
Abstract
Bile acids are cholesterol-derived surfactants that circulate actively between the liver and ileum and that are classically recognized for emulsifying dietary lipids to facilitate absorption. More recent studies, however, have revealed new functions of bile acids; as pleotropic signaling metabolites that regulate diverse metabolic and inflammatory pathways in multiple cell types and tissues through dynamic interactions with both germline-encoded host receptors and the microbiota. Accordingly, perturbed bile acid circulation and/or metabolism is now implicated in the pathogenesis of cholestatic liver diseases, metabolic syndrome, colon cancer, and inflammatory bowel diseases (IBDs). Here, we discuss the three-dimensional interplay between bile acids, the microbiota, and the mucosal immune system, focusing on the mechanisms that regulate intestinal homeostasis and inflammation. Although the functions of bile acids in mucosal immune regulation are only beginning to be appreciated, targeting bile acids and their cellular receptors has already proven an important area of new drug discovery.
Similar content being viewed by others
Introduction
Immune cells at mucosal surfaces, including the skin, lung, and gut, are charged with the rapid detection and elimination of pathogenic microorganisms, while also maintaining tolerance toward commensal bacteria and innocuous antigens.1,2,3 This challenge is particularly onerous for immune cells in intestinal mucosae, where both resident and circulating immune cells are continuously exposed to a barrage of foreign antigens and metabolites derived from the commensal flora, food, and host metabolism.4,5,6 To meet this challenge, humans and rodents deploy diverse arsenals of immune cells to survey and safeguard the intestinal tract, including conventional CD4+Foxp3+ T regulatory (Treg) cells, CD4+RORγt+ (Th17) cells, CD8+ tissue-resident memory (Trm) cells, intraepithelial lymphocytes (IELs), mucosa-associated invariant T (MAIT) cells, innate lymphoid cells (ILCs), secretory (s)IgA+ plasma cells, and specialized antigen-presenting cells (APCs), such as CD103+ dendritic cells (DCs) and CX3CR1+ phagocytes.7,8,9,10,11,12,13,14,15,16 This network of mucosal immune cells acts in concert with each other, as well as with the intestinal epithelium, to enforce barrier function, prevent mucosal infections and maintain a symbiotic relationship with the commensal flora;17,18,19,20,21 disruption of this balance, whether due to genetic or environmental insults, precipitates chronic intestinal inflammation characteristic of the inflammatory bowel diseases (IBDs), Crohn’s disease (CD), and ulcerative colitis (UC).22,23 Local dysregulation of mucosal immune responses in the gut is also now recognized for having important and far-reaching impacts on immune tolerance and hypersensitivity in distal tissues, including joints, the lung, and the central nervous system (CNS).24,25,26,27,28
Much of our recent understanding of intestinal immune regulation has centered on the microbiota. However, bile acids (BAs) represent another unique, dynamic, and fundamental feature of gastrointestinal physiology. Historically considered simple emulsifying agents produced by the liver to facilitate the absorption and/or elimination of dietary fats in the intestinal lumen, more recent studies have revealed that BAs are also pleotropic, hormone-like signaling metabolites that regulate mucosal homeostasis and inflammation via direct interactions with both germline-encoded cellular receptors and luminal bacteria.29,30,31 BAs shape microbial colonization in the gut due to intrinsic bacteriostatic activities, but are also metabolized by many commensal bacteria in the intestinal lumen.32,33,34 Thus, the size and composition of an individual’s microbiome dictates that of one’s circulating BA pool. In addition, BAs interact directly with a variety of transmembrane and nuclear receptors expressed in hepatocytes, intestinal epithelial cells (IECs), as well as innate and adaptive immune cells to regulate mucosal immune function.35,36,37 Finally, it is essential to understand that BAs produced in the liver and metabolized in the intestine are maintained in vivo through a highly efficient and tightly orchestrated enterohepatic circulatory system, in which BAs synthesized in hepatocytes are actively transported into bile ducts, stored in the gall bladder, deposited into the duodenum following food intake, reabsorbed in the ileum, and returned to the liver via portal circulation (Fig. 1).38 Each step of enterohepatic circulation is directly responsive to dietary patterns, linked by hormone-like signaling events, and acts in synergy with the others to maintain a functional BA pool that is both capable of meeting digestive demands and not toxic to the gastrointestinal tract.39 Indeed, BAs are highly pro-inflammatory and cytotoxic when dysregulated; this is due to their detergent-like activities and best exemplified in cholestatic liver diseases (e.g., biliary atresia), where mutations in hepatic BA transporters prompt BA accumulation in the liver, leading to chronic inflammation, hepatocellular necrosis and liver failure.40,41,42 Disrupted BA reabsorption in the ileum also precipitates BA malabsorption (BAM)/BA diarrhea (BAD), an under-appreciated and often mis-diagnosed condition in which reduced BA reabsorption—due to genetic mutations (type 2/primary BAM), ileal resection (type 1), or gastrointestinal disease (type 3)—results in abnormally high BA concentrations and water secretion in the colon.43 Still, steady-state signaling through BA receptors is now recognized as essential for proper regulation of glucose and lipid metabolism, insulin sensitivity, as well as intestinal immunity.23,29 Even extra-intestinal functions of BAs have begun to emerge, ranging from the ability of BAs to directly promote hematopoietic stem/progenitor cell (HSPC) expansion in the fetal liver,44 to BA-dependent signaling in the CNS that regulates neuroinflammation and neurodegeneration.45 Thus, whereas much remains to be learned about the multifaceted functions of BAs in vivo, existing data already paint a picture that BAs, like the microbiota, represent an important and dynamic aspect of human health and disease.

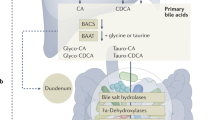
Bile acid biosynthesis and circulation. Primary bile acids (BAs; e.g., cholic acid [CA] and chenodeoxycholic acid [CDCA]) are synthesized by hepatocytes via two pathways (i.e., classical, alternative) of orchestrated cholesterol catabolism mediated by the cytochrome P450 (CYP) enzymes CYP7A1, CYP8B1, CYP27A1, and CYP7B1. Bile acid-CoA:amino acid N-acyltransferase (BAAT) catalyzes the conjugation of primary BAs to glycine or taurine (G/T), which enables their active transport into bile ducts and storage in the gall bladder. The majority (90–95%) of BAs postprandially secreted into the small intestine are actively reabsorbed in the terminal ileum and circulate back to the liver. Both re-circulating BAs and fibroblast growth factor (FGF)-15/19—induced by BA-dependent farsenoid X receptor (FXR) activation in ileal epithelial cells and secreted into portal circulation—independently restrict de novo BA synthesis in the liver by suppressing expression of CYP7A1, the rate-limiting enzyme in the classical BA biosynthetic pathway
The old: bile acid synthesis, enterohepatic circulation and microbial metabolism
Two major classes of BAs exist in mammals: primary and secondary. Whereas primary BAs are synthesized via cholesterol catabolism in hepatocytes, secondary BAs are derivatives of primary BAs generated by microbial metabolism in the intestine (Fig. 1).34,39 The most abundant primary BAs in humans are cholic acid (CA) and chenodeoxycholic acid (CDCA), whereas rodent hepatocytes produce CA together with the 6β-hydroxylated form of CDCA, termed muricholic acid (MCA). In both humans and rodents, hepatic BA biosynthesis proceeds via two overlapping, but non-redundant pathways, termed the ‘classical’ and ‘alternative/acidic’ pathways (Fig. 1).46 Cytochrome P450 (CYP) enzymes are key regulators of both biosynthetic pathways, and evolutionary divergence between human and rodent CYP genes underlies the distinctive patterns of hepatic BA production in these species; mouse Cyp2c70 efficiently converts CDCA to MCA, whereas its human ortholog, CYP2C9, does not.47 The relative contribution of the classical and alternative/acidic pathways to primary BA production in various physiological settings remains incompletely understood, though the classical pathway—defined by the activities of the cholesterol hydrolases CYP7A1, CYP8B1, and CYP27A1—is thought to account for the majority (~75%) of primary BAs that are produced at steady-state.39,46 The alternative pathway, by contrast, is considered a compensatory pathway that is upregulated during periods of hepatic stress or disease and results in the near-exclusive production of CDCA. Irrespective of the originating pathway, the final step of BA biosynthesis in hepatocytes is the conjugation of primary BAs to the amino acids, taurine or (less frequently) glycine; this is executed by the BA-CoA:amino acid N-acyltransferase (BAAT) enzyme, and allows BAs to retain their amphipathic structure, which is critical for their lipid emulsifying activities in the acidic environment of the duodenum (Fig. 1).48 In addition, amino acid conjugation renders BAs membrane impermeant, and necessitates active transport by a host of BA transporters expressed in the liver and ileum.49
BA reabsorption in the ileum follows a tightly orchestrated pathway of trans-cellular transport. Luminal BAs reaching the ileum are first bound and transported across the apical surface of enterocytes by the apical sodium-dependent BA transporter (ASBT; encoded by SLC10A2).50 Upon entry into the cytoplasm, BAs are rapidly bound and chaperoned to the basolateral surface by the ileal BA-binding protein (IBABP; encoded by FABP6), where they are transported across the basolateral membrane—and into the underlying mucosal tissue—by the heterodimeric organic solute transporter α/β (OSTα/β; encoded by SLC51A/B) complex.51 BAs reabsorbed in the ileum accumulate in the mucosa and ultimately diffuse into fenestrated capillaries for portal re-circulation to the liver. This enterohepatic circulation of BAs is completed approximately six to eight times each day in humans, depending on dietary patterns, and maintains a circulating BA pool of between 4 and 6 g in healthy adults.38,52
Both the hepatic synthesis of BAs and their reabsorption in the ileum are linked by an intricate network of hormone-like signaling events in hepatocytes and IECs. De novo BA biosynthesis in hepatocytes is subject to direct feedback inhibition by the existing pool of BAs re-circulating to the liver from the ileum; this involves direct binding of BAs to the nuclear receptor (NR), farsenoid X receptor (FXR; encoded by NR1H4), FXR-dependent trans-activation of small heterodimer partner (SHP; encoded by NR0B2), and SHP-mediated repression of CYP7A1 expression, the rate-limiting enzyme in hepatic BA biosynthesis.46,53 BA-dependent activation of FXR in ileal epithelial cells further suppresses hepatic BA synthesis through an endocrine pathway involving the hormone-like fibroblast growth factor (FGF)-15/19 (FGF-15 in mice; FGF-19 in humans); BA-activated FXR drives FGF-15/19 expression in IECs, which in turn is secreted into portal circulation, transits to the liver, binds to its heterodimeric receptor on hepatocytes (FGFR4/βKlotho) and restricts hepatic BA synthesis by suppressing CYP7A1 expression (Fig. 1).54 Intriguingly, FXR not only drives expression of FGF-15/19 in IECs, but it also directs expression of the signaling subunit of the FGF-15/19 receptor, βKlotho, in hepatocytes, thereby conditioning the liver for feedback regulation by FGF-15/19.55 Such tight and integrated control over BA synthesis and circulation serves as a rheostat to maintain a functional, but not toxic, circulating BA pool.
The small portion of BAs (5–10%) that escape reabsorption in the ileum are mostly unconjugated (see below) and enter the large intestine for bacterial metabolism; these are either passively reabsorbed in the colon and re-enter the circulating BA pool, or are excreted in the feces (Fig. 1). Many bacterial taxa in the mammalian gut (e.g., Firmicutes, Bacteroides, Eubacterium, and Clostridium) express bile salt hydrolase (BSH) enzymes that deconjugate taurine- and glycine-conjugated BAs.34 As only conjugated BAs are actively recycled in the ileum, this is one mechanism by which microbial metabolism directly impacts ileal BA reabsorption. Indeed, one cause of type 3 BAM is bacterial overgrowth in the small intestine, which prompts excessive BA deconjugation and reduced ileal reabsorption.56 Other enteric bacteria express hydroxysteroid dehydrogenase (HSDH) enzymes, including Bacteroides, Clostridium, Eubacterium, Lactobacillus, and Escherichia, which convert (through 7-dehydroxylation) primary BA precursors into secondary BA products. The most common secondary BAs are deoxycholic acid (DCA; the 7-dehydroxylation product of CA) and lithocholic acid (LCA; the 7-dehydroxylation product of CDCA). However, recent advances in mass spectrometric BA analysis have now led to the identification and characterization of more than 20 distinct secondary BA species in humans and rodents.57 This diversity of primary BAs and their secondary metabolites underscores the complex and dynamic nature by which BAs can influence mucosal immune responses, as each BA species has unique physiochemical properties (e.g., hydrophobicities, critical micelle concentrations, membrane permeabilities), as well as varying affinities for host receptors and transporters (see below).
BAs also reciprocally regulate microbial colonization in the intestinal tract, due to both direct bacteriostatic activities and BA-dependent signaling in IECs. For example, the constitutively high concentrations of BAs in the small intestinal lumen (1–10 mM) approaches most critical micelle concentrations; this not only facilitates emulsification of dietary lipids, but also leads to direct lysis of bile-sensitive bacteria. Accordingly, both clinical and experimentally induced liver injury decreases BA secretion and leads to bacterial overgrowth in the small bowel.58,59,60,61 Bile duct ligation drives a similar elevation in small bowel bacterial levels,61,62,63,64 whereas feeding rodents either bile or conjugated BAs during states of relative BA-insufficiency reduces small bowel bacterial growth to normal levels.65,66,67 However, the potent antimicrobial activity of BAs observed in vivo is not evident at physiological concentrations in vitro.33,68,69,70,71 Therefore, two additional mechanisms have been proposed to explain the antimicrobial activities of BAs in vivo. First, BAs present in bile fluid in vivo exist as mixed micelles, together with phospholipids, long-chain fatty acids, and bilirubin; the bacteriostatic functions of BAs are enhanced in the presence of other bile constituents, particularly long-chain fatty acids.72,73 Second, recent studies have revealed that BA-dependent FXR activation in IECs promotes antimicrobial peptide expression.35,74,75 Thus, the antimicrobial effects of BAs in vivo, as well as their immunoregulatory functions (described below), likely involve a number of synergistic mechanisms.
In contrast to the small intestine, BA concentrations are orders of magnitude lower in the colon (10–50 μM), due to active reabsorption in the ileum, and this favors bacterial colonization and BA metabolism. Still, reports of BA-bug interactions in the colon that regulate mucosal inflammation are beginning to emerge. For example, mice fed a milk-fat diet display elevated levels of taurine (t)-conjugated CA in feces, as well as overgrowth of Bilophila wadsworthia, a pathobiont associated with human ulcerative colitis.76,77 Expansion of B. wadsworthia is causative in a mouse model of colitis induced in IL-10-deficient mice by either a milk-fat diet or a tCA-supplemented diet.76 Future studies interrogating the interplay between BAs and the microbiota—and how these interactions are altered by dietary modification—should lead to important new insights in the understanding and treatment of IBDs.
The new: bile acids as active signaling metabolites
The interactions between BAs and the microbiota, as well as their basic functions in dietary fat absorption, have been recognized for many decades. However, it was not until the turn of this century that interest in BA metabolism was re-invigorated by the discovery that the human genome encodes dedicated BA receptors. The first BA receptor described was FXR,78 as detailed above, FXR is now recognized as a master regulator of both BA biosynthesis in the liver and BA-dependent endocrine signaling in the ileum. However, additional cell surface and nuclear BA receptors have been identified over the last 15 years, most notably the membrane-type bile acid receptor (M-BAR), which is also named Takeda G-protein receptor 5 (TGR5) and encoded by the GPBAR locus.79 In addition, other nuclear receptors—pregnane X receptor (PXR; encoded by NR1I2) and vitamin D receptor (VDR; encoded by NR1I1)—are directly bound and modulated by specific endogenous BA species.80,81 Further, constitutive androstane receptor (CAR; encoded by NR1I3) is activated through an indirect mechanism following cellular BA exposure.82,83 As a rule, these receptors are broadly expressed in both parenchymal and hematopoietic compartments, particularly within the gastrointestinal tract, and display preferential affinities for distinct BA species.84,85 Whereas FXR and TGR5 are considered dedicated BA receptors, which bind to BAs with high-affinity and transduce an array of metabolic and anti-inflammatory signaling pathways, PXR, VDR, and CAR reflect non-specific BA “sensors”, which detoxify BAs through the induction of CYPs, BA sulfotransferases (SULTs), and BA transporters during periods of BA overload (Fig. 2).86,87 Importantly, both therapeutic BAs (e.g., ursodeoxycholic acid [UDCA]) and semi-synthetic BA receptor agonists have shown efficacy in animal models of cholestatic liver diseases, type 2 diabetes (T2D) and inflammatory diseases.41,88 In addition, the semi-synthetic FXR agonist, obeticholic acid (OCA; a.k.a., INT-747)—a 6α-ethyl derivative of CDCA—is now in clinical development for the treatment of non-alcoholic fatty liver disease (NAFLD) and T2D.89 The development of small molecule BA receptor modulators has proven key in the evolution of our understanding that BAs are active, and therapeutically relevant, signaling metabolites.

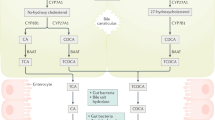
Differences in bile acids and bacteria underscore unique immunoregulatory microenvironments in the colon and small intestine. a High (millimolar) concentrations of conjugated primary (1°) bile acids (BAs) in the ileum restrict bacterial growth and are actively reabsorbed by specialized enterocytes expressing the apical sodium-dependent bile acid transporter (ASBT), the cytoplasmic BA chaperone, ileal bile acid-binding protein (IBABP), and the basolateral organic solute transporter (OST)-α/β complex. Following reabsorption, BAs accumulate in the mucosa, interact with mucosal immune cells and enter portal blood for re-circulation to the liver. Conjugated 1° BAs are high-affinity farsenoid X receptor (FXR) agonists; BA-dependent FXR activation in enterocytes concomitantly promotes secretion of fibroblast growth factor (FGF)-15/19 (to suppress hepatic BA biosynthesis) and expression of antimicrobial peptides (AMPs) to enforce the epithelial barrier. BAs also activate FXR in resident dendritic cells (DCs) and macrophages to limit Toll-like receptor 4 (TLR4)-dependent pro-inflammatory cytokine expression and suppress NLRP3 inflammasome activation. OSTα/β (in enterocytes) and the xenobiotic transporter, multidrug resistance protein 1 (MDR1) (in mucosal Th17, Th1 cells) suppress oxidative stress induced by high BA concentrations in the ileum. b The minority (~ 5%) of conjugated 1° BAs that escape ileal reabsorption enter the colon, where reduced (micromolar) BA concentrations facilitate bacterial colonization and BA metabolism into unconjugated secondary (2°) BAs. Unlike conjugated 1° BAs, unconjugated 2° BAs passively diffuse across the epithelial surface into mucosae. Colonic BAs, in particular lithocholic acid (LCA), activates pregnane X receptor (PXR) in epithelial cells to promote TGFβ expression and suppress TLR4-dependent pro-inflammatory cytokines production. Both PXR, as well as the related xenobiotic-sensing nuclear receptor, constitutive androstane receptor (CAR), also respond to LCA in enterocytes by repressing NF-κB activation and activating expression of drug metabolizing enzymes (DMEs), which in turn promotes the detoxification and clearance of excess BAs. LCA, as well as deoxycholic acid (DCA) are high-affinity agonists of the cell-surface BA receptor, Takeda G-protein receptor 5 (TGR5), expressed on colonic DCs and macrophages; DCA-dependent TGR5 activation limits TNF and IL-12 secretion by DCs, blocks NLRP3 inflammasome activation in M1 macrophages and promotes differentiation of IL-10-secreting M2 macrophages. These anti-inflammatory functions of BA-dependent TGR5 signaling act in concert with bacterial metabolites (e.g., short chain fatty acids (SCFAs)) to enforce the development, recruitment and expansion of Foxp3+ T regulatory (Treg) cells in the colon
Farsenoid X receptor
Interaction between BAs and the FXR ligand-binding domain (LBD) promotes FXR:DNA-binding to FXR response elements (FXREs), both as a monomer and as a heterodimer with liver X receptors (LXRs) (encoded by NR1H3 and NR1H2). As typical for ligand-bound nuclear receptors, FXR undergoes a conformational change upon BA binding, which displaces negative co-regulators (NCoRs) and recruits co-activators (NCoAs).90,91 Further, as the preferred substrate for FXR is tCDCA, optimal FXR activation—at least in the liver—requires active BA internalization by transporters.92 Functionally, BA-dependent FXR activation in IECs inhibits ASBT expression (SLC10A2) and promotes the expression of both IBABP (FABP6) and OSTα/β (SLC51A/B) to enforce efficient BA trans-cellular export.49,74 However, a growing body of literature also indicates that FXR activity is critical for mucosal immune homeostasis and is generally decreased during chronic intestinal inflammation.
FXR-deficiency increases, whereas small molecule FXR agonist treatment suppresses, mucosal inflammation in several mouse models of colitis, including dextran sulfate sodium (DSS)- and 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis.93 Mechanistically, colonic tissue from colitic mice treated with the FXR agonist INT-747 display reduced pro-inflammatory cytokine (e.g., IL-1β, IL-6) and chemokine (e.g., CCL2) expression,93 and these phenotypes have been attributed to BA- and FXR-dependent transcriptional responses in IECs that enforce barrier function and antimicrobial peptide production and that limit bacterial translocation across the epithelial barrier (Fig. 2). However, INT-747-dependent FXR activation also represses TLR4-induced pro-inflammatory gene expression in IECs,94 and restricts inflammatory cytokine and chemokine expression in cultured human CD14+ monocytes and DCs.93 Thus, FXR appears to limit mucosal inflammatory responses via synergistic activities in both IECs and innate immune cells. Whether FXR activation in mucosal immune cells involves active internalization of conjugated BAs or passive membrane diffusion of unconjugated BAs remains unclear and will be an important area of future investigation.
Mechanistically, BA-dependent FXR activation has been reported to repress NF-κB activity by preventing nuclear co-receptor clearance from NF-κB-binding sites in the Tnf and Il1b locus.95,96 In addition, FXR activation by INT-747 in mice with chemically induced colitis appears to exert systemic anti-inflammatory effects, including elevated serum IL-10 levels, retention of DCs in the spleen and increased Treg cell numbers.97 These effects have been linked to reduced mucosal expression of Madcam1—the endothelial ligand for α4β7 integrin-dependent leukocyte extravasation into the colonic lamina propria—as well as CXCL3, the ligand for CCR2 that is expressed on inflammatory macrophages and DCs and that mediates leukocyte homing to inflamed peripheral tissues.97,98 By contrast, colons of colitic mice treated with INT-747 display elevated CCL25 expression, which, together with reduced CXCL3 expression, may favor CCR9-dependent recruitment of Treg cells to inflamed mucosal tissues.97 BA-dependent FXR activation also directly trans-activates expression of the glucocorticoid receptor (GR; encoded by NR3C1), a widely recognized anti-inflammatory receptor of corticosteroids that remain gold-standard therapeutic modalities in IBDs.99 Finally, a recent study has shown that FXR suppresses assembly of NLRP3-containing inflammasomes by physically interacting with both NLRP3 and Caspase-1 (Fig. 2).100 In contrast to most other FXR functions, which require BA-dependent activation of FXR transcriptional activity, this study showed that BAs act as a danger-associated molecular pattern (DAMP) in macrophages to activate inflammasomes together with ATP, and that FXR-dependent suppression of inflammasome assembly occurs in the absence of BA binding.100
Consistent with these anti-inflammatory functions of FXR in the intestine, FXR transcriptional activity is consistently impaired during chronic mucosal inflammation, in both mice and human IBD patient biopsies.96,101 On one hand, inflammatory cytokines (e.g., TNF, IL-1β) have been shown to promote a physical association between FXR and the NF-κB p50 and p65 subunits, which limits FXR transcriptional activity.96 At the same time, chronic mucosal inflammation is associated with type 1 BAM, which by definition restricts BA-dependent FXR activity in IECs and mucosal phagocytes.50,74
An important consideration for understanding FXR activity in complex inflammatory diseases, such as IBDs, is the preferential binding of discrete BA species to FXR. Both CA and CDCA (i.e., primary BAs) are potent FXR agonists, whereas the more hydrophobic secondary BAs (e.g., DCA, LCA) generated through microbial metabolism in the gut, display reduced affinities for FXR.30,78 There are also important functional differences between the FXR-regulating activities of human- and rodent-specific BAs; whereas CDCA (preferentially produced in humans) is a potent FXR activator, β-MCA (the rodent-specific derivative of CDCA) acts as an FXR antagonist.102 Thus, understanding the full scope of BA-dependent FXR functions in mucosal immune regulation will require meticulous attention to the circulating BA pool in both experimental animal models and human patients, and how these parameters change in response to diet, inflammation and dysbiosis.
Transmembrane G-protein-coupled bile acid receptor
The discovery of FXR as a dedicated BA receptor established the notion that BAs are active signaling metabolites. However, BA-dependent FXR activity is seen primarily in parenchymal cells of the gastrointestinal tract, namely hepatocytes and ileal IECs, that express BA transporters (e.g., NCTP in hepatocytes; ASBT in IECs) and that are specialized to internalize conjugated BAs during enterohepatic circulation. Thus, it was not until the discovery that conjugated BAs could also transduce signals through a dedicated transmembrane BA receptor, coined TGR5/M-BAR, that it became clear additional cell types may be permissive to BA-dependent signaling in vivo.103 Indeed, TGR5 is highly expressed not only in hepatocytes and IECs, but also in several hematopoietic cell lineages, most notably monocytes and macrophages;79,104 a growing body of literature has now established that BA-dependent TGR5 signaling potently suppresses pro-inflammatory macrophage function, both systemically and in the intestinal mucosa (Fig. 2).105,106
TGR5 activation by either endogenous BAs or the synthetic TGR5 agonist 6α-ethyl-23(S)-methylcholic acid (S-EMCA/INT-777) suppresses lipopolysaccharide (LPS)-induced inflammatory cytokine expression, whereas these responses are both elevated and unaffected by BAs in macrophages lacking TGR5.107,108 TGR5 activates adenylate cyclase, which drives increased cyclic AMP (cAMP) production and leads to activation of the cAMP-responsive transcription factor, cAMP response element binding protein (CREB). BA- and TGR5-dependent CREB activation, in turn, suppresses TLR4-mediated NF-κB transcriptional activation of multiple pro-inflammatory cytokine genes (e.g., TNF, IL1A, IL1B, IL6, and IL8), and this is reversed by treatment with adenylate cyclase inhibitors.107 In addition, in vivo activation of TGR5 by another selective small molecule agonist, BAR501, has been shown to switch mucosa-associated macrophage phenotypes from M1 (pro-inflammatory) to M2 (tissue-protective) during chemically induced colitis.109 In this setting, TGR5 activation promotes epidermal growth factor receptor (EGFR)–SRC kinase (SRC) signaling, as well as STAT3 phosphorylation/activation, which suppresses pro-inflammatory cytokine (e.g., TNF, IFN-β, IL-6, and IL-12) expression and promotes Treg cell recruitment to inflamed colonic tissue.109 TGR5-dependent cAMP production has also been shown to suppress activation of the NLRP3 inflammasome, a multiprotein complex that activates capase-1-dependent processing of IL-1β and IL-18. Here, TGR5-dependent cAMP production leads to protein kinase A (PKA) activation, and subsequent phosphorylation of NLRP3 at Ser 291, resulting in ubiquitination and degradation of the inflammasome.110 These synergistic anti-inflammatory pathways downstream of TGR5 not only acutely suppress innate immune responses, but also influence the downstream priming of inflammatory T cell responses. Specifically, BA-dependent TGR5 activation directs the differentiation of human monocytes into tolerogenic DCs that secrete low levels of TNF and IL-12, cytokines that are required for the priming of pro-inflammatory Th1 responses commonly elevated in IBD patients.111 In addition, mucosa-associated macrophages isolated from Crohn’s disease patient biopsies express high levels of TGR5 and ex vivo treatment of these cells with BAs leads to reduced inflammatory cytokine expression, including TNF.108 Thus, whereas BA-dependent TGR5 signaling, like that of BA-dependent FXR activation, generally suppresses mucosal inflammatory responses, perturbed BA circulation and/or metabolism during chronic intestinal inflammation may limit endogenous TGR5 activation. In addition, altered microbial metabolism of primary BA precursors into secondary BA products during states of dysbiosis, at least conceptually, stands to have an important influence on the balance between FXR and TGR5 activation. Unlike FXR, TGR5 displays the highest affinity for the secondary BAs, LCA, and DCA, whereas primary BAs—that act as potent FXR agonists—show lower affinities for TGR5.74,103,112 Together, these data illustrate that the dedicated BA receptors, FXR and TGR5, are critical regulators of BA metabolism, BA circulation, as well as intestinal immune function, which has cemented their status as possible therapeutic targets for the treatment of mucosal inflammatory diseases (Fig. 2).
Pregnane X receptor, constitutive androstane receptor and vitamin D receptor
In contrast to the anti-inflammatory functions of “tonic” BA signaling through FXR and TGR5, BAs are also lipophilic detergents that damage cellular membranes and have potent cytotoxic and pro-inflammatory effects at high concentrations. Accordingly, mammals use at least three additional nuclear receptors—PXR, VDR, and CAR—to act as low-affinity BA “sensors” and to promote BA detoxification and protect tissues from BA-driven injury.113 It is worth noting that the term BA ‘sensors’ is used when discussing PXR, VDR, and CAR because, unlike FXR and TGR5, these nuclear receptors are also activated by food- and bacteria-derived metabolites (i.e., xenobiotics), where they can also influence mucosal immunity and homeostasis.113,114,115
Pro-inflammatory functions of BAs are classically recognized in cholestatic liver diseases, where BA accumulation in the liver following loss of hepatic BA efflux activity incites chronic inflammation, hepatocellular necrosis and liver failure.65,116,117 More recent studies, however, indicate that BA reabsorption in the ileum is also potentially pathogenic, and that mechanisms exist in both ileal IECs and mucosal immune cells to actively prevent BA-driven intestinal injury. First, BA accumulation in ileal IECs lacking the basolateral BA transporter, OSTα, leads to villous blunting and IEC proliferation in the ileum that is rescued by ablating ASBT-dependent BA import.118 In addition, circulating subsets of pro-inflammatory CD4+ T helper (TH) cells—including IL-17A-secreting Th17 and IFNγ-producing Th1 cells—adapt to high local BA concentrations in the ileum by upregulating the xenobiotic transporter, multidrug resistance protein 1 (MDR1, encoded by ABCB1 in humans, Abcb1a and Abcb1b in mice) (Fig. 2).119 Here, TH cells deficient in MDR1 display localized dysfunction in the ileal mucosa, and transfer Crohn’s disease-like ileitis in lymphopenic (i.e., Rag1−/−) hosts, which is again rescued by genetic or pharmacologic blockade of ileal BA reabsorption.120 Notably, BA-induced injury, whether in OSTα-deficient IECs or MDR1-deficient T cells, is associated with oxidative stress and overexpression of inflammatory cytokines, akin to the phenotype of hepatocytes in cholestatic livers.116,117,118,119
PXR, VDR, and CAR all serve to protect cells from BA-driven injury through the transcriptional activation of drug- and BA-detoxifying enzymes—cytochrome P450 enzymes (i.e., CYPs), BA sulfotransferases (i.e., SULTs), and uridine 5′-diphospho-glucuronosyltransferase (i.e., UGTs)—as well as transporters, such as multidrug resistance-associated protein 3 (MRP3, encoded by ABCC3) (Fig. 2).85,121,122 Here again, protective functions of PXR, VDR, and CAR against BA-driven injury have been largely characterized in settings of cholestasis; all three of these nuclear receptors protect mice from LCA-induced liver toxicity.80,81,83,86 Still, there is increasing evidence that PXR, VDR, and CAR are also protective in settings of chronic intestinal inflammation. Notably, mucosal biopsies from IBD patients display reduced expression of PXR, VDR, and CAR target genes, and IBD-associated polymorphisms in each of these loci have been reported.123,124,125 Pharmacologic activation of CAR and PXR has been shown to modulate the gut microbiome, reducing Bifidobacterium, Anaeroplasma, and Dorea species, and promoting an increase in primary conjugated BAs.126 Still, BA-dependent functions of these nuclear receptors during normal physiology—particularly for PXR and CAR—are only beginning to be interrogated, and it is currently unclear whether PXR, VDR, and CAR enforce mucosal homeostasis through their functions in hepatocytes, IECs, mucosal immune cells, or some combination thereof.
PXR selectively binds LCA and has emerged as an intrinsic regulator of IEC function under homeostatic conditions.81 Intestines of PXR-deficient mice show decreased villus to crypt ratio, increased neutrophil infiltration, and enhanced basal MPO activity.114 In addition, small bowel inflammation and increased epithelial expression of NF-κB target genes, including IL-1β, TNF, and iNOS, have been reported in PXR knockout mice. Reciprocally, activation of PXR with the xenobiotic agonist, pregnenolone-16α-carbonitrile (PCN),127 confers protection against DSS-induced colitis and promotes mucosal healing, which likely involves a series of cytoprotective mechanisms in IECs (Fig. 2).128 First, PXR activation has been reported to stimulate p38 MAP kinase activity and IEC motility, thereby facilitating mucosal wound healing.129 Second, PXR activation promotes TGFβ expression in IECs, which limits TNF, IL-8, CCL5, and CCL20 expression.128,130 Third, PXR activation with rifaximin, a gut-specific human PXR agonist, promotes an inhibitory interaction between PXR and NF-κB p65, which in turn suppresses LPS-driven inflammatory gene expression.131 This function of PXR is particularly noteworthy, considering that rifaximin is approved for use in several chronic gastrointestinal diseases and has shown clinical efficacy in moderate to severe Crohn’s disease.132 Finally, LCA-dependent PXR activation has been shown to reduce TLR4 mRNA stability, diminish TLR4 signaling and protect mice from experimental necrotizing enterocolitis (NEC);133 PXR activation by bacterial-derived indole metabolites has also been reported to modulate TLR4 signaling and maintain epithelial barrier function.114 Whether PXR regulates mucosal immune cell function intrinsically remains an open question, but at least one previous study has suggested that PXR regulates the pro-inflammatory function of Kupffer cells, a specialized lineage of liver-resident macrophages implicated in numerous inflammatory liver diseases.134
The role of VDR in BA-dependent mucosal immune regulation has been obscured by the more classical endogenous VDR agonists, such as 1,25-dihydroxyvitamin D3 (a.k.a., calcitriol).135 Still, vitamin D insufficiency is an established risk factor in IBDs, and reduced VDR expression is commonly observed in mucosal biopsies from IBD patients.123 Consistent with these human data, mice in which VDR is conditionally ablated in IECs display reduced colonic antimicrobial activity, gut dysbiosis and an increased susceptibility to chemically induced colitis.136,137 By contrast, reconstituting VDR-deficient IECs with a human VDR transgene renders mice highly resistant to both chemically and T cell transfer-induced colitis.138 In this scenario, VDR activity suppresses IκBα degradation, thus limiting NF-κB activation and attenuating p53 upregulated modulator of apoptosis (PUMA)-mediated IEC apoptosis.138 Like PXR, VDR preferentially binds LCA, and is thought to accelerate BA detoxification in IECs through the induction of CYP3A expression.80 However, it is not clear from these studies whether the protective functions of VDR in IECs is due to interaction with 1,25-dihydroxyvitamin D3, LCA, or both. Nonetheless, other studies have shown directly that LCA-dependent VDR activation suppresses pro-inflammatory cytokine expression in IECs during experimental colitis, and that the anti-inflammatory effects of LCA in these models are abolished in mice lacking VDR.136,138
Of course, VDR is recognized as an important regulator of both innate and adaptive immune cell function, negatively regulating monocyte-derived macrophage activation, inhibiting DC maturation, promoting Treg cell differentiation and suppressing pro-inflammatory Th1 and Th17 responses.139,140,141 However, the vast majority of these studies have focused on the immunoregulatory functions of VDR following activation by 1,25-dihydroxyvitamin D3, and it remains unclear whether VDR-dependent transcriptional activity is the same or different when bound by 1,25-dihydroxyvitamin D3 or LCA. Still, one recent study interrogated LCA-dependent VDR functions in CD4+ TH cells directly. Here, the authors identified unconjugated LCA as a potent suppressor of mouse and human TH cell activation.142 Reduced TH cell activation and Th1 cell differentiation in the presence of LCA was associated with lower ERK-1/2 phosphorylation and decreased expression of Th1-promoting transcription factors, namely T-BET, STAT1, and STAT4; all of these anti-inflammatory effects of LCA were sensitive to shRNA-mediated VDR depletion.142
CAR is a PXR-related xenobiotic-sensing nuclear receptor, and as such is also recognized for its role in drug and BA metabolism in the liver. Unlike PXR and VDR, however, there is no experimental evidence that BAs are direct ligands of CAR. Rather, CAR is phosphorylated at steady-state and sequestered in a multiprotein cytoplasmic complex comprised of heat shock protein 90 (Hsp90), Hsp70 and the cytoplasmic CAR retention protein (CCRP).143 Release of CAR from this inhibitory complex, and subsequent nuclear translocation, is triggered by either direct binding of small molecule ‘super-agonists’ (e.g., the phenobarbital derivative agonist, 4-bis[2-(3,5-dichloropyridyloxy)]benzene [TCPOBOP])144 or by a poorly understood indirect pathway that stems from transmembrane receptors and that induces protein phosphatase 2A (PP2A)-mediated CAR dephosphorylation; this indirect activation of CAR occurs in the presence of numerous pharmaceutical compounds (e.g., phenobarbital) as well as hydrophobic BAs, such as LCA.86,143,145,146,147,148 Another distinctive feature of CAR (vis-à-vis most other nuclear receptors) is that CAR displays constitutive (i.e., ligand-independent) transcriptional activity, due to a truncated C-terminal activation function 2 (AF2) motif in the LBD that stables a transcriptionally active conformation.149 Nuclear CAR trans-activates target gene expression by binding to cognate DNA regulatory elements either as a monomer or as a heterodimer with retinoid X receptor (RXRα; encoded by NR2B1),149,150 whereas constitutive CAR activity is inhibited by the binding of androstanes, such as 5α-Androstan-3β-ol.151
As with PXR and VDR, polymorphisms in the CAR (NR1I3) locus have been associated with IBDs, and the expression of CAR and a number of its target genes have been found to be reduced in inflamed mucosal biopsies from CD and UC patients.152 In mice, the small molecule CAR agonist, TCPOBOP, reduces severity of DSS-induced colitis, which is associated with reductions in both monocyte and macrophage mucosal infiltration and in pro-inflammatory cytokine expression (Fig. 2).153 Like PXR, CAR is thought to regulate IEC function in the context of wound healing; the selective human CAR agonist 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO)154 increases IEC migratory activity.152 In addition, a series of recent studies suggest that CAR-dependent transcriptional activity in IECs is influenced by the enteric flora. For example, dysbiosis induced by a high-fat diet leads to elevated expression of CAR target genes in the mouse intestine.155 Second, introduction of a normal microbiome into germ free (GF) mice suppresses the expression of CAR, as well as CAR-regulated CYPs, in IECs in a TLR2-dependent manner.115 Rawls and colleagues reported a similar finding, in which intestines of GF mice display increased transcriptional activity associated with xenobiotic metabolism, compared with conventionally housed specific pathogen-free (SPF) counterparts.156 Together, these data support a model in which microbe-mediated BA deconjugation decreases intestinal reabsorption, leading to reduced levels of mucosa-associated BAs and less need to detoxify them. Of course, while these studies raise intriguing new concepts in the interplay between CAR, the microbiota and mucosal homeostasis, much remains to be learned about whether these observations are due to CAR functions in IECs, mucosal immune cells and/or hepatocytes. In addition, it will be important to understand if microbiota-dependent alterations in mucosal CAR activity reflect direct responses to BAs or microbial metabolites. Clearly, future studies will be important to elucidate the diverse and cell type-specific functions, not only of dedicated BA receptors, but also of non-specific BA sensors during mucosal homeostasis and inflammation (Fig. 2).
The implications: targeting bile acids and their receptors in mucosal inflammatory diseases
BAs and their receptors have emerged as important targets for the treatment of human metabolic and inflammatory diseases. Ursodeoxycholic acid (UDCA) is an endogenous hydrophilic secondary BA produced at low levels in both humans and rodents, and has been used pharmacologically for many years in the treatment of a range of cholestatic disorders, including biliary atresia, to stimulate bile flow and protect hepatocytes from membrane-damaging BAs, such as LCA.40,88,157 Indeed, a series of recent reports indicate that UDCA is also protective in mouse models of IBDs. For example, UDCA ameliorates DSS-induced colitis in mice, and this is associated with expansion of anti-inflammatory enteric bacterial species, including cluster XIVa Clostridium and Akkermansia muciniphila, which are generally depleted in IBD patients.158,159 Whether the therapeutic effects of UDCA in rodent colitis models is translatable to human IBD patients, and the extent to which this activity involves direct effects on hepatic BA production, the gut flora, and/or the mucosal immune system remains to be elucidated. Nonetheless, these data raise the possibility that elevated BA accumulation in the intestinal mucosa, due to either altered BA metabolism or increased intestinal permeability, contribute to IBD-associated mucosal inflammation. Another BA derivative, NorUDCA, has marked anti-inflammatory and anti-fibrotic effects in PSC and NASH patients.160 The FXR agonist, obeticholic acid (OCA), represents the first-in-class synthetic BA receptor modulator, and has shown efficacy in clinical trials of primary biliary sclerosis (PBC), primary sclerosing cholangitis (PSC), and non-alcoholic steatohepatitis (NASH).66,89,161 Small molecule agonists of the dedicated transmembrane BA receptor, TGR5, are also in active clinical development and have shown promise in suppressing inflammation in mouse models of atherosclerosis, T2D and IBD; these have yet to enter clinical testing.79 Vitamin D3 supplementation has shown efficacy in treating some IBD patients, supporting further clinical studies of the molecular links between VDR and intestinal inflammation. PXR and CAR are also ligand-regulated nuclear receptors that appear to safeguard the intestinal mucosa during periods of BA overload. However, given that PXR and CAR also have established functions in hepatic drug metabolism and stimulating these nuclear receptors could trigger drug–drug interactions,162 further studies are needed to understand the risk:benefit ratios of systemically targeting these nuclear receptor pathways and to develop approaches that enable topical delivery of PXR and/or CAR agonists to sites of intestinal inflammation. Indeed the success of treating human IBDs with rifaximin,163,164,165 which activates PXR locally in the intestine but is poorly absorbed into circulation, provides a clinical proof-of-concept for targeting BA sensors in the intestinal mucosa. Finally, manipulating enterohepatic BA circulation, with either well-established BA sequestrants (e.g., cholestyramine) or more recently developed small molecule ASBT inhibitors, is another avenue for clinical intervention in circumstances where elevated BA levels in the intestinal mucosa trigger tissue injury.166,167 However, considering that low-level BA-dependent signaling (i.e., through FXR and TGR5) is generally protective, it is possible that the most effective BA-directed therapies for mucosal inflammatory disorders will involve a combination of intestinal BA reabsorption inhibitors and FXR/TGR5 agonists. Ultimately, while the use and utility of BA-directed therapies in human IBDs remains to be seen, the emergence of these modalities offer several points of clinical intervention and will continue to play a key role in the march toward a better, more comprehensive understanding of BAs in mucosal immune regulation.
Concluding remarks
BAs have emerged as important and pleotropic signaling metabolites involved in the regulation of metabolism and inflammation through interactions with both microbiota and host receptors. The dynamic three-dimensional interplay between BAs, the microbiome and the mucosal immune system represents an important new frontier in the field of Mucosal Immunology (Fig. 3). Still, focused efforts are needed to dissect the dynamics and functional consequences of the circulating BA pool in health and mucosal inflammatory disease, as well as to elucidate the relative functions of both dedicated BA receptors and non-specific BA sensors. If successful, these insights stand to generate a truly integrated view of mucosal inflammation that, in turn, can be leveraged for the design of more rational, personalized, and effective human therapies.

Three-dimensional interplay between bile acids, the microbiota, and the mucosal immune system. The mucosal immune system prevents bacterial translocation across the intestinal epithelium and regulates microbiota composition by sensing pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) and induction of barrier-enforcing cytokines and antimicrobial peptides (AMPs). Reciprocally, enteric commensals can produce immuno-modulatory metabolites (i.e., short chain fatty acids, SCFA, and indole derivatives) that shape mucosal immune function directly. Gut bacteria also metabolize primary bile acids (BAs) into more hydrophobic secondary BAs and can deconjugate BAs, which together influences the size and composition of the circulating BA pool. High concentrations of BAs, particularly those observed in the lumen of the small intestine, can directly lyse some enteric bacteria, thereby limiting small bowel bacterial colonization. BAs also directly shape mucosal immune function via interactions with both dedicated BA receptors (i.e., farsenoid X receptor /FXR, Takeda G-protein receptor 5/TGR5) and non-specific BA sensors (i.e., pregnane X receptor/PXR, vitamin D receptor/VDR, constitutive androstane receptor/CAR). Whether and how mucosal immune function impacts the synthesis and circulation of BAs is currently unknown and will be an important area of future investigation
References
Belkaid, Y. & Hand, T. W. Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014).
Belkaid, Y. & Segre, J. A. Dialogue between skin microbiota and immunity. Science 346, 954–959 (2014).
O’Dwyer, D. N., Dickson, R. P. & Moore, B. B. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J. Immunol. 196, 4839–4847 (2016).
Agace, W. W. & McCoy, K. D. Regionalized development and maintenance of the intestinal adaptive immune landscape. Immunity 46, 532–548 (2017).
Mowat, A. M. & Agace, W. W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 14, 667–685 (2014).
Postler, T. S. & Ghosh, S. Understanding the holobiont: how microbial metabolites affect human health and shape the immune system. Cell. Metab. 26, 110–130 (2017).
Zeissig, S. & Blumberg, R. S. Commensal microbiota and NKT cells in the control of inflammatory diseases at mucosal surfaces. Curr. Opin. Immunol. 25, 690–696 (2013).
Coombes, J. L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Suzuki, K. et al. Gut cryptopatches: direct evidence of extrathymic anatomical sites for intestinal T lymphopoiesis. Immunity 13, 691–702 (2000).
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Spits, H. et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Cerovic, V., Bain, C. C., Mowat, A. M. & Milling, S. W. Intestinal macrophages and dendritic cells: what’s the difference? Trends Immunol. 35, 270–277 (2014).
Veenbergen, S. & Samsom, J. N. Maintenance of small intestinal and colonic tolerance by IL-10-producing regulatory T cell subsets. Curr. Opin. Immunol. 24, 269–276 (2012).
Kjer-Nielsen, L. et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723 (2012).
Mora, J. R. et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006).
Konjar, S., Ferreira, C., Blankenhaus, B. & Veldhoen, M. Intestinal barrier interactions with specialized CD8 T cells. Front. Immunol. 8, 1281 (2017).
Kaser, A. & Blumberg, R. S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 140, 1738–1747 (2011).
Peterson, L. W. & Artis, D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14, 141–153 (2014).
Hooper, L. V. Epithelial cell contributions to intestinal immunity. Adv. Immunol. 126, 129–172 (2015).
Perez-Lopez, A., Behnsen, J., Nuccio, S. P. & Raffatellu, M. Mucosal immunity to pathogenic intestinal bacteria. Nat. Rev. Immunol. 16, 135–148 (2016).
Cerf-Bensussan, N. & Gaboriau-Routhiau, V. The immune system and the gut microbiota: friends or foes? Nat. Rev. Immunol. 10, 735–744 (2010).
Maloy, K. J. & Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Tsuei, J., Chau, T., Mills, D. & Wan, Y. J. Bile acid dysregulation, gut dysbiosis, and gastrointestinal cancer. Exp. Biol. Med. (Maywood). 239, 1489–1504 (2014).
Yoo, B. B. & Mazmanian, S. K. The enteric network: interactions between the immune and nervous systems of the gut. Immunity 46, 910–926 (2017).
Powell, N., Walker, M. M. & Talley, N. J. The mucosal immune system: master regulator of bidirectional gut-brain communications. Nat. Rev. Gastroenterol. Hepatol. 14, 143–159 (2017).
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323 (2009).
Marsland, B. J., Trompette, A. & Gollwitzer, E. S. The gut-lung axis in respiratory disease. Ann. Am. Thorac. Soc. 12, S150–S156 (2015). Suppl 2.
Wu, H. J. et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827 (2010).
de Aguiar Vallim, T. Q., Tarling, E. J. & Edwards, P. A. Pleiotropic roles of bile acids in metabolism. Cell. Metab. 17, 657–669 (2013).
Lefebvre, P., Cariou, B., Lien, F., Kuipers, F. & Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89, 147–191 (2009).
Fiorucci, S. & Distrutti, E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol. Med. 21, 702–714 (2015).
Swann, J. R. et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl Acad. Sci. USA 108, 4523–4530 (2011).
Begley, M., Gahan, C. G. & Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 29, 625–651 (2005).
Ridlon, J. M., Kang, D. J. & Hylemon, P. B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 47, 241–259 (2006).
Inagaki, T. et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl Acad. Sci. USA 103, 3920–3925 (2006).
Zollner, G., Marschall, H. U., Wagner, M. & Trauner, M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol. Pharm. 3, 231–251 (2006).
Zhu, C., Fuchs, C. D., Halilbasic, E. & Trauner, M. Bile acids in regulation of inflammation and immunity: friend or foe? Clin. Exp. Rheumatol. 34, 25–31 (2016).
Roberts, M. S., Magnusson, B. M., Burczynski, F. J. & Weiss, M. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin. Pharmacokinet. 41, 751–790 (2002).
Russell, D. W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 72, 137–174 (2003).
Paumgartner, G. Medical treatment of cholestatic liver diseases: From pathobiology to pharmacological targets. World J. Gastroenterol. 12, 4445–4451 (2006).
Chiang, J. Y. L. Bile acid metabolism and signaling in liver disease and therapy. Liver Res 1, 3–9 (2017).
Fischer, S., Beuers, U., Spengler, U., Zwiebel, F. M. & Koebe, H. G. Hepatic levels of bile acids in end-stage chronic cholestatic liver disease. Clin. Chim. Acta 251, 173–186 (1996).
Phillips, F., Muls, A. C., Lalji, A. & Andreyev, H. J. Are bile acid malabsorption and bile acid diarrhoea important causes of loose stool complicating cancer therapy? Colorectal. Dis. 17, 730–734 (2015).
Sigurdsson, V. et al. Bile acids protect expanding hematopoietic stem cells from unfolded protein stress in fetal liver. Cell. Stem. Cell. 18, 522–532 (2016).
Mertens, K. L., Kalsbeek, A., Soeters, M. R. & Eggink, H. M. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front. Neurosci. 11, 617 (2017).
Chiang, J. Y. Bile acids: regulation of synthesis. J. Lipid Res. 50, 1955–1966 (2009).
Takahashi, S. et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 57, 2130–2137 (2016).
Monte, M. J., Marin, J. J., Antelo, A. & Vazquez-Tato, J. Bile acids: chemistry, physiology, and pathophysiology. World J. Gastroenterol. 15, 804–816 (2009).
Dawson, P. A., Lan, T. & Rao, A. Bile acid transporters. J. Lipid Res. 50, 2340–2357 (2009).
Xiao, L. & Pan, G. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: the apical sodium-dependent bile acid transporter (SLC10A2/ASBT). Clin. Res. Hepatol. Gastroenterol. 41, 509–515 (2017).
Ballatori, N. et al. OST alpha-OST beta: a key membrane transporter of bile acids and conjugated steroids. Front. Biosci. 14, 2829–2844 (2009).
Hofmann, A. F. The enterohepatic circulation of bile acids in mammals: form and functions. Front. Biosci. 14, 2584–2598 (2009).
Lu, T. T. et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 6, 507–515 (2000).
Kliewer, S. A. & Mangelsdorf, D. J. Bile acids as hormones: the FXR-FGF15/19 pathway. Dig. Dis. 33, 327–331 (2015).
Fu, T. et al. FXR primes the liver for intestinal FGF15 signaling by transient induction of beta-klotho. Mol. Endocrinol. 30, 92–103 (2016).
Camilleri, M. Bile Acid diarrhea: prevalence, pathogenesis, and therapy. Gut Liver 9, 332–339 (2015).
Wahlstrom, A., Sayin, S. I., Marschall, H. U. & Backhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell. Metab. 24, 41–50 (2016).
Ilan, Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 18, 2609–2618 (2012).
Wigg, A. J. et al. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 48, 206–211 (2001).
Wyke, R. J. Problems of bacterial infection in patients with liver disease. Gut 28, 623–641 (1987).
Clements, W. D. et al. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut 39, 587–593 (1996).
Hackstein, C. P. et al. Gut microbial translocation corrupts myeloid cell function to control bacterial infection during liver cirrhosis. Gut 66, 507–518 (2017).
Hartmann, P., Haimerl, M., Mazagova, M., Brenner, D. A. & Schnabl, B. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology 143, 1330–1340 e1331 (2012).
Fouts, D. E., Torralba, M., Nelson, K. E., Brenner, D. A. & Schnabl, B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 56, 1283–1292 (2012).
Maillette de Buy Wenniger, L. & Beuers, U. Bile salts and cholestasis. Dig. Liver. Dis. 42, 409–418 (2010).
Hegade, V. S., Speight, R. A., Etherington, R. E. & Jones, D. E. Novel bile acid therapeutics for the treatment of chronic liver diseases. Therap. Adv. Gastroenterol. 9, 376–391 (2016).
Lorenzo-Zuniga, V. et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 37, 551–557 (2003).
Corpechot, C. Primary biliary cirrhosis and bile acids. Clin. Res. Hepatol. Gastroenterol. 36, S13–S20 (2012).
Watanabe, M., Fukiya, S. & Yokota, A. Comprehensive evaluation of the bactericidal activities of free bile acids in the large intestine of humans and rodents. J. Lipid Res. 58, 1143–1152 (2017).
Sannasiddappa, T. H., Lund, P. A. & Clarke, S. R. In vitro antibacterial activity of unconjugated and conjugated bile salts on Staphylococcus aureus. Front. Microbiol. 8, 1581 (2017).
Kurdi, P., Kawanishi, K., Mizutani, K. & Yokota, A. Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J. Bacteriol. 188, 1979–1986 (2006).
Tremblay, S. et al. Bile acid administration elicits an intestinal antimicrobial program and reduces the bacterial burden in two mouse models of enteric infection. Infect. Immun. 85, https://doi.org/10.1128/IAI.00942-16 (2017).
le Maire, M., Champeil, P. & Moller, J. V. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta 1508, 86–111 (2000).
Ding, L., Yang, L., Wang, Z. & Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 5, 135–144 (2015).
Parseus, A. et al. Microbiota-induced obesity requires farnesoid X receptor. Gut 66, 429–437 (2017).
Devkota, S. et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 487, 104–108 (2012).
Baron, E. J. et al. Bilophila wadsworthia, gen. nov. and sp. nov., a unique gram-negative anaerobic rod recovered from appendicitis specimens and human faeces. J. Gen. Microbiol. 135, 3405–3411 (1989).
Makishima, M. et al. Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 (1999).
Duboc, H., Tache, Y. & Hofmann, A. F. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig. Liver. Dis. 46, 302–312 (2014).
Makishima, M. et al. Vitamin D receptor as an intestinal bile acid sensor. Science 296, 1313–1316 (2002).
Staudinger, J. L. et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl Acad. Sci. USA 98, 3369–3374 (2001).
Kim, K. H. et al. Constitutive androstane receptor differentially regulates bile acid homeostasis in mouse models of intrahepatic cholestasis. Hepatol Commun 3, 147–159 (2019).
Saini, S. P. et al. A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol. Pharmacol. 65, 292–300 (2004).
Guo, G. L. et al. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 278, 45062–45071 (2003).
Li, T. & Chiang, J. Y. Nuclear receptors in bile acid metabolism. Drug Metab. Rev. 45, 145–155 (2013).
Zhang, J., Huang, W., Qatanani, M., Evans, R. M. & Moore, D. D. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J. Biol. Chem. 279, 49517–49522 (2004).
Han, S., Li, T., Ellis, E., Strom, S. & Chiang, J. Y. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol. Endocrinol. 24, 1151–1164 (2010).
Paumgartner, G. & Beuers, U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 36, 525–531 (2002).
Mudaliar, S. et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582 e571 (2013).
Steinmetz, A. C., Renaud, J. P. & Moras, D. Binding of ligands and activation of transcription by nuclear receptors. Annu. Rev. Biophys. Biomol. Struct. 30, 329–359 (2001).
Glass, C. K. & Rosenfeld, M. G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14, 121–141 (2000).
Nakanishi, T. & Tamai, I. Genetic polymorphisms of OATP transporters and their impact on intestinal absorption and hepatic disposition of drugs. Drug. Metab. Pharmacokinet. 27, 106–121 (2012).
Gadaleta, R. M. et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60, 463–472 (2011).
Vavassori, P., Mencarelli, A., Renga, B., Distrutti, E. & Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 183, 6251–6261 (2009).
Wang, Y. D. et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48, 1632–1643 (2008).
Gadaleta, R. M. et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim. Biophys. Acta 1812, 851–858 (2011).
Massafra, V. et al. Splenic dendritic cell involvement in FXR-mediated amelioration of DSS colitis. Biochim. Biophys. Acta 1862, 166–173 (2016).
Connor, S. J. et al. CCR2 expressing CD4+ T lymphocytes are preferentially recruited to the ileum in Crohn’s disease. Gut 53, 1287–1294 (2004).
Renga, B. et al. FXR mediates a chromatin looping in the GR promoter thus promoting the resolution of colitis in rodents. Pharmacol. Res. 77, 1–10 (2013).
Garcia-Irigoyen, O. & Moschetta, A. A novel protective role for FXR against inflammasome activation and endotoxemia. Cell. Metab. 25, 763–764 (2017).
Nijmeijer, R. M. et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS. ONE. 6, e23745 (2011).
Reschly, E. J. et al. Evolution of the bile salt nuclear receptor FXR in vertebrates. J. Lipid Res. 49, 1577–1587 (2008).
Kawamata, Y. et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440 (2003).
Perino, A. & Schoonjans, K. TGR5 and immunometabolism: insights from physiology and pharmacology. Trends Pharmacol. Sci. 36, 847–857 (2015).
Wang, Y. D., Chen, W. D., Yu, D., Forman, B. M. & Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology 54, 1421–1432 (2011).
McMahan, R. H. et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J. Biol. Chem. 288, 11761–11770 (2013).
Pols, T. W. et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell. Metab. 14, 747–757 (2011).
Yoneno, K. et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 139, 19–29 (2013).
Biagioli, M. et al. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J. Immunol. 199, 718–733 (2017).
Guo, C. et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45, 944 (2016).
Ichikawa, R. et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 136, 153–162 (2012).
Maruyama, T. et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 298, 714–719 (2002).
Halilbasic, E., Claudel, T. & Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 58, 155–168 (2013).
Venkatesh, M. et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 41, 296–310 (2014).
Lundin, A. et al. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol. 10, 1093–1103 (2008).
Cai, S. Y. et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2, e90780 (2017).
Hofmann, A. F. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 159, 2647–2658 (1999).
Ferrebee, C. B. et al. Organic solute transporter alpha-beta protects ileal enterocytes from bile acid-induced injury. Cell. Mol. Gastroenterol. Hepatol. 5, 499–522 (2018).
Cao, W. et al. The xenobiotic transporter Mdr1 enforces T cell homeostasis in the presence of intestinal bile acids. Immunity 47, 1182–1196 e1110 (2017).
Burk, O. et al. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol. Pharmacol. 67, 1954–1965 (2005).
Xu, C., Li, C. Y. & Kong, A. N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 28, 249–268 (2005).
Tolson, A. H. & Wang, H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 62, 1238–1249 (2010).
Simmons, J. D., Mullighan, C., Welsh, K. I. & Jewell, D. P. Vitamin D receptor gene polymorphism: association with Crohn’s disease susceptibility. Gut 47, 211–214 (2000).
Dring, M. M. et al. The pregnane X receptor locus is associated with susceptibility to inflammatory bowel disease. Gastroenterology 130, 341–348 (2006). quiz 592.
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
Dempsey, J. L. et al. Pharmacological activation of PXR and CAR down-regulates distinct bile acid-metabolizing intestinal bacteria and alters bile acid homeostasis. Toxicol. Sci. https://doi.org/10.1093/toxsci/kfy271 (2018).
Kliewer, S. A., Goodwin, B. & Willson, T. M. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr. Rev. 23, 687–702 (2002).
Shah, Y. M., Ma, X., Morimura, K., Kim, I. & Gonzalez, F. J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1114–G1122 (2007).
Terc, J., Hansen, A., Alston, L. & Hirota, S. A. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. Eur. J. Pharm. Sci. 55, 12–19 (2014).
Cheng, J., Shah, Y. M. & Gonzalez, F. J. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol. Sci. 33, 323–330 (2012).
Mencarelli, A. et al. Inhibition of NF-kappaB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur. J. Pharmacol. 668, 317–324 (2011).
Cheng, J. et al. Therapeutic role of rifaximin in inflammatory bowel disease: clinical implication of human pregnane X receptor activation. J. Pharmacol. Exp. Ther. 335, 32–41 (2010).
Huang, K. et al. Targeting the PXR-TLR4 signaling pathway to reduce intestinal inflammation in an experimental model of necrotizing enterocolitis. Pediatr. Res. 83, 1031–1040 (2018).
Wallace, K. et al. The PXR is a drug target for chronic inflammatory liver disease. J. Steroid. Biochem. Mol. Biol. 120, 137–148 (2010).
Cantorna, M. T., Zhu, Y., Froicu, M. & Wittke, A. Vitamin D status, 1,25-dihydroxyvitamin D3, and the immune system. Am. J. Clin. Nutr. 80, 1717S–1720S (2004).
Kim, J. H. et al. Implication of intestinal VDR deficiency in inflammatory bowel disease. Biochim. Biophys. Acta 1830, 2118–2128 (2013).
Kong, J. et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G208–G216 (2008).
Liu, W. et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J. Clin. Invest. 123, 3983–3996 (2013).
Prietl, B., Treiber, G., Pieber, T. R. & Amrein, K. Vitamin D and immune function. Nutrients 5, 2502–2521 (2013).
Hewison, M. Vitamin D and the immune system: new perspectives on an old theme. Endocrinol. Metab. Clin. North. Am. 39, 365–379 (2010).
Chun, R. F., Liu, P. T., Modlin, R. L., Adams, J. S. & Hewison, M. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front. Physiol. 5, 151 (2014).
Pols, T. W. H. et al. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS ONE. 12, e0176715 (2017).
Mackowiak, B. & Wang, H. Mechanisms of xenobiotic receptor activation: direct vs. indirect. Biochim. Biophys. Acta 1859, 1130–1140 (2016).
Tzameli, I., Pissios, P., Schuetz, E. G. & Moore, D. D. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell. Biol. 20, 2951–2958 (2000).
Mutoh, S. et al. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci. Signal. 6, ra31 (2013).
Sueyoshi, T., Kawamoto, T., Zelko, I., Honkakoski, P. & Negishi, M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J. Biol. Chem. 274, 6043–6046 (1999).
Honkakoski, P., Zelko, I., Sueyoshi, T. & Negishi, M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell. Biol. 18, 5652–5658 (1998).
Kawamoto, T. et al. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol. Cell. Biol. 19, 6318–6322 (1999).
Suino, K. et al. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol. Cell 16, 893–905 (2004).
Frank, C., Gonzalez, M. M., Oinonen, C., Dunlop, T. W. & Carlberg, C. Characterization of DNA complexes formed by the nuclear receptor constitutive androstane receptor. J. Biol. Chem. 278, 43299–43310 (2003).
Forman, B. M. et al. Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature 395, 612–615 (1998).
Hudson, G. M. et al. Constitutive androstane receptor regulates the intestinal mucosal response to injury. Br. J. Pharmacol. 174, 1857–1871 (2017).
Uehara, D. et al. Constitutive androstane receptor and pregnane X receptor cooperatively ameliorate DSS-induced colitis. Dig. Liver Dis., https://doi.org/10.1016/j.dld.2018.10.008 (2018).
Maglich, J. M. et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 278, 17277–17283 (2003).
Steegenga, W. T. et al. Maternal exposure to a Western-style diet causes differences in intestinal microbiota composition and gene expression of suckling mouse pups. Mol. Nutr. Food Res. 61, https://doi.org/10.1002/mnfr.201600141 (2017).
Camp, J. G. et al. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res. 24, 1504–1516 (2014).
Beuers, U., Boyer, J. L. & Paumgartner, G. Ursodeoxycholic acid in cholestasis: potential mechanisms of action and therapeutic applications. Hepatology 28, 1449–1453 (1998).
Png, C. W. et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 105, 2420–2428 (2010).
Van den Bossche, L. et al. Ursodeoxycholic acid and its taurine- or glycine-conjugated species reduce colitogenic dysbiosis and equally suppress experimental colitis in mice. Appl. Environ. Microbiol. 83, https://doi.org/10.1128/AEM.02766-16 (2017).
Fickert, P. et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J. Hepatol. 67, 549–558 (2017).
Nevens, F. et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N. Engl. J. Med. 375, 631–643 (2016).
Anzenbacher, P. & Anzenbacherova, E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 58, 737–747 (2001).
Shafran, I. & Burgunder, P. Adjunctive antibiotic therapy with rifaximin may help reduce Crohn’s disease activity. Dig. Dis. Sci. 55, 1079–1084 (2010).
Prantera, C. et al. Antibiotic treatment of Crohn’s disease: results of a multicentre, double blind, randomized, placebo-controlled trial with rifaximin. Aliment. Pharmacol. Ther. 23, 1117–1125 (2006).
Muniyappa, P., Gulati, R., Mohr, F. & Hupertz, V. Use and safety of rifaximin in children with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 49, 400–404 (2009).
Rao, A. et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci. Transl. Med. 8, 357ra122 (2016).
West, R. J. & Lloyd, J. K. The effect of cholestyramine on intestinal absorption. Gut 16, 93–98 (1975).
Acknowledgements
We thank members of the Sundrud lab for critical discussions and for review of the manuscript. This work was supported by grants from the National Institutes of Health (NIH; 1R01AI143821-01 and U01AI30830, sub-contract) and the Crohn’s and Colitis Foundation (CCF; Senior Research Award # 422515) to M.S.S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, M.L., Takeda, K. & Sundrud, M.S. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol 12, 851–861 (2019). https://doi.org/10.1038/s41385-019-0162-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-019-0162-4
This article is cited by
-
Integrated annotation prioritizes metabolites with bioactivity in inflammatory bowel disease
Molecular Systems Biology (2024)
-
Bile acid metabolites enhance expression of cathelicidin antimicrobial peptide in airway epithelium through activation of the TGR5-ERK1/2 pathway
Scientific Reports (2024)
-
Altered microbial bile acid metabolism exacerbates T cell-driven inflammation during graft-versus-host disease
Nature Microbiology (2024)
-
Gut microbiota-derived ursodeoxycholic acid alleviates low birth weight-induced colonic inflammation by enhancing M2 macrophage polarization
Microbiome (2023)
-
Akkermansia muciniphila protects mice against an emerging tick-borne viral pathogen
Nature Microbiology (2023)
