissues and peripheral blood samples from CD patients.32 ShipKO mice exhibit a myeloproliferative disorder and a short lifespan due to eosinophilic crystalline pneumonia. By 4 weeks of age, they develop a robust segmental inflammatory response in the ileum, which extends from the intestinal wall to the mesentery, and progressively leads to villus atrophy, crypt elongation, muscle layer hyperplasia, and fibrosis.30,32,33 Inflammation in this model is mediated by hematopoietic-specific SHIP deficiency, as generation of bone marrow chimeras revealed that Ship deletion in the hematopoietic and not the stromal cellular compartment is required for disease induction.30 Studies attempting to pinpoint the exact cellular type driving disease induction have shown that adoptive transfer of ShipKO splenocytes induced the disease in immunocompetent hosts, whereas purified T and natural killer cell transfer showed no effect.30 In addition, although there was evidence suggesting that macrophages are responsible for disease induction, as SHIP-deficient macrophages had enhanced PI3K signaling and increased IL-1β and IL-18 production, myeloid-specific Ship deletion failed to cause intestinal pathology similar to ShipKO mice.34 Further studies are required to delineate the specific cellular and molecular mechanisms that drive disease in this model.
The IEC-dependent Casp8 IECKO, Fadd IECKO and Setdb1 IECKO models are associated with IEC-specific death pathways
After the generation of the IEC-related NCADΔ and Xbp1IECKO, two independent ileitis models were generated, both involving IEC-specific deletion of critical molecular components required for the regulation of cell death signaling pathways.35 IEC-specific ablation of Fadd (FaddIECKO), Fas-associated via death domain, an adaptor protein that couples caspase-8 (Casp8) to death receptor signaling pathways, led to spontaneous ileitis and colitis.36 Ileitis in this model is characterized by PC loss and massive infiltration of immune cells in the ileal wall due to receptor-interacting serine/threonine Kinase 3 (RIPK3)-dependent epithelial cell death mechanisms. At the same year, Gunther et. al. utilized the Villin-cre line to generate Casp8IECKO mice that lack Casp8, a cysteine caspase that acts as a critical regulator of cell death and inflammatory pathways.37 These mice acquire an inflammatory phenotype across the intestinal wall of the terminal ileum, with the inflammation onset occurring in the LP and progressing in the intestinal muscle layer. The pathology involves villous erosions and epithelial cell RIPK3/mixed lineage kinase domain-like (MLKL)-mediated necroptosis.38 It should be noted that mice with inducible IEC-specific deletion of cFlip (a catalytically inactive caspase-8 homolog that regulates the levels of Casp8 activation) were previously shown to develop spontaneously lethal intestinal inflammation in the colon and SI. The mice exhibited overactivation of Casp8 and high levels of apoptosis in intestinal crypts, revealing that a fine balance between the active and inactive form of Casp8 is required for the maintenance of intestinal homeostasis.39
In addition, a recently described ileitis model is also associated with IEC death by inducing genome instability-related necroptosis through the epithelial-specific deletion of SET Domain Bifurcated 1 (Setdb1) protein (Setdb1IECKO), a histone-lysine N-methyltransferase involved in transcriptional repression and recognition of endogenous retroviruses (ERVs).40,41 Human data support the generation of such a mouse model, as decreased SEDTB1 levels particularly in the intestinal stem cells (ISCs) of IBD patients have been reported,40 while another study described overrepresentation of rare loss of function SETDB1 variants among IBD patients.41 Both of them demonstrated that constitutive or inducible IEC-specific deletion of Setdb1 in mice led to severe inflammation in the colon and terminal ileum in the first days of life or a few days post induction, respectively, inducing fast lethality. Endoscopically, the mice display bowel wall thickening, oedema, and ulcerations while inflammatory cells are expanded in the LP. The phenotype is recapitulated by the ISC-specific deletion of Setdb1, in line with the reduced expression of Setdb1 in the ISCs of IBD patients.40 In addition to inflammation, the epithelium of Setdb1IECKO is destroyed, the villi are atrophic, and the crypts become deformed. Interestingly, mice with insufficient deletion of Setdb1 in epithelial cells develop adenomas in the terminal ileum, similar to the NCADΔ mice, providing an experimental model for studying inflammation-induced tumorigenesis that occur in a subset of CD patients.40
Driving mechanisms in CD patients and ileitis mouse models
The gut microbiota involvement in ileitis initiation and progression
Microbiota and host interactions have been recognized as critical factors participating in the pathogenesis of IBD. Gut microbes have a fundamental role in shaping the immune system of the human host, maintaining immune homeostasis and metabolizing substrates to become accessible for the host.42 Microbiota start colonizing the human gut after birth, and continuously evolve and develop during the first decade of life.43 The variety and the number of microorganisms ranges within the different intestinal parts with the ileum having the highest microbial diversity. The majority of the microbiota is represented by bacterial species and, to a lesser extent, fungi, viruses, and other microorganisms.44 Host responses to microbial signals are of paramount importance in CD pathogenesis, as they could propagate disease progression and inflammation-induced microbial alterations, and the existing animal models have aided to illuminate their contribution.
Dysbiosis, a general term used to describe the impaired microbial balance often associated to disease, has been identified in the majority of IBD patients.45 Various studies utilizing the 16S rDNA sequencing technology have been performed the last years to highlight the microbiota alterations of IBD patients. Despite the utilization of next-generation sequencing technology and its widespread application in microbiome studies, the lack of quantitative data interpretation and the inefficiency of the method to represent accurate bacterial species taxonomy are important limitations impeding the detailed depiction of bacterial diversity.46 Although the existence of an IBD “dysbiotic signature” has not been defined, several phylogenetic differences in the gut microbiota during IBD have been described. Most of the studies report a markedly reduced microbial alpha and beta diversity both in fecal- and mucosal-associated microbes.47,48 Specific taxa with anti-inflammatory functions (such as Bacteroides and Lachnospiraceae) are reduced, while potentially aggressive strains (such as Proteobacteria and adherent Escherichia coli) are expanded among patients.49,50 In addition, postoperative bacterial composition alterations in ileal mucosa-associated microbes were shown to have predictive value for recurrence after surgery.51 Importantly, combinatorial administration of metronidazole and azathioprine, a widely used antibiotic and an immunosuppressive drug respectively, resulted in lower postoperative recurrence rate.52 In agreement with human CD data, most of the reported ileitis models display dysbiotic features. TnfΔΑRE mice develop progressive dysbiosis following inflammation, showing reduced alpha and beta diversity with a significant decline in the Bacteroidetes relative abundance in the inflamed terminal ileum.53 Microbiota samples from the inflamed ileum of ShipKO mice also revealed reduced beta—but not alpha—diversity with most notably the decrease in the Bacteroidales order.54 The same study suggests that these mice display slight dysbiotic phenomena preceding the onset of inflammation. Collectively, there is substantial evidence suggesting that CD-associated microbial alterations caused by the excessive immune response occur in the majority of ileitis models and could further impact disease progression. To assess the contribution of microbiota in the development of ileitis, various studies were performed including animal models raised under germ-free (GF) conditions or treated with broad-spectrum antibiotic cocktails, while their disease progression was monitored. GF TnfΔARE or TnfΔARE mice treated early with antibiotics were completely free of ileitis53,55 and as it was suggested the absence of microbiota colonization hampered TNF upregulation in the ileum of TnfΔARE mice preventing the onset of inflammation.53 This finding highlights the crucial function of microbiota-derived innate signals for the initiation of proinflammatory responses in the ileum. In addition, SAMP1/YitFc and ShipKO models show a partial dependency on microbiota, as antibiotic treatment markedly attenuated ileitis.54,56,57 Conversely, GF Casp8IECKO and FaddIECKO mice develop similar ileal inflammation to control specific-pathogen-free (SPF) mice,36,58 whereas the opposite effect is seen in colonic inflammation. Specifically, microbial depletion in FaddIECKO prevented colitis development, while Casp8IECKO mice exhibited inflammation in the colon only when raised under conventional (CV) conditions, and not under SPF or GF conditions.58 The discrepancy in the microbiota requirement for ileitis progression between Casp8IECKO/ FaddIECKO and the aforementioned models suggests that gut microbes contribute differentially to the distinct disease initiating mechanisms. Thus, epithelial cell death induced in the ileum by Fadd or Casp8 deficiency, accompanied by the subsequent breakdown of epithelial barrier integrity and inflammatory cell influx, does not seem to rely on microbiota-related signals. The topology-specific contribution of microbiota in the colonic, but not ileal, inflammation in the same genetic models can be attributed to the divergent physiology between ileum and colon, including different antimicrobial-related mechanisms, microbiota richness, and immunological responses both during homeostasis and inflammation.59 Human studies have attempted to manipulate the microbiota composition by antibiotic administration in patients suffering from CD showing, however, modest efficacy.60 Potential escape or enrichment of proinflammatory microbial strains could be responsible for the propagation of inflammation in nonresponders to antibiotic treatment. For example, the Debaryomyces hansenii fungi, which is enriched in the ileal and colonic inflamed areas of CD patients, was introduced in murine colon damage models and resulted in impaired mucosal healing through myeloid IFNAR/CCL5 axis.61 This suggests that depleting pathogenic bacterial species by antibiotics could allow the expansion of pathogenic mycobiota strains, such as Debaryomyces hansenii that impede mucosal regeneration, and highlights the role of mycobiota in the pathogenesis and future therapeutic strategies of CD.
Besides antibiotic administration, clinical trials for fecal microbiota transplantation involving CD patients have been initiated without still providing strong evidence for their efficacy.62 An interesting study revealed that transfer of dysbiotic microbiota from TnfΔΑRE mice to GF TnfΔΑRE, but not to GF wild-type littermates, caused ileitis.55 As further shown, the transfer of TnfΔΑRE microbiota increased the expression levels of TNF in the host, which acts as the pathogenic driver in this model. In this regard, it can be concluded that colonization of susceptible genetic hosts with dysbiotic microbiota can lead to the development of intestinal inflammation. However, the aforementioned study lacks a long-term follow-up of the GF TnfΔΑRE phenotype reconstituted with WT microbiota, as it is possible that they could develop a delayed inflammatory response. A similar microbiota-transmissible intestinal inflammation has been previously reported by co-housing colitic Tbet–/–Rag2–/– (TRUC) mice with immunocompromised (Rag2–/–) and immunocompetent (wild-type) mice and subsequent transfer of colitis.63 TRUC mice develop TNF-dependent intestinal inflammation, but the inflammation is topologically restricted in the colon, in sharp contrast to the ileitis induced by TNF overexpression in TNFΔΑRE mice. However, the lack of adaptive immune responses in the TRUC mice, as well as the identification of colitogenic strains including Klebsiella pneumoniae, Proteus mirabilis, Helicobacter Typhlonius strains, suggests that the different immunological background may favor the development of inflammation in distinct locations and reveals that the horizontal transmission of colitis in TRUC mice is a result of pathogenic rather than dysbiotic microbial community transfer.64,65
Overall, it seems that there is an interdependent, dynamic relationship between dysbiosis and inflammation in CD supported by ileitis models. Inflammation promotes dysbiotic phenomena, while microbial imbalance further propagates inflammatory response. Data from ileitis models and human studies suggest that targeting the microbiota axis can contribute, but not fully restore, intestinal homeostasis. Yet, resolving inflammation can reverse dysbiosis, as resolution of inflammation by anti-TNF treatment administration in CD patients reversed the intestinal microbial diversity to approximate healthy controls.66,67 In another study using the double deficient Xbp1/Atg16l1IECKO mice, amelioration of inflammation was achieved by genetic deletion of Ern1. However, the mice developed similar dysbiotic phenomena to diseased control mice, suggesting that resolutions of inflammation do not necessarily affect microbial componsition.68 These findings propose that developing strategies to simultaneously target inflammation and dysbiosis could efficiently facilitate ileitis attenuation. However, additional studies are necessary to identify the underlying mechanisms that lead to disruption of the microbial homeostasis, as well as the relative contributions of the dysbiotic microbiota to initiation and evolution of ileitis under different disease contexts.
Gut microbiota-derived metabolites alterations during ileitis progression
Gut microbes interact mutualistically with the human host through their metabolic products affecting both health and disease. These microbial products can be the result of de novo production by microbes, digestion of dietary fibers, or modification of host-derived metabolites. Several studies have revealed alterations in the microbial metabolome of IBD patients, which is associated with the dysbiotic nature of the disease.69 Using an integrative multi-omic approach, Franzosa et al. studied the microbial and metabolic changes in fecal samples from CD, UC, and healthy subjects, and demonstrated a loss in the metabolite diversity analogous to the decrease in microbial heterogeneity.70 In particular, it was shown that short-chain fatty acids (SCFAs; such as butyrate, propionate, and acetate) and tryptophan metabolites, which possess known immunomodulatory functions, were eliminated in IBD patients providing mechanistic insight into the interaction of the perturbed microbial species with the host. Moreover, the decline in the levels of fecal SCFAs is in line with the reduction of Bacteroidetes and Firmicutes members, the major producers of SCFAs.71 Faecalibacterium prausnitzii, member of the Firmucites plylum and producer of butyrate, has low abundance in the ileum of CD patients and has been associated with increased recurrence after surgery.72 Interestingly, SCFA reduction in the fecal content of CD patients has also been associated with poor response to anti-TNF therapy, suggesting it can be used as a predictive marker to identify patients who are unlikely to achieve remission following anti-TNF therapy.66 Most of the mouse studies have utilized colitis models to elucidate the contribution of the different metabolic products during IBD and limited data are available regarding ileitis models. Focusing on ileitis, TnfΔΑRE mice show an altered metabolic profile, mainly related to lipid metabolism, with increased polyunsaturated fatty acids (such as arachidonic acid) and decreased levels of triglycerides and glycerphospholids (PCaa),73 similar to CD patients.74,75 This altered fatty acid metabolism has not been linked directly to dysbiosis, but rather to host defects, such as excessive immune infiltration and deregulated IEC metabolism.
A large number of clinical trials have been conducted to study the efficiency of probiotics in the treatment of IBD with various results.76 In this line, supplementation of TnfΔΑRE mice with Lactocepin, a Lactobacillus paracasei-derived protease that selectively degrades CXCL10 and other proinflammatory chemokines, attenuated immune infiltration in the ileum.77 Interestingly, Lactobacillus paracasei is one of the bacterial strains included in the VSL#3 probiotic, which exhibited beneficial results in IBD patients.78 Young SAMP1/Yit mice also responded well upon VSL#3 administration, which prevented the onset of inflammation.79 Taken together, an interesting interplay is revealed between microbiota metabolites and the host during CD-ileitis that regulates intestinal inflammatory responses and potentially can be exploited for therapeutic purposes.
Intestinal epithelial barrier dysfunction in CD and ileitis models
The intestinal epithelium is a cell monolayer comprised of specialized epithelial cell types firmly connected by adherens and tight junctions. It forms a dynamic physical and biochemical barrier between the luminal microbes and the host immune system, serving as a crucial regulator of intestinal homeostasis. Mucosal destruction and epithelial cell compositional alterations are major defects observed in IBD patients that correlate with increased risk of relapse. Whether or not the barrier dysfunction is causal to IBD remains an open question. Family studies have revealed that up to 40% of first-degree relatives of CD patients have a higher paracellular permeability in their SI compared to healthy controls.80,81 These data suggest that genetic susceptibility related to epithelial barrier function can act in combination with other genetic or environmental factors to shift the threshold toward the development of intestinal inflammation. Epithelial changes involved in the development of mouse model ileitis and human are discussed below and summarized in Fig. 2.

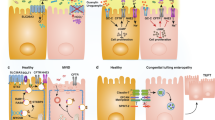
Intestinal epithelial cells (IECs) undergo various functional alterations in ileitis mouse models. a Paneth cell (PC) dysfunction and impaired antimicrobial responses that contribute to the establishment of dysbiosis are induced either by activation of PC necroptosis (reported for Casp8IECKOand FaddIECKO) or differentiation of intestinal stem cells (ISCs) to a defective PC-like phenotype (TnfΔΑRE mice). b Distinct cell death pathways are activated in ileitis models. Genome instability (Setdb1IECKO, FaddIECKO) and TNF signaling (FaddIECKO, Casp8IECKO) induce RIPK3/MLKL-dependent necroptosis and RIPK3/MLKL-mediated pyroptosis in the IECs. c Decreased expression of tight junctions (ShipKO and SAMP1/Yit) leads to the deregulation of paracellular transport through the intestinal epithelium, while loss of adherens junctions in NCADΔ model disrupts IEC adhesion and initiation of inflammation. d Defective autophagy and ER stress responses in the IECs are actively implicated in pathogenesis of Xbp1IECKO. e Immune-associated functions of IECs, including the recognition of microbial signals (reported for the Casp8IECKO model) and the expression of proinflammatory mediators, such as TNF (in the TnfΔΑRE model), contribute to the perpetuation of inflammation. AMPs antimicrobial peptides, TNFR1 tumor necrosis factor 1, RIPK3 receptor interacting serine/threonine kinase 3, ZBP1 Z-DNA binding protein 1, TLRs toll-like receptors, MLKL mixed lineage kinase domain-like, GSDMD Gasdermin D, ER endoplasmic reticulum. Created with BioRender.com.
Epithelial cell death as the causative factor for ileitis development in mouse models
Intestinal barrier integrity is maintained by a synchronous regulation of IEC death and proliferation rates. Deregulation of the balance between these two cellular processes can lead to disruption of intestinal homeostasis predisposing to inflammation and/or driving tumorigenesis. Early studies have described persistent cell death in the epithelium of CD patients82,83,84 implicating this process in the pathogenesis of IBD. Genetic deletion of Casp8 or Fadd, in Casp8IECKO and FaddIECKO models, disrupted IEC homeostasis causing an excessive inflammatory response governed by distinct molecular mechanisms between colon and ileum. Thus, although disruption of RIPK3/MLKL-mediated necroptosis fully rescued the development of colitis in both models and ileitis in Casp8IECKO mice, it did not fully prevent the inflammation in the ileum of FaddIECKO mice.38 Furthermore, it was shown that IEC death and inflammation in the FaddIECKO model required both Casp8-dependent pyroptosis-like cell death and MLKL-dependent necroptosis.38 These results indicate that the induction of different death pathways can be the causative event for the inflammation developed in these models. Yet, the triggering mechanisms of necroptosis in the context of CD remain under investigation. TLR signaling was shown to mediate necroptosis in IECs deficient for Casp8, through TNF-dependent and -independent pathways.85 These results are challenged by the fact that microbial-induced signals are dispensable for the ileitis development in FaddIECKO and Casp8IECKO mice contrary to colitis, which is microbiota-dependent.36,58 Therefore, distinct mechanisms drive necroptosis in the colon and ileum of FaddIECKO and Casp8IECKO models. TNF is a major inducer of apoptotic and necroptotic pathways and could act as a mediator of epithelial cell death during ileitis. Nonetheless, TNF overexpression in the TnfΔARE model is not sufficient to induce epithelial cell death,21 suggesting that compensatory mechanisms in the IECs confer protection from the TNF-induced cell death. In addition, inhibition of TNF signaling in both FaddIECKO and Casp8IECKO models did not prevent the development of ileitis, as it was found to have redundant role in inducing IEC necroptosis. Interestingly, RIPK3 induction either by Z-DNA binding protein 1 (ZBP1) activation, which recognizes z-DNA and z-RNA structures, or by TNFR1 signaling was shown to contribute to IEC necroptosis and inflammation in these models.38 Similarly to the causal implications of Casp8-mediated signaling in murine ileitis, human data from CASP8 and RIPK1 mutation carriers suggest the involvement of these pathways in the development of very early onset (VEO) colitis, yet through IEC death-independent mechanisms.86 Receptor-interacting serine/threonine-protein kinase 1 (RIPK1), a protein with scaffold and kinase properties, is a crucial molecular player in inflammatory and cell death pathways.87 Gene mutations resulting in diminished expression of CASP886 or RIPK187,88 were identified in patients with VEO. In contrast to mice where IEC-RIPK1 deficiency induced necroptosis and mild inflammation in the ileum and colon,88 human RIPK1 mutations did not lead to aberrant IEC death.89 Moreover, TNF treatment was not sufficient to induce epithelial cell death in healthy and CASP8-mutated human organoids.90 Intriguingly, CASP8- and RIPK1-mutated macrophages showed an enhanced response to LPS by increased inflammasome activation and IL-1b production.90,91 Overall, the discrepancy between human IBD and murine models in the absence of Casp8 or RIPK1 suggests the potential existence of compensatory mechanisms in the ileal epithelium of human patients and paves the way for new functional studies implicating the hematopoietic-specific Casp8/RIPK1 axis during IBD.
Besides the highly appreciated FaddIECKO and Casp8IECKO models, a novel concept that introduces genome instability as the driver of necroptosis-induced inflammation was described using the Setdb1IECKO mice.40,41 This model involves necroptosis, as RIPK3 or MLKL IEC-specific deficiency attenuated the inflammation, in contrast to the IEC-specific Casp8 depletion that worsened the disease.40 Genetic depletion of Setdb1 in ISCs, a repressor of ERVs, reactivated ERVs to produce dsRNA and induce viral mimicry. ZBP1 recognized this genomic RNA promoting necroptosis of ISCs and triggering intestinal inflammation.40,41 Taken together, induction of inflammatory cell death pathways, including necroptosis and pyroptosis, by distinct signals has been determined as the driving mechanism for several ileitis models highlighting its importance in the pathophysiology of human disease. Targeting IEC death for the treatment of CD has not yet been attempted in clinical practice, as new approaches to target specific and not indiscriminate cell death are required.
Defective epithelial paracellular and transcellular transport in human and mouse ileitis
Luminal ions and small molecules utilize either diffusion mechanisms to passively cross intestinal epithelium or paracellular transport, a more controlled transport process strictly regulated by the presence of adherens junctions, tight junctions, and desmosomes between the adjacent IECs. Besides paracellular mechanisms, dietary and microbial antigens evade LP through transcellular transport mechanisms as well. Enterocytes and primarily the follicle-associated intestinal epithelium (FAE), a specialized type of epithelial cells, exploit endocytosis to uptake and present luminal antigens and macromolecules (>600 Da) to subepithelial immune cells.92 Increased paracellular permeability and remodeling of tight junctions is reported in patients suffering from CD.93 The NCADΔ model is the earliest generated ileitis model highlighting defective paracellular intestinal barrier permeability as the initiating pathogenic mechanism. However, expression of a dominant negative N-cadherin mutant with disrupted extracellular regions and subsequent loss of enterocytic E-cadherin expression caused impaired epithelial cell migration and apoptosis, in addition to the loss of epithelial cell adhesion.10 Increased paracellular transport is also prominent in the SAMP1/Yit model irrespective of the presence of microbiota and precedes the first signs of inflammation, indicating that it might be the driver mechanism of the inflammatory response.12 In support of this hypothesis, changes in the epithelial tight junctions are detectable early before the onset of disease including altered expression of claudin 2 and occludin. These data support the notion that changes in epithelial cell permeability of small molecules could actively promote chronic inflammation during CD. In addition, ex vivo assays have revealed that CD patients exhibit high transcellular transport through the FAE in the ileum.94,95,96 However, more studies in animal models are needed to assess the exact mechanism of luminal antigen translocation and their contribution in ileitis pathogenesis.
Paneth cell dysfunction as a predominant feature of human CD and ileitis models
PCs are highly specialized granulocytic epithelial cells located at the bottom part of the crypts with important antimicrobial functions, markedly reduced in the ileum of CD patients and ileitis models.97 A plethora of high CD risk-related gene variants affect antimicrobial PC responses, including genes linked to autophagy (ATG16L1), microbial recognition (NOD2), ER stress (XBP1), and WNT signaling (TCF4).98 As already mentioned, Xbp1IECKO mice develop spontaneously inflammation in the SI and they have been used for the study of PCs and their involvement in ileitis. Strikingly, ablation of Xbp1 restrictedly in the PCs recapitulated the phenotype of Xbp1IECKO mice, suggesting that defective ER stress responses in the PCs is the initiating mechanism of inflammation.27 Although XBP1 was not detected in the ISCs, deletion of Xbp1 from the intestinal epithelium induced ISC expansion and increased epithelial cell turnover, defects that are governed by distinct molecular mechanisms.99 The increased number of ISCs was mediated by overactivation of the ER sensor, inositol-requiring enzyme 1a (IRE1a), while the enhanced turnover was mediated by STAT3 activation.99 This suggests that ER stress sensed by the PCs can cause ISC dysfunction and IEC hyperproliferation through independent mechanisms. More mechanistically, the UPR pathway, mediated by the activation of CSNK2B/ATF6/ACSL1 axis, induces a proinflammatory signature (NF-κB activation, TNF production) in the epithelium of Xbp1IECKO, as well as for CD patients, proposing novel druggable targets that can suppress the inflammatory mediators produced by the epithelium.100 Apart from ER stress-induced inflammation, ER stress and autophagy have been identified as highly interconnected cellular pathways, as it was eloquently demonstrated in the Xbp1IECKO mice by induction of autophagy in IECs. Compared to single Xbp1IECKO mice, additional blockade of autophagy by Atg7 or Atg16l1 deletion in the intestinal epithelium of Xbp1IECKO mice caused exacerbated inflammation that extended to the submucosa and muscular layer.29 These data reveal a novel compensatory mechanism between autophagy and the ER response that occurs during CD-ileitis and is mediated by IRE1a inhibition in the IECs.68 It should also be noted that Atg16l1IECKO mice, which exhibit defective autophagy, show only late signs of ileitis (at 35 weeks of age), unlike the Xbp1IECKO that develop established ileitis early on (18 weeks).68
Different models highlight different pathways with respect to PC dysfunction suggesting either that PCs exhibit increased cell death, or that the differentiation process of ISCs is impaired. The Casp8IECKO model is characterized by excessive PC necroptosis, a prominent characteristic in CD patients.37 This is mediated by interferon-lambda (IFN-λ), a type III interferon, upregulated in the epithelium of CD inflamed areas.101 Particularly, overexpression of IFN-λ in mice was sufficient to induce necroptotic pathways in epithelial cells located in the crypts, as well as epithelial cells apoptosis in the upper part of the villi. This effect was dependent on epithelial-specific STAT1 expression and was rescued by administration of JAK/STAT inhibitors. However, it is unclear whether IFN-λ-dependent PC depletion suffices for the development of chronic inflammation, as seen in Casp8IECKO mice, since IFN-λ overexpression caused only minor signs of inflammation in the ileum. Contrary to Casp8IECKO, a study utilizing the TnfΔΑRE model supported the idea that PC number reduction is not due to cell death induction, but rather to epithelial cell differentiation defects.102 In particular, it was shown that besides the previously reported PC depletion, patients and TnfΔARE mice display reduced Lgr5+ ISCs in the crypts correlating to disease progression. The few remaining ISCs showed metabolic dysfunction related to mitochondrial structural and functional alterations, eventually leading to their differentiation toward a non-functional PC-like phenotype with reduced granularity. In support of this hypothesis, pediatric CD patients showed altered IEC dynamics with enrichment of transit amplifying cells, an intermediate cell type between ISCs and differentiated cells, and a marked reduction in terminally differentiated IECs, such as PCs.103 Apart from the intrinsic IEC defects, interferon-gamma (IFN-γ) has been shown to directly induce extrusion and degranulation of PCs ex vivo, in organoids, suggesting that the proinflammatory signals can participate in induction of defective PCs.104 In this regard, TnfΔARE mice with IFN-γ depletion show a full restoration of PC number.53 Collectively, there is compelling evidence that disruption of PC homeostasis is a fundamental mechanism for CD-ileitis pathophysiology with different scenarios regarding their causal involvement in disease development.
The inflammatory milieu of CD and ileitis models
CD as a chronic inflammatory disorder implicates complex immunological responses that are mediated not only by the classical immune cells, but also by stromal cells (epithelial, endothelial, and mesenchymal cells) that actively participate in the inflammatory process. The currently available treatments target the proinflammatory environment through different strategies aiming to induce remission in patients. Recently, a study using cytometry by time of flight (CYTOF) and single-cell RNA sequencing (scRNA-seq) technologies revealed that CD and UC patients with colonic inflammation exhibit substantial differences in the immune cell composition of the periphery and the mucosa. This heterogeneity can be exploited for the development of targeted therapeutics between the two IBD forms, as well as for a more accurate stratification of newly diagnosed patients.105 CD-ileitis mouse models successfully illuminate main aspects of the human disease immunopathology and have shed light on the underlying mechanisms (Fig. 3).

Cross-talk of immune cells with the stromal microenvironment, immune cell activation, and trafficking between the ileum and the proximal lymph nodes orchestrate inflammatory responses in ileitis mouse models. a by epithelial cells or resident T cells and macrophages activate lamina propria (LP) fibroblasts, which secrete pro-inflammatory mediators to attract immune cells. Extravasating myeloid cells and lymphocytes are circulating either in the LP or segregated in granuloma structures and tertiary lymphoid organs (TLOs). Microbiota antigen sampling of dysbiotic microbiota, particularly within the Peyer’s Patches (PP), primes T cells that differentiate into distinct effector subtypes. b Alternatively, antigen presenting cells transport luminal antigens to the proximal lymph nodes, where they prime naïve T cells that get activated and c home the intestine via adhesion cascade interactions with the blood endothelial cells. E-cad E-cadherin, αεβ7 integrin alpha Ε/beta7, α4β7 integrin alpha4/beta7, GZMB granzyme B, TLO tertiary lymphoid organ, α4β1 integrin alpha4/beta1, PSGL1 p-selectin glycoprotein ligand, VCAM-1 vascular cell adhesion protein 1, ICAM-1 intracellular adhesion molecule 1, MadCAM1 mucosal vascular addressin cell adhesion molecule 1, LFA-1 lymphocyte function-associated antigen 1, CCL21 chemokine (C-C motif) ligand 21, CCL19 chemokine (C-C motif) ligand 19, CCL25 chemokine (C-C motif) ligand 25, CCR9 C-C motif chemokine receptor 9, CCR7: C-C motif chemokine receptor 7. Created with BioRender.com.
Myeloid cell functions in CD-ileitis
Defective innate immunity has been directly linked to the pathogenesis of IBD with the identification of several SNPs influencing innate immune cell functions, such as SNPs on NOD2, IRGM, and NLRP3 genes.106 Different concepts explaining how dysfunction in innate immunity can initiate inflammation have been proposed over the years. These include inappropriate recognition of commensal microbiota, impaired microbial clearance or insufficient resolution of inflammation.107,108 In support of these concepts, selective development of granulomas in CD, and not in UC, suggests a hyper-reaction of mononuclear phagocytic cells in response to an inflammatory stimulus and a subsequent inability to eliminate it. Although the presence of granulomas in CD patients is associated with a higher disease severity,109 there is still no substantial evidence for their function. Most of the CD animal models (TnfΔΑRE, Xbp1IECKO, SAMP/Yit, ShipKO) have been reported to develop granulomas in the deeper intestinal layers providing an ideal preclinical platform for the study of this histologic feature. In general, loss of immune tolerance accompanied by an enhanced proinflammatory phenotype of classical innate immune cells is observed in IBD patients, such as macrophages and dendritic cells (DCs).110 A recent study by Martin et al. uncovered through scRNA-seq technology111 the immune cell heterogeneity in CD patients and validated previous studies112 showing that at least two macrophage populations exist in the LP: the resident macrophages and the inflammatory macrophages. The resident macrophages were marked by the expression of the mannose receptor CD206 and lacked a proinflammatory gene signature, whereas the inflammatory macrophages were highly responsive to NF-κB signaling and produced various chemokines (CCL3, CCL20, CXCL10, CXCL2) and cytokines (TNF, IL-6, IL-1b, IL-23) to induce the activation of the adaptive immune response. TNF was profoundly expressed by inflammatory macrophages suggesting that this cell type is among the major TNF sources in the LP. Indeed, specific TNF overexpression by the myeloid cells suffices for the development of inflammation in the ileum.113 The importance of myeloid cells in ileitis was also demonstrated in the ShipKO model, a monocytic/granulocytic infiltrating model, where the ability of myeloid cells to orchestrate an inflammatory reaction was demonstrated without subsequent lymphocytic expansion.30 Depletion of macrophages and anti-IL-1β therapy attenuated the inflammation in the LP suggesting that therapies targeting the macrophages or macrophage mediators, such as IL-1β, can be efficacious in treating ileitis.114 Curiously, unlike murine studies that emphasize on the epithelial-specific Casp8 and RIPK1 functions, increased IL-1β production by macrophages is prominent in IBD patients with Casp8 and RIPK1 mutations, suggesting that further studies in mouse models are required to decipher the associated mechanisms involved in human disease. Pharmacological blocking of another IL-1 family member cytokine, the IL-1α, importantly attenuated ileitis severity in the SAMP1/YitFc model. Notably, the effect of IL-1α was proposed to be mediated by shaping directly or indirectly the intestinal microbiome, which in turn enhanced inflammation.115 The contribution of other innate immune cell types, such as DCs, neutrophils, and ILCs, in the initiation and progression of CD has been primarily studied in colitis models and there is little experimental evidence of their function in CD. Nonetheless, a study described an immunoregulatory subset of DCs expressing CD103 (E-cadherin receptor), which was found to be reduced in the TnfΔΑRE mice.116 Expansion of this subset by in vivo administration of FLT3 ligand induced amelioration of inflammation by activating T regulatory cell (Treg) responses. Although there is an increasing body of evidence suggesting that innate immunity is critical in the pathogenesis of CD-ileitis, further investigation is required for the identification of specific inflammatory or regulatory cell subtypes and cellular mediators with active role in the disease pathology.
The complex adaptive immune responses of human and mouse CD-ileitis
T cells are effector adaptive immune cells expanded locally and systemically in CD patients. CD has been initially considered as a T-helper 1 (Th1)-mediated disease, contrary to UC that has been predominantly characterized by a T-helper 2 (Th2) type response. However, the identification of new T cell subsets [Tregs, T-helper 17 (Th17)] with active roles in CD pathogenesis resulted in a paradigm shift from the binary Th1 or Th2 classification. Several studies using the TnfΔΑRE and SAMP1/Yit mouse models have uncovered the central role of T cells in the initiation and propagation of ileitis, as adoptive transfer of CD4+ T cells from diseased mice to immunocompromised hosts induced inflammation in the ileum.117,118 Nonetheless, adoptive T cell transfer experiments should be interpreted with caution, as the immunodeficient background of the hosts (RagKO, SCID mice) is not relevant to the context of CD.
IL-12, IL-6, TGF-β, and IL-23 cytokines are mainly produced by macrophages and DCs, which act as a connective bridge between the adaptive and innate immune systems to prime and induce Th1 and Th17 responses. Targeting these cytokines with antibody-based approaches has been attempted in different clinical trials. For example, double inhibition of IL-12 and IL-23 using a monoclonal antibody targeting their common subunit (IL-12p40) or single inhibition of IL-23 with antibody against IL-23p19 are effective in causing remission in a subset of both CDs119,120 and UC patients.121 Although it has been reported that genetic deletion of IL-12p40 in the TnfΔARE model induces attenuation of inflammation,113 the mechanisms of IL-12 and IL-23 functions have not yet been elucidated in ileitis mouse models. Studies have also been investigating the role of known effector cytokines produced by CD4+ T cells, such as TNF, IFN-γ, and IL-17. Although IFN-γ promotes the progression of inflammation in the TnfΔΑRE and SAMP1/Yit models,113.122 clinical data regarding the potential use of antibodies against IFN-γ have generated inconsistent results.123 Furthermore, targeting IL-17 was considered as a promising therapeutic strategy; however, clinical studies showed that inhibition of IL-17 worsened inflammation in patients, rather than suppressing it. This can be attributed to the critical role of Th17 cells for the intestinal homeostasis, as they regulate the immune responses against commensals124 and maintain epithelial barrier integrity.125 Although IL-17 has been reported to be upregulated in CD-ileitis mouse models, it has not yet been determined if IL-17 blockade deteriorates ileitis, as in the case of human patients. Interestingly, reduced presence of IL-17A+ T cells is observed in the mucosa of CD compared to UC patients, suggesting that targeting proinflammatory Th17-dependent mechanisms could be a promising therapeutic candidate for UC, but not for CD.105 In agreement with this study, single-cell -omic technologies revealed a reduction of IL-17A+-expressing CD4+ T cells in the LP of CD patients, in contrast to the increased abundance of intraepithelial CD4+ IL-17A+ T cells, suggesting spatially regulated Th17 responses during CD.126 Unlike the UC traditionally characterized as Th2-mediated disease, the previously unappreciated role of Th2 immunity in CD-ileitis was revealed using the SAMP1/Yit model. Specifically, blockade of IL-4 dampened inflammation, while adoptive transfer of IL-4-producing CD4+ cells in immunocompromised mice was sufficient to induce ileitis.122 Enhanced activation of proinflammatory T cell function is accompanied by expansion of Tregs in CD patients127 and mouse models.128,129 Depletion of Tregs by administration of an anti-CD25 treatment in the SAMP1/Yit model enhanced the severity of the disease.129 Tregs were also associated with sex differences observed in ileitis patients, as exogenous estrogen (E2) administration in male mice expanded the Treg population in the mesenteric lymph nodes (mLNs) and attenuated ileitis.130
Despite the mouse studies harnessing CD4+ T cell transfer experiments in immunocompromised hosts to study the effector functions of CD4+ T cells during intestinal inflammation, there is now increasing evidence that the expanded CD8+ effector T cells in the ileal LP of CD patients might also have a functional role during CD.126 Experimental data using the TnfΔΑRE model have shown a novel role of CD8+ T lymphocytes for ileitis progression. In particular, functional impairment of CD8+ T cell responses by beta-2 microglubulin depletion ameliorated the intestinal inflammation, in contrast to CD4+ T cell deficiency that caused a slight exacerbation of the pathology.113 It is noteworthy to mention that these results do not exclude the importance of CD4+ T cell responses in this model, as Treg (CD4+) deficiency might have compensated the loss of proinflammatory CD4+ T cell responses. In line with this, the effector TCRαβ+CD8αβ+ subset of intraepithelial lymphocytes (IELs) profoundly produces IFN-γ, TNF, and the cytotoxic mediator granzyme B, and it exhibits high relative abundance to IELs at later stages of the disease.131 Interestingly, high ER stress was identified as the triggering mechanism of granzyme B expression in CD8αβ+, but not in CD8αα+, regulatory IELs,132 which are markedly reduced early in the development of ileitis.133 Strikingly, scRNA-seq and CYTOF analysis validated that intraepithelial effector and resident CD8+ subtypes were decreased in the ileum of CD patients. In contrast to their disease-promoting functions, a regulatory subset of CD8+ T cells (CD44lowCD103high) expressing TGF-β was identified in spleen and mLNs of TnfΔΑRE, as able to suppress CD4+ T cell effector functions.134 A distinct gene signature present in CD8+, but not in CD4+, T cells was also found predictive of a high relapse risk in CD patients, suggesting that indeed these cells are likely to be highly involved in disease-promoting mechanisms.135
Another immune cell population massively expanded in the ileum of CD patients and most of the CD mouse models is the B cells, which either infiltrate the inflamed areas or segregate in the gut-associated lymphoid tissue, including the Peyer patches and TLOs. B cells serve as key mediators of humoral immunity by producing antibodies, presenting antigens to T cells and expressing proinflammatory cytokines. TLOs are organized lymphoid structures with germinal centers, comprised mainly by B cells and to a lesser extent by CD4+ T cells and DCs, massively expanding in the inflamed ileum and mesenterium of CD patients and ileitis mouse models.13,136,137,138 Although their increase correlates to the progression of chronic inflammation and the development of dysbiosis, their direct role in disease has been poorly characterized. Besides TLOs, mouse models exhibit a marked increase in the number of B cells in the LP and mLNs. Adoptive co-transfer of B cells together with CD4+ T cells from SAMP1/Yit mLNs to SCID mice enhanced the ability of CD4+ T cells to induce ileitis and exacerbated the disease.139 Pharmacological inhibition of B cells has been tested only in UC, where it has been proved insufficient to cause remission, suggesting that B cells do not play a dominant role in the pathogenesis of IBD.140 In agreement, genetic depletion of B cells in the TnfΔARE model did not affect the progression of ileitis.113
Overall, it is evident that adaptive immunity is required in the majority of CD-ileitis models for the full perpetuation of intestinal inflammation, emphasizing its significance for human disease. However, due to the complex nature of adaptive immune responses, there is a compelling need to deconvolute the function of the lymphocytic mosaic during IBD. Recent lines of evidence challenge the immunological dogma and propose that the functionality of intestinal T cells does not rely on the cytokine repertoire they are producing during infection, but rather to the specific antigen-induced responses imprinted on their transcriptomic profile.141 It remains to examine if this new concept applies for chronic inflammatory diseases, such as IBD, providing new T cell-specific therapeutic targets. Such approaches will lead to further delineation of how lymphocytes drive inflammation and pave the way for a new personalized medical care for IBD patients.
Gut immune cell homing through the endothelium during inflammatory ileitis
Immune cells establish a dynamic interaction with the endothelium that facilitates their efficient trafficking between the gut and the lymphoid tissues to propagate or regulate inflammation. Recently, an activated subset of endothelial cells, marked by the expression of atypical chemokine receptor 1 (ACKR1), was identified by scRNA-seq in ileal samples of CD patients.111 ACKR1 binds to various chemokines, such as CCL2 and CLL5, to mediate chemokine transcytosis and facilitate monocyte trafficking and extravasation through the endothelium.142 In particular, heterodimeric integrins containing β7 integrin mediate efficient trafficking and homing of lymphocytes in the intestine via the binding of β7 to either α4 or αΕ (CD103) and subsequently forming the α4β7 and αΕβ7, respectively. α4β7 mediates the trafficking of lymphocytes through blood circulation into the intestinal mucosa through its interaction with mucosal addressin cell adhesion molecule-1 (MAdCAM-1), expressed predominantly by the endothelium, while the upregulation of αΕβ7 on the surface of lymphocytes guides and retains them close to the intestinal epithelium through E-cadherin binding. In addition, Vedolizumab, a selective humanized antibody for α4β7, has been approved for achieving remission to CD patients.143 Interestingly, Vedolizumab administration in IBD patients with concomitant HIV infection attenuated intestinal CD4+ T cell infiltration, as well as TLO numbers and B-cell follicle numbers in the terminal, suggesting that β7 blockade may also act by preventing lymphoid aggregate formation and B-cell responses.144 TnfΔARE mice with β7 deficiency are rescued from ileitis supporting the hypothesis that blockade of lymphocytic migration to the gut can efficiently suppress the initiation of inflammation.131 In contrast, antibody-mediated blockade of MAdCAM/α4β7 pathway or L-selectin was not sufficient to attenuate ileitis alone, but only as a combinatorial treatment in the SAMP1/YitFc model.145 This suggests that not only α4β7 but other integrins as well can efficiently facilitate the gut homing of lymphocytes. Indeed, α4β1 integrin expression is enhanced in patients treated with Vedolizumab suggesting that redundant functions characterize the molecules involved in the homing of gut T cells.146 A redundancy in the adhesion pathways that T cells use to access inflamed areas is further supported by the fact that double blockade of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), but not either one alone, prevented ileitis onset in the SAMP1/Yit adoptive T cell transfer model.147 Furthermore, blockade of P-selectin glycoprotein ligand 1 (PSGL1), which was found to be upregulated in the endothelial cells of SAMP1/Yit mice, ameliorated the inflammation revealing a new endothelial-related adhesive molecules that could be involved in CD.148
An important chemokine secreted by the endothelium that mediates lymphocyte gut tropism, analogous to MAdCAM-1 expression, is the CCL25 that interacts with CCR9+ migrating lymphocytes. CCR9+ circulating T cells and CCL25 expression in the SI are elevated in CD patients, while the efficiency of anti-CCR9 treatment in CD is still under investigation.149,150 To further explore whether targeting of the CCL25/CCR9 axis could be used as a therapeutic intervention, studies in animal models have been performed; however, contradictory results have been generated. Apostolaki et al. showed that this chemokine-receptor pair is dispensable for the ileitis progression in the TnfΔARE model by genetic depletion of Ccr9 and antibody-based CCL25 blocking.131 In contrast, two follow-up studies reported either exacerbated inflammation by genetic deficiency of Ccr9151 or amelioration of the disease by administration of a specific CCR9 antagonist in the TnfΔΑRE mice.152 It is possible to speculate that differences in the strategy of CCR9 deletion and/or potential changes in the microbiota composition might have led to these conflicting results. Furthermore, in the SAMP1/Yit model, the CCL25/CCR9 axis was shown to be essential only in the early stages for inflammation.153 Moreover, CCR7 1 has been considered as an essential chemokine/receptor pair for the trafficking of T cells to the LNs and TLOs through its interaction with CCL19/CCL2. Genetic or pharmacological inhibition of Ccr7 in the TnfΔARE mice inhibited immune cell egress toward draining LNs, retained them in the LP and aggravated inflammation.137,154
Collectively, these results reveal that targeting the trafficking of lymphocytes through the endothelium can inhibit the ingress or egress of immune cells into or from the secondary lymphoid organs, and can efficiently suppress chronic intestinal inflammation. Research efforts should focus on pinpointing the specific interactions, as well as defining the redundant ones between immune and endothelial cells that affect the gut homing of inflammatory cells.
Intestinal mesenchymal cells as key cellular mediators for ileitis development
Intestinal mesenchymal cells (IMCs), including mainly fibroblasts, myofibroblasts, pericytes, and smooth muscle cells, have gained great interest the last years, as they serve important functions for villous formation, epithelial cell homeostasis and immune cell regulation.155 Α recent study uncovered the previously unexplored mesenchymal cell spatial diversity along the crypt-villous axis in the murine SI and defined a functional zonation of these cells inside the LP.156 In human CD, scRNA-seq studies revealed that fibroblast subsets have distinct gene expression signature during disease that regulate immune response, epithelial cell differentiation, and apoptosis.103,157 Expression of proinfammatory cytokines (such as IL-6), chemokines (including CCL2 and CCL7 for monocyte recruitment), and neutrophil chemo-attractants (such as CXCL1, CXCL2, CXCL8, and CXCL5) was observed in the activated fibroblast cluster from CD patients,103,157 validating previously reported results.158 The close proximity of PODOPLANIN+ activated fibroblasts with a subset of inflammatory macrophages in ileal tissues of CD patients suggests potential significant interactions for disease pathogenesis, which involves proinflammatory signals expressed and/or received by the fibroblasts.111 Intriguingly, the intestinal fibroblasts have been identified as the TNF responder cell types in the TnfΔARE mice, capable of initiating ileitis in this model. Fibroblast-specific genetic restriction of TNFR1 expression suffices for the development of inflammation highlighting the critical and causal role of fibroblasts during intestinal inflammation.22 CD patients with NOD2 mutations were shown to have an increased activation of fibroblasts and macrophages, as well as upregulated expression of profibrotic (IL-11, Wt1) and proinflammatory (IL-6, Oncostatin M; OSM) mediators regulated by STAT3. This NOD2 mutation-driven gene signature was induced post-treatment in patients with anti-TNF refractory CD, suggesting that mutations in NOD2 can instruct the activation of macrophages and fibroblasts in TNF-nonresponders. Targeting the gp130 (required for IL-6, IL-11 and OSM signaling) could prevent the dual stromal-myeloid activation and offer a therapeutic option for patients with primary or secondary non-response to anti-TNF therapy.159 Although ileitis in SAMP1/YitFc mice is TNF-dependent,160 they offer an appropriate tool to further study the mechanism of NOD2 loss of functions mutations as this model is irresponsive to NOD2-mediated signaling.161
In addition to the altered regulation of immune response, a remodeling of the transcriptional profile of specific fibroblast clusters was observed in paediatric CD patients exhibiting differential expression of morphogen molecules (WNTs and BMPs).103 This indicates that the fibroblast-epithelial cell crosstalk is impaired during CD and epithelial barrier dysfunction in the ileum could be a result of defective morphogenic signal gradient derived from fibroblasts. In addition, pro-apoptotic signals for epithelial cells (TNFSF10-TNFRSF10B) can be derived from activated fibroblasts suggesting that IMCs could also modulate IEC cell death.103 Taking advantage of the various CD-ileitis animal models, further studies may provide mechanistic insights on how IMCs participate in CD and bring forward strategies to target this stromal cell population in the inflammatory intestinal microenvironment.
Another severe and unexplored aspect of CD is the development of fibrotic strictures during or after the onset of inflammation. Fibrosis is characterized by the expansion of myofibroblasts and extensive ECM deposition across the bowel wall. Currently, there is no available treatment for CD patients suffering from fibrotic manifestations, as the mechanism of how progressive inflammation leads to intestinal fibrosis remains elusive. The limited number of IBD mouse models that develop fibrosis reflecting human disease has contributed to the understanding of the driving mechanisms of intestinal fibrosis, albeit modestly. SAMP1/YitFc model is the first described ileitis model displaying fibrotic strictures in the hypertrophic muscularis propria and perianal fistula formation.13 However, due to the difficulties in breeding and maintaining these mice, no further studies have been conducted for the elucidation of the underlying fibrotic mechanisms in this model. ShipKO mice exhibit fibrotic features, such as expansion of the collagen layer in the submucosa and between the circular and longitudinal muscular layer, which are partially restored by early blockade of arginase activity.33 Collectively, the elucidation of the fibrotic mechanisms in CD still remains a challenge, as there is a great need for better mouse models reflecting this pathogenic aspect.
Conclusion
CD is governed by complex, yet distinguishable from UC, pathophysiological mechanisms. There is an urgent need to define the factors that differentiate the topology, the clinical features, and the progression of the two clinical subtypes. This can up to a certain point be addressed by comparing the spontaneous ileitis and colitis models in light of their triggering mechanisms, molecular mediators, and cellular interactions. Distinct initiating mechanisms between CD-ileitis and colitis models suggest that, although chronic inflammation is shared between the two diseases, the genetic and environmental insults that drive the disease are different. Activation of epithelial cell death pathways involving various molecular cell death executors, unresolved ER stress, and deregulation of immune cell homeostasis elicit inflammation in the majority of CD-ileitis animal models without colonic manifestation, indicating that CD and UC are driven by distinct etiopathogenic factors. Strikingly, animal models such as the FaddIECKO, Casp8IECKO, and the Setdb1IECKO mice have both ileal and colonic inflammation suggesting that common causal mechanisms may be shared between the two subtypes. Although ileitis and colitis in these models are progressing in very different ways, this does not exclude the possibility of a common cause and a subsequent divergence during disease development. This could be attributed to the different pathophysiology of the ileum and colon, as increased microbiota diversity localizes in the ileum and different epithelial cell secretory lineages, such as the PCs, are unique for the SI. Future studies on these models can delineate how the microenvironment in each topological intestinal part can contribute to the heterogeneity detected in UC and CD patients. In addition to the substantial differences between CD and UC, there is also great heterogeneity among CD patients that leads to individual differences in drug responses. Ileitis models harbor specific perturbations in genes encoding crucial regulators of intestinal homeostasis that drive the development of ileal inflammation through independent mechanisms. This variety of triggering factors in mouse models can potentially reflect different subtypes of CD patients; patients with dominant characteristics of epithelial cell defects (IEC or ISC death, ER stress in the PCs, increased epithelial permeability) and patients with a primary inflammatory signature (involving both immune and stromal cells). However, challenging aspects of the disease, such as the fibrotic features, still remain unresolved. In addition, mouse models have not yet managed to recapitulate other topologically distinct subtypes of CD, which involve inflammation in the upper GI tract (stomach, esophagus, mouth). Future experimental work in already established or new animal models will hopefully provide new insights into the complex nature of IBD and reveal the distinct driving mechanisms between colitis and ileitis ultimately leading to new strategies toward more targeted therapeutics.
References
Lamb, C. A. et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 68, s1–s106 (2019).
Odze, R. Diagnostic problems and advances in inflammatory bowel disease. Mod. Pathol. 16, 347–358 (2003).
Hendrickson, B. A., Gokhale, R. & Cho, J. H. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin. Microbiol. Rev. 15, 79–94 (2002).
Levine, J. S. & Burakoff, R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol. Hepatol. 7, 235–241 (2011).
Neurath, M. F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 14, 269–278 (2017).
Gisbert, J. P. & Chaparro, M. Predictors of primary response to biologic treatment [anti-TNF, vedolizumab, and ustekinumab] in patients with inflammatory bowel disease: from basic science to clinical practice. J. Crohns Colitis 14, 694–709 (2020).
Kaplan, G. G. The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 12, 720–727 (2015).
Neurath, M. F. Animal models of inflammatory bowel diseases: illuminating the pathogenesis of colitis, ileitis and cancer. Dig. Dis. 30, 91–94 (2012).
Haberman, Y. et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 124, 3617–3633 (2014).
Hermiston, M. L. & Gordon, J. I. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science 270, 1203–1207 (1995).
Matsumoto, S. et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut 43, 71–78 (1998).
Olson, T. S. et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J. Exp. Med. 203, 541–552 (2006).
Rivera-nieves, J. et al. Emergence of perianal fistulizing disease in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis. Gastroenterology 124, 972–982 (2003).
Kozaiwa, K. et al. Identification of a quantitative trait locus for ileitis in a spontaneous mouse model of crohn’s disease: SAMP1/YitFc. Gastroenterology 125, 477–490 (2003).
Pietropaoli, D. et al. Occurrence of spontaneous periodontal disease in the SAMP1/ YitFc murine model of Crohn disease. J. Periodontol. 85, 1799–1805 (2014).
Kontoyiannis, D., Pasparakis, M., Pizarro, T. T., Cominelli, F. & Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU- rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398 (1999).
Powrie, F. et al. Inhibition of Thl responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562 (1994).
Neurath, M. F. et al. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur. J. Immunol. 27, 1743–1750 (1997).
Targan, S. R. et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor α for Crohn’s Disease. N. Engl. J. Med. 337, 1029–35 (1997).
Present, D. H. et al. Infliximab for the treatment of fistulas in patients with Crohn’s disease. N. Engl. J. Med. 340, 1398–1405 (1999).
Roulis, M., Armaka, M., Manoloukos, M., Apostolaki, M. & Kollias, G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc. Natl Acad. Sci. U.S.A. 108, 5396–5401 (2011).
Armaka, M. et al. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 205, 331–337 (2008).
Cominelli, F., Arseneau, K. O., Rodriguez-Palacios, A. & Pizarro, T. T. Uncovering pathogenic mechanisms of inflammatory bowel disease using mouse models of Crohn’s disease–like ileitis: what is the right model? CMGH 4, 19–32 (2017).
Sakkou, M. et al. Mesenchymal TNFR2 promotes the development of polyarthritis and comorbid heart valve stenosis. JCI Insight 3, (2018).
Lee, A.-H., Iwakoshi, N. N. & Glimcher, L. H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 (2003).
Calfon, M. et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002).
Kaser, A. et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756 (2008).
Hampe, J. et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211 (2007).
Adolph, T. E. et al. Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276 (2013).
Kerr, W. G., Park, M. Y., Maubert, M. & Engelman, R. W. SHIP deficiency causes Crohn’s disease-like ileitis. Gut 60, 177–188 (2011).
Hibbs, M. L., Raftery, A. L. & Tsantikos, E. Regulation of hematopoietic cell signaling by SHIP-1 inositol phosphatase: growth factors and beyond. Growth Factors 36, 213–231 (2018).
Ngoh, E. N. et al. Activity of SHIP, which prevents expression of interleukin 1β, is reduced in patients with Crohn’s disease. Gastroenterology 150, 465–476 (2016).
McLarren, K. W. et al. SHIP-deficient mice develop spontaneous intestinal inflammation and arginase-dependent fibrosis. Am. J. Pathol. 179, 180–188 (2011).
Maxwell, M. J. et al. SHIP-1 deficiency in the myeloid compartment is insufficient to induce myeloid expansion or chronic inflammation. Genes Immun. 15, 233–240 (2014).
Muzio, M. et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85, 817–827 (1996).
Welz, P. S. et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334 (2011).
Günther, C. et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011).
Schwarzer, R., Jiao, H., Wachsmuth, L., Tresch, A. & Pasparakis, M. FADD and caspase-8 regulate gut homeostasis and inflammation by controlling MLKL- and GSDMD-mediated death of intestinal epithelial cells. Immunity 52, 978–993.e6 (2020).
Wittkopf, N. et al. Cellular FLICE-like inhibitory protein secures intestinal epithelial cell survival and immune homeostasis by regulating caspase-8. Gastroenterology 145, 1369–1379 (2013).
Wang, R. et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 580, 386–390 (2020).
Juznić, L. et al. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut 70, 485–498 (2021).
Ni, J., Wu, G. D., Albenberg, L. & Tomov, V. T. Gut microbiota and IBD: causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 14, 573–584 (2017).
Derrien, M., Alvarez, A. S. & de Vos, W. M. The gut microbiota in the first decade of life. Trends Microbiol. 27, 997–1010 (2019).
Sartor, R. B. & Wu, G. D. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 152, 327–339.e4 (2017).
Tamboli, C. P., Neut, C., Desreumaux, P. & Colombel, J. F. Dysbiosis in inflammatory bowel disease. Gut 53, 1–4 (2004).
Johnson, J. S. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10, 1–11 (2019).
Manichanh, C. et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211 (2006).
Joossens, M. et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637 (2011).
Frank, D. N. et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl Acad. Sci. U.S.A. 104, 13780–13785 (2007).
Frank, D. N. et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 179–184 (2011).
Sokol, H. et al. Prominence of ileal mucosa-associated microbiota to predict postoperative endoscopic recurrence in Crohn’s disease. Gut 69, 462–472 (2020).
D’Haens, G. R. et al. Therapy of metronidazole with azathioprine to prevent postoperative recurrence of Crohn’s disease: a controlled randomized trial. Gastroenterology 135, 1123–1129 (2008).
Roulis, M. et al. Host and microbiota interactions are critical for development of murine Crohn’s-like ileitis. Mucosal Immunol. 9, 787–797 (2016).
Dobranowski, P. A., Tang, C., Sauvé, J. P., Menzies, S. C. & Sly, L. M. Compositional changes to the ileal microbiome precede the onset of spontaneous ileitis in SHIP deficient mice. Gut Microbes 10, 578–598 (2019).
Schaubeck, M. et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 65, 225–237 (2016).
Bamias, G. et al. Commensal bacteria exacerbate intestinal inflammation but are not essential for the development of murine ileitis. J. Immunol. 178, 1809–1818 (2007).
Bamias, G. et al. Down-regulation of intestinal lymphocyte activation and Th1 cytokine production by antibiotic therapy in a murine model of Crohn’s disease. J. Immunol. 169, 5308–5314 (2002).
Stolzer, I. et al. Environmental microbial factors determine the pattern of inflammatory lesions in a murine model of Crohn’s disease–like inflammation. Inflamm. Bowel Dis. 26, 66–79 (2020).
Atreya, R. & Siegmund, B. Location is important: differentiation between ileal and colonic Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 0123456789, (2021).
Feller, M. et al. Long‐term antibiotic treatment for crohn’s disease: systematic review and meta‐analysis of placebo‐controlled trials. Clin. Infect. Dis. 50, 473–480 (2010).
Jain, U. et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science 371, 1154–1159 (2021).
Colman, R. J. & Rubin, D. T. Fecal microbiota transplantation as therapy for inflammatory bowel disease: a systematic review and meta-analysis. J. Crohns Colitis 8, 1569–1581 (2014).
Garrett, W. S. et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45 (2007).
Garrett, W. S. et al. Enterobacteriaceae Act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300 (2010).
Powell, N. et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 37, 674–684 (2012).
Aden, K. et al. Metabolic functions of gut microbes associate with efficacy of tumor necrosis factor antagonists in patients with inflammatory bowel diseases. Gastroenterology 157, 1279–1292.e11 (2019).
Ribaldone, D. G. et al. Adalimumab therapy improves intestinal dysbiosis in Crohn’s disease. J. Clin. Med. 8, 1646 (2019).
Tschurtschenthaler, M. et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease-like ileitis. J. Exp. Med. 214, 401–422 (2017).
Lavelle, A. & Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 17, 223–237 (2020).
Franzosa, E. A. et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 4, 293–305 (2019).
Russo, E. et al. Immunomodulating activity and therapeutic effects of short chain fatty acids and tryptophan post-biotics in inflammatory bowel disease. Front. Immunol. 10, 2754 (2019).
Sokol, H. et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl Acad. Sci. U.S.A. 105, 16731–16736 (2008).
Baur, P. et al. Metabolic phenotyping of the Crohn’s disease-like IBD etiopathology in the TNF ΔARE/WT mouse model. J. Proteome Res. 10, 5523–5535 (2011).
Morita, H., Nakanishi, K., Dohi, T., Yasugi, E. & Oshima, M. Phospholipid turnover in the inflamed intestinal mucosa: arachidonic acid-rich phosphatidyl/plasmenyl-ethanolamine in the mucosa in inflammatory bowel disease. J. Gastroenterol. 34, 46–53 (1999).
Scoville, E. A. et al. Serum polyunsaturated fatty acids correlate with serum cytokines and clinical disease activity in Crohn’s disease. Sci. Rep. 9, 1–11 (2019).
Prantera, C. Probiotics for Crohn’s disease: What have we learned? Gut 55, 757–759 (2006).
Von Schillde, M. A. et al. Lactocepin secreted by Lactobacillus exerts anti-inflammatory effects by selectively degrading proinflammatory chemokines. Cell Host Microbe 11, 387–396 (2012).
Fedorak, R. N. et al. The probiotic vsl#3 has anti-inflammatory effects and could reduce endoscopic recurrence after surgery for crohn’s disease. Clin. Gastroenterol. Hepatol. 13, 928–935.e2 (2015).
Pagnini, C. et al. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc. Natl Acad. Sci. U.S.A. 107, 454–459 (2010).
May, G. R., Sutherland, L. R. & Meddings, J. B. Is small intestinal permeability really increased in relatives of patients with Crohn’s disease? Gastroenterology 104, 1627–1632 (1993).
Fries, W. et al. Intestinal permeability and genetic determinants in patients, first-degree relatives, and controls in a high-incidence area of crohn’s disease in Southern Italy. Am. J. Gastroenterol. 100, 2730–2736 (2005).
Dourmashiin, R. R. et al. Epithelial patchy necrosis in Crohn’s disease. Hum. Pathol. 14, 643–648 (1983).
Di Sabatino, A. et al. Increased enterocyte apoptosis in inflamed areas of Crohn’s disease. Dis. Colon Rectum 46, 1498–1507 (2003).
Günther, C., Neumann, H., Neurath, M. F. & Becker, C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut 62, 1062–1071 (2013).
Günther, C. et al. Caspase-8 controls the gut response to microbial challenges by Tnf-α-dependent and independent pathways. Gut 64, 601–610 (2015).
Charbit-Henrion, F., Parlato, M., Malamut, G., Ruemmele, F. & Cerf-Bensussan, N. Intestinal immunoregulation: lessons from human mendelian diseases. Mucosal Immunol. 1–21 (2021).
Mifflin, L., Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 19, 553–571 (2020).
Dannappel, M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014).
Cuchet-Lourenço, D. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018).
Lehle, A. S. et al. Intestinal inflammation and dysregulated immunity in patients with inherited caspase-8 deficiency. Gastroenterology 156, 275–278 (2019).
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl Acad. Sci. U.S.A. 116, 970–975 (2019).
Salim, S. Y. & Söderholm, J. D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 362–381 (2011).
Zeissig, S. et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56, 61–72 (2007).
Söderholm, J. D. et al. Increased epithelial uptake of protein antigens in the ileum of Crohn’s disease mediated by tumour necrosis factor α. Gut 53, 1817–1824 (2004).
Keita, Å. V. et al. Increased uptake of non-pathogenic E. coli via the follicle-associated epithelium in longstanding ileal Crohn’s disease. J. Pathol. 215, 135–144 (2008).
Keita, Å. V. et al. Enhanced E. coli LF82 translocation through the follicle-associated epithelium in Crohn’s disease is dependent on long polar fimbriae and CEACAM6 expression, and increases paracellular permeability. J. Crohn’s Colitis 14, 216–229 (2020).
Vandussen, K. L. et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of crohn’s disease. Gastroenterology 146, 200–209 (2014).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Niederreiter, L. et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J. Exp. Med. 210, 2041–2056 (2013).
Stengel, S. T. et al. Activating transcription factor 6 mediates inflammatory signals in intestinal epithelial cells upon endoplasmic reticulum stress. Gastroenterology 159, 1357–1374.e10 (2020).
Günther, C. et al. Interferon lambda promotes paneth cell death via STAT1 signaling in mice and is increased in inflamed ileal tissues of patients with Crohn’s disease. Gastroenterology 157, 1310–1322.e13 (2019).
Khaloian, S. et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 69, 1939–1951 (2020).
Elmentaite, R. et al. Single-cell sequencing of developing human gut reveals transcriptional links to childhood Crohn’s disease. Dev. Cell 55, 771–783 (2020).
Farin, H. F. et al. Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell-derived IFN-γ. J. Exp. Med. 211, 1393–1405 (2014).
Mitsialis, V. et al. Single-cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative colitis and Crohn’s disease. Gastroenterology 159, 591–608.e10 (2020).
Corridoni, D., Chapman, T., Ambrose, T. & Simmons, A. Emerging mechanisms of innate immunity and their translational potential in inflammatory bowel disease. Front. Med. 5, 32 (2018).
Casanova, J. L. & Abel, L. Revisiting Crohn’s disease as a primary immunodeficiency of macrophages. J. Exp. Med. 206, 1839–1843 (2009).
Steinbach, E. C. & Plevy, S. E. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm. Bowel Dis. 20, 166–175 (2014).
Hong, S. W. et al. Clinical significance of granulomas in Crohn’s disease: a systematic review and meta‐analysis. J. Gastroenterol. Hepatol. 35, 364–373 (2020).
Caër, C. & Wick, M. J. Human intestinal mononuclear phagocytes in health and inflammatory bowel disease. Front. Immunol. 11, 410 (2020).
Martin, J. C. et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178, 1493–1508.e20 (2019).
Lissner, D. et al. Monocyte and M1 macrophage-induced barrier defect contributes to chronic intestinal inflammation in IBD. Inflamm. Bowel Dis. 21, 1297–1305 (2015).
Kontoyiannis, D. et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J. Exp. Med. 196, 1563–1574 (2002).
Ngoh, E. N. et al. Activity of SHIP, which prevents expression of interleukin 1β, is reduced in patients with Crohn’s disease. Gastroenterology 150, 465–476 (2016).
Menghini, P. et al. Neutralization of IL-1α ameliorates Crohn’s diseaselike ileitis by functional alterations of the gut microbiome. Proc. Natl Acad. Sci. U.S.A. 116, 26717–26726 (2019).
Collins, C. B. et al. Flt3 ligand expands CD103+ dendritic cells and FoxP3+ T regulatory cells, and attenuates Crohn’s-like murine ileitis. Gut 61, 1154–1162 (2012).
Collins, C. B. et al. CD44 deficiency attenuates chronic murine ileitis. Gastroenterology 135, 1993–2002 (2008).
Kosiewicz, M. M. et al. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn’s disease. J. Clin. Invest. 107, 695–702 (2001).
Sandborn, W. J. et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 367, 1519–1528 (2012).
Feagan, B. G. et al. Risankizumab in patients with moderate to severe Crohn’s disease: an open-label extension study. Lancet Gastroenterol. Hepatol. 3, 671–680 (2018).
Sands, B. E. et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med 381, 1201–1214 (2019).
Bamias, G. et al. Proinflammatory effects of TH2 cytokines in a murine model of chronic small intestinal inflammation. Gastroenterology 128, 654–666 (2005).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT00072943 (2003).
Omenetti, S. et al. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 cells. Immunity 51, 77–89.e6 (2019).
Lee, J. S. et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 43, 727–738 (2015).
Jaeger, N. et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun. 12, 1921 (2021).
Maul, J. et al. Peripheral and intestinal regulatory CD4+CD25high T cells in inflammatory bowel disease. Gastroenterology 128, 1868–1878 (2005).
Collins, C. B. et al. Retinoic acid attenuates ileitis by restoring the balance between T-helper 17 and t regulatory cells. Gastroenterology 141, 1821–1831 (2011).
Ishikawa, D. et al. Tregs are dysfunctional in vivo in a spontaneous murine model of Crohn’s disease. Mucosal Immunol. 6, 267–275 (2013).
Goodman, W. A. et al. Loss of estrogen-mediated immunoprotection underlies female gender bias in experimental Crohn’s-like ileitis. Mucosal Immunol. 7, 1255–1265 (2014).
Apostolaki, M. et al. Role of β7 integrin and the chemokine/chemokine receptor pair CCL25/CCR9 in modeled TNF-dependent Crohn’s disease. Gastroenterology 134, 2025–2035 (2008).
Chang, J.-S. et al. Endoplasmic reticulum stress response promotes cytotoxic phenotype of CD8αβ+ intraepithelial lymphocytes in a mouse model for Crohn’s disease-like Ileitis. J. Immunol. 189, 1510–1520 (2012).
Lutter, L., Hoytema van Konijnenburg, D. P., Brand, E. C., Oldenburg, B. & van Wijk, F. The elusive case of human intraepithelial T cells in gut homeostasis and inflammation. Nat. Rev. Gastroenterol. Hepatol. 15, 637–649 (2018).
Ho, J. et al. A CD8+/CD103 high T cell subset regulates TNF-mediated chronic murine ileitis. J. Immunol. 180, 2573–2580 (2008).
Lee, J. C. et al. Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J. Clin. Invest. 121, 4170–4179 (2011).
Sura, R., Colombel, J. F. & Van Kruiningen, H. J. Lymphatics, tertiary lymphoid organs and the granulomas of Crohn’s disease: an immunohistochemical study. Aliment. Pharmacol. Ther. 33, 930–939 (2011).
McNamee, E. N. et al. Ectopic lymphoid tissue alters the chemokine gradient, increases lymphocyte retention and exacerbates murine ileitis. Gut 62, 53–62 (2013).
Bénézech, C. et al. Inflammation-induced formation of fat-associated lymphoid clusters. Nat. Immunol. 16, 819–828 (2015).
Olson, T. S. et al. Expanded B cell population blocks regulatory T cells and exacerbates ileitis in a murine model of Crohn disease. J. Clin. Invest. 114, 389–398 (2004).
Leiper, K. et al. Randomised placebo-controlled trial of rituximab (anti-CD20) in active ulcerative colitis. Gut 60, 1520–1526 (2011).
Kiner, E. et al. Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat. Immunol. 22, 216–228 (2021).
Pruenster, M. et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat. Immunol. 10, 101–108 (2009).
Sandborn, W. J. et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 369, 711–721 (2013).
Uzzan, M. et al. Anti-47 therapy targets lymphoid aggregates in the gastrointestinal tract of HIV-1–infected individuals. Sci. Transl. Med. 10, (2018).
Rivera-Nieves, J. et al. L-selectin, α 4 β 1, and α 4 β 7 integrins participate in CD4+ T cell recruitment to chronically inflamed small intestine. J. Immunol. 174, 2343–2352 (2005).
Zundler, S. et al. The 4β1 homing pathway is essential for ileal homing of Crohn’s disease effector T cells in vivo. Inflamm. Bowel Dis. 23, 379–391 (2017).
Burns, R. C. et al. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology 121, 1428–1436 (2001).
Rivera-Nieves, J. et al. Critical role of endothelial P-selectin glycoprotein ligand 1 in chronic murine ileitis. J. Exp. Med. 203, 907–917 (2006).
Papadakis, K. A. et al. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn’s disease. Gastroenterology 121, 246–254 (2001).
Wendt, E. & Keshav, S. CCR9 antagonism: potential in the treatment of inflammatory bowel disease. Clin. Exp. Gastroenterol. 8, 119–130 (2015).
Wermers, J. D., McNamee, E. N., Wurbel, M. A., Jedlicka, P. & Riveranieves, J. The Chemokine receptor CCR9 is required for the T-cell-mediated regulation of chronic ileitis in mice. Gastroenterology 140, 1526–1535 (2011).
Walters, M. J. et al. Characterization of CCX282-B, an orally bioavailable antagonist of the CCR9 chemokine receptor, for treatment of inflammatory bowel disease. J. Pharmacol. Exp. Ther. 335, 61–69 (2010).
Rivera-Nieves, J. et al. Antibody blockade of CCL25/CCR9 ameliorates early but not late chronic murine ileitis. Gastroenterology 131, 1518–1529 (2006).
McNamee, E. N. et al. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohn’s-like murine ileitis. J. Leukoc. Biol. 97, 1011–22 (2015).
Koliaraki, V., Prados, A., Armaka, M. & Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 21, 974–982 (2020).
Bahar Halpern, K. et al. Lgr5+ telocytes are a signaling source at the intestinal villus tip. Nat. Commun. 11, 1–12 (2020).
Martin, J. C. et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to Anti-TNF therapy. Cell 178, 1493–1508.e20 (2019).
Kerami, Z. et al. Effect of interleukin-17 on gene expression profile of fibroblasts from Crohn’s disease patients. J. Crohn’s Colitis 8, 1208–1216 (2014).
Nayar, S. et al. A myeloid–stromal niche and gp130 rescue in NOD2-driven Crohn’s disease. Nature 593, 275–281 (2021).
Marini, M. et al. TNF-α neutralization ameliorates the severity of murine Crohn’s-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc. Natl Acad. Sci. U.S.A. 100, 8366–8371 (2003).
Corridoni, D. et al. Dysregulated NOD2 predisposes SAMP1/YitFc mice to chronic intestinal inflammation. Proc. Natl Acad. Sci. U.S.A. 110, 16999–17004 (2013).
Vidrich, A. et al. Altered epithelial cell lineage allocation and global expansion of the crypt epithelial stem cell population are associated with ileitis in SAMP1/YitFc mice. Am. J. Pathol. 166, 1055–1067 (2005).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT00269841 (2005).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT00195715 (2005).
US National Library of Medicine. ClinicalTrials.go https://www.clinicaltrials.gov/ct2/show/NCT00152425 (2005).
US National Library of Medicine. ClinicalTrials.go https://www.clinicaltrials.gov/ct2/show/NCT00032786 (2002).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT00032799 (2002).
Gorfu, G. et al. 7 integrin deficiency suppresses B cell homing and attenuates chronic ileitis in SAMP1/YitFc mice. J. Immunol. 185, 5561–5568 (2010).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT01369329 (2011).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT01369342 (2011).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT01369355 (2011).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT01316939 (2011).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT03283085 (2017).
US National Library of Medicine. ClinicalTrials.go https://www.clinicaltrials.gov/ct2/show/NCT00048113 (2002).
Karuppuchamy, T. et al. Sphingosine-1-phosphate lyase inhibition alters the S1P gradient and ameliorates Crohn’s-like ileitis by suppressing thymocyte maturation. Inflamm. Bowel Dis. 26, 216–228 (2020).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT02531113 (2015).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT02675153 (2016).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT00630643 (2008).
US National Library of Medicine. ClinicalTrials.go https://clinicaltrials.gov/ct2/show/NCT03185000 (2017).
US National Library of Medicine. ClinicalTrials.go https://www.clinicaltrials.gov/ct2/show/NCT01632462 (2012).
Acknowledgements
We would like to thank Chritina Eftychi and Aikaterini Nanou for critically reading the manuscript. This work was supported by the FP7 Advanced ERC Grant MCs-inTEST (grant 340217).
Author information
Authors and Affiliations
Contributions
LI and GK contributed in conceptualization and writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Iliopoulou, L., Kollias, G. Harnessing murine models of Crohn’s disease ileitis to advance concepts of pathophysiology and treatment. Mucosal Immunol 15, 10–26 (2022). https://doi.org/10.1038/s41385-021-00433-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-021-00433-3
