
Abstract
Junctional adhesion molecule-A (JAM-A) is a transmembrane glycoprotein expressed on leukocytes, endothelia, and epithelia that regulates biological processes including barrier function and immune responses. While JAM-A has been reported to facilitate tissue infiltration of leukocytes under inflammatory conditions, the contributions of leukocyte-expressed JAM-A in vivo remain unresolved. We investigated the role of leukocyte-expressed JAM-A in acute peritonitis induced by zymosan, lipopolysaccharide (LPS), or TNFα using mice with selective loss of JAM-A in myelomonocytic cells (LysM-Cre;Jam-afl/fl). Surprisingly, in LysM-Cre;Jam-afl/fl mice, loss of JAM-A did not affect neutrophil (PMN) recruitment into the peritoneum in response to zymosan, LPS, or TNFα although it was significantly reduced in Jam-aKO mice. In parallel, Jam-aKO peritoneal macrophages exhibited diminished CXCL1 chemokine production and decreased activation of NF-kB, whereas those from LysM-Cre;Jam-afl/fl mice were unaffected. Using Villin-Cre;Jam-afl/fl mice, targeted loss of JAM-A on intestinal epithelial cells resulted in increased intestinal permeability along with reduced peritoneal PMN migration as well as lower levels of CXCL1 and active NF-kB similar to that observed in Jam-aKO animals. Interestingly, in germ-free Villin-Cre;Jam-afl/fl mice, PMN recruitment was unaffected suggesting dependence on gut microbiota. Such observations highlight the functional link between a leaky gut and regulation of innate immune responses.
Similar content being viewed by others

Introduction
Junctional adhesion molecule-A (JAM-A or F11R) is a transmembrane glycoprotein that is expressed on the surface of a variety of cells including endothelia and epithelia, as well as on subsets of leukocytes including monocytes, lymphocytes, polymorphonuclear neutrophils (PMNs), platelets, and dendritic cells.1,2,3,4 Given its broad cellular and tissue expression pattern, a diversity of functions has been described for JAM-A. In epithelia and endothelia, JAM-A is enriched at tight junctions where it homodimerizes in cis as well as in trans to serve as a platform to recruit intracellular signaling molecules that regulate key functions including paracellular permeability to macromolecules, cell proliferation, and cell migration.5,6,7,8
In immune cells, JAM-A has been reported to facilitate leukocyte diapedesis in various inflammatory models. Function blocking JAM-A antibodies and genetic depletion of JAM-A in mice (Jam-aKO) have been shown to reduce leukocyte extravasation in inflammatory meningitis, thioglycollate-induced peritonitis, skin inflammation, or ischemia-reperfusion of heart and liver.3,9,10,11 In addition, there is considerable evidence that JAM-A-mediated effects on leukocyte recruitment are stimulus-specific.10,12,13 On a molecular level, the mechanism by which JAM-A mediates leukocyte trafficking is not completely elucidated. It has been shown that inflammatory conditions result in JAM-A redistribution in endothelial cells from the intercellular area to the apical surface facilitating interactions in trans with JAM-A on leukocytes or with leukocyte integrin lymphocyte function-associated antigen 1 (LFA-1).14 However, others have reported that endothelial intercellular adhesion molecule (ICAM)-1 preferentially binds LFA-1 rather than JAM-A, suggesting that JAM-A/LFA-1 interaction might be cell-context or stimulus-dependent.15 In addition, non-redundant functional roles for JAM-A in a variety of cells including platelets, dendritic cells, and PMN have been reported.16,17,18 Pertinent to this study, it has been shown that loss of JAM-A negatively regulates polarized migration of PMN, while having a positive effect on dendritic cell trafficking.9,19,20
Despite a number of previous reports, the role of leukocyte-expressed JAM-A in regulating PMN migration remains unclear. In this study, we investigated contributions of JAM-A in PMN recruitment using acute peritonitis induced by intraperitoneal (i.p.) administration of zymosan, lipopolysaccharide (LPS), or tumor necrosis factor alpha (TNFα) in mice after selective deletion of JAM-A in myeloid cells, including PMN (LysM-Cre;Jam-afl/fl). Surprisingly, no defect in PMN recruitment into the peritoneal cavity was observed in LysM-Cre;Jam-afl/fl mice compared to controls in response to zymosan, LPS, or TNFα. However, Jam-aKO mice displayed reduced recruitment of PMN into the peritoneum in response to zymosan, LPS, or TNFα, suggesting non-myeloid or extrinsic contributions of JAM-A in regulating PMN migration. Experiments directed at identifying the mechanism for reduced PMN recruitment in Jam-aKO mice revealed decreased production of PMN chemokine CXCL1 as well as impaired activation of nuclear factor- kB (NF-kB) from peritoneal macrophages derived from Jam-aKO mice in response to zymosan or LPS. Conversely, peritoneal macrophages derived from LysM-Cre;Jam-afl/fl mice displayed unaltered response compared to normal controls, consistent with leukocyte-independent JAM-A function(s) contributing to defective PMN recruitment in Jam-aKO mice. Given that Jam-aKO mice have enhanced intestinal permeability that is associated with increased bacterial translocation and adaptive immune compensation,21,22 we investigated whether a leaky gut in Jam-aKO mice contributed to the observed alteration in peritoneal macrophage response. Indeed, mice with selective JAM-A deficiency in the intestinal epithelium (Villin-Cre;Jam-afl/fl) had increased intestinal permeability, reduced peritoneal infiltration of PMN in response to zymosan or LPS, diminished levels of CXCL1 and impaired activation of NF-kB. Interestingly, we observed that peritoneal macrophages from germ-free Villin-Cre;Jam-afl/fl mice showed unaltered PMN migration into the peritoneal cavity and normal CXCL1 production. Taken together, these findings reveal a central role for intestinal barrier function in regulating peripheral innate immune responses that are dependent on microbial colonization of the gut.
Results
Leukocyte-expressed JAM-A is not necessary for PMN migration in vivo and in vitro
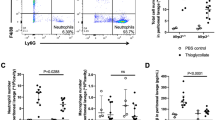
We used in vivo models of acute peritonitis to study PMN recruitment in mice with targeted deletion of JAM-A in myeloid cells (LysM-Cre;Jam-afl/fl). Selective loss of JAM-A expression was confirmed in bone marrow-derived PMN and monocytes (Supplemental Fig. 1). Unexpectedly, PMN migration into the peritoneal cavity 2 h after intraperitoneal (i.p.) injection of zymosan was comparable in LysM-Cre+;Jam-afl/fl mice and in littermate Jam-afl/fl controls (Fig. 1a). By contrast and consistent with a previous report,9 PMN infiltration into the inflamed peritoneal cavity after injection of zymosan was significantly reduced in Jam-aKO mice compared to Jam-aWT mice (Fig. 1b). Furthermore, PMN migration into the peritoneum was significantly reduced in Jam-aKO mice in response to i.p. injection of LPS or TNFα while PMN recruitment was unchanged in LysM-Cre+;Jam-afl/fl mice compared to controls (Fig. 1c, d). These results suggest that the defect in PMN recruitment in Jam-aKO mice was not stimulus-dependent and indicate a leukocyte-independent role for JAM-A in regulation of PMN recruitment in vivo.

JAM-A expression is not necessary for PMN migration in response to various pro-inflammatory stimuli. a, b Number of PMN in the peritoneal lavage in mice untreated (control) or 2 h post-injection with zymosan by flow cytometry. a Jam-afl/fl versus LysM-Cre+;Jam-afl/fl mice. b Jam-aWT versus Jam-aKO mice. Symbols represent individual mice. Data are pooled from four independent experiments and represent means ± SEM. ***P < 0.001 by two-way analysis of variance (ANOVA) with a Bonferroni multiple comparison post hoc test. ns: not significant. c, d Number of PMN in the peritoneal lavage 2 h post-injection with LPS (c) or 4 h post-injection with TNFα (d) by flow cytometry. Each data point represents individual mice from two independent experiments. Data represent means ± SEM. **P < 0.01 by two-way ANOVA with a Bonferroni multiple comparison post hoc test. ns: not significant. e, f Chemotactic migration of bone marrow neutrophils across collagen type I coated polycarbonate filters (5-μm pore size) towards a gradient of CXCL1 or LTB4. e Jam-afl/fl versus LysM-Cre+;Jam-afl/fl mice. f Jam-aWT versus Jam-aKO mice. Results are represented as the percentage of PMN that migrated into the bottom chambers that has been normalized to controls (Jam-a WT and Jam-afl/fl, respectively). Each dot represents the value for an individual experiment (N = 6 independent experiments ± SEM). Differences are not significant (ns) by two-way ANOVA with a Bonferroni multiple comparison post hoc test. PMN polymorphonuclear neutrophils, JAM-A Junctional adhesion molecule-A, LPS lipopolysaccharide
Since we did not observe reduced PMN recruitment into the inflamed peritoneum in LysM-Cre;Jam-afl/fl mice, we next determined if leukocyte-expressed JAM-A was necessary for PMN migration toward an inflammatory stimuli using an in vitro chemotaxis assay and gradients of the potent PMN chemoattractants CXCL123 or Leukotriene B4 (LTB4).24 Bone marrow-derived PMN were stimulated to migrate across collagen-coated transwell filters in response to CXCL1 or LTB4. Migration of PMN isolated from LysM-Cre;Jam-afl/fl (Fig. 1e) and Jam-aKO mice (Fig. 1f) was comparable to PMN from control mice. Altogether, these observations indicate that PMN-expressed JAM-A does not influence migration in response to chemoattractants in vitro and in vivo. Since Jam-aKO mice displayed reduced recruitment of PMN into the peritoneum in response to several pro-inflammatory stimuli, collectively these observations indicate non-myeloid or extrinsic contributions of JAM-A in regulating PMN migration.
CXCL1 generation is impaired in macrophages derived from Jam-a-deficient mice
Since it is well appreciated that zymosan is actively phagocytosed by peritoneal macrophages resulting in release of pro-inflammatory cytokines and chemokines that promote leukocyte (e.g., PMN and monocytes) recruitment,25,26 we investigated whether loss of JAM-A might alter generation of PMN chemokines during zymosan-induced peritonitis. We first examined whether there was altered distribution of immune cell populations in the peritoneal cavity of Jam-aKO mice compared to wild-type (WT) controls at basal condition. Jam-aKO mice and WT controls displayed similar numbers of macrophages, mast cells, T lymphocytes and B lymphocytes, indicating that immune cell populations are not changed within the peritoneal cavity of Jam-aKO mice (Supplemental Fig. 2). Given these observations, we evaluated production levels of major inflammatory mediators that promote PMN trafficking in peritoneal lavage fluid 2 h after injection of zymosan. In analyses of peritoneal lavage fluids, only CXCL1 was found significantly reduced in vivo in Jam-aKO mice compared to Jam-aWT mice (Fig. 2a and Supplemental Fig. 3a–c). Similarly, there was a significant decreased zymosan-stimulated generation of CXCL1 compared to CXCL2, interleukin (IL)-6, and TNFα in culture supernatants derived from peritoneal macrophages isolated from Jam-aKO mice and treated with zymosan ex vivo (Fig. 2c and Supplemental Fig. 3d–f). While a small decrease in TNFα was observed ex vivo, only CXCL1 showed consistent and significant decreases in vivo and ex vivo. Therefore, CXCL1 was used as a marker of peritoneal macrophage function in response to zymosan-induced peritonitis. CXCL1 levels were then assessed in peritoneal exudates as well as in culture supernatants from JAM-A-deficient peritoneal macrophages isolated from LysM-Cre;Jam-afl/fl mice (Supplemental Fig. 4) after administration of zymosan. CXCL1 levels were unaffected in LysM-Cre+;Jam-afl/fl and Jam-afl/fl control mice in vivo and ex vivo (Fig. 2b, d). Collectively, these results suggest that peritoneal macrophages derived from Jam-aKO mice after zymosan administration have reduced CXCL1 generation that may contribute to the decreased intraperitoneal PMN recruitment observed Jam-aKO mice. However, since CXCL1 generation by JAM-A-deficient macrophages derived from LysM-Cre;Jam-afl/fl mice was not impaired, these findings are consistent with non-myeloid or extrinsic contributions of JAM-A in regulating PMN migration in response to zymosan.

CXCL1 production is reduced in Jam-aKO mice while unaffected in LysM-Cre+;Jam-afl/fl mice. a, b Quantitative measurement of CXCL1 expression in vivo by ELISA in peritoneal exudates from untreated mice or after 2 h post-injection of zymosan. a Jam-aWT versus Jam-aKO mice. Symbols represent individual mice. Data are pooled from four independent experiments and represent means ± SEM. ***P < 0.001 by two-way analysis of variance (ANOVA) with a Bonferroni multiple comparison post hoc test. ns: not significant. b Jam-afl/fl versus LysM-Cre+;Jam-afl/fl mice. Symbols represent individual mice and data are pooled from four independent experiments. Data represent means ± SEM. Differences are not significant by two-way ANOVA with a Bonferroni multiple comparison post hoc test. c, d Quantitative measurement of CXCL1 expression by ELISA in cell media supernatants of isolated peritoneal macrophages from mice either untreated or treated ex vivo with zymosan for 1 h. c Jam-aWT versus Jam-aKO mice mice. Symbols represent individual mice from three independent experiments. Data represent means ± SEM. ***P < 0.001 by two-way ANOVA with a Bonferroni multiple comparison post hoc test. d Jam-afl/fl versus LysM-Cre+;Jam-afl/fl mice. Symbols represent individual mice from two independent experiments. Data represent means ± SEM. Differences are not significant (ns) by two-way ANOVA with a Bonferroni multiple comparison post hoc test
Impaired macrophage-dependent neutrophil recruitment is secondary to increased intestinal permeability in Jam-a-deficient mice
Since it has been shown that Jam-a-deficient mice have increased intestinal permeability and bacterial translocation but do not develop spontaneous colitis due to compensatory adaptive immune responses,21,22 we hypothesized that the leaky gut in Jam-aKO mice may play a role in regulating the above response in peritoneal macrophages. To test this hypothesis, we first determined that intestinal barrier function in LysM-Cre;Jam-afl/fl mice as assessed by permeability to 4 kDa FITC-dextran, was not impaired in LysM-Cre+;Jam-afl/fl mice (Supplemental Fig. 5). Then, we conducted experiments with mice with selective deletion of JAM-A in the intestinal epithelium (Villin-Cre;Jam-afl/fl mice). Despite lacking intestinal epithelial JAM-A, these mice retain normal expression of JAM-A in endothelia and immune cells (Fig. 3a, b). Analogous to Jam-aKO mice and consistent with a recent report,27 Villin-Cre+;Jam-afl/fl mice have a leaky intestinal barrier as shown by enhanced permeability to 4 kDa FITC-dextran (Fig. 3c). Peritonitis experiments revealed that targeted loss of JAM-A on intestinal epithelial cells (IECs) resulted in reduced PMN recruitment into the peritoneal cavity in response to zymosan or LPS in Villin-Cre+;Jam-afl/fl (Fig. 3d). To determine the effects of a leaky intestinal epithelial barrier on macrophage function, we isolated peritoneal macrophages from Villin-Cre;Jam-afl/fl mice and analyzed CXCL1 protein and messenger RNA (mRNA) levels in response to zymosan. While comparable JAM-A mRNA levels were observed in peritoneal macrophages from Villin-Cre+;Jam-afl/fl mice compared to controls (Supplemental Fig. 6), we observed significantly reduced CXCL1 mRNA and protein generation in macrophages isolated from Villin-Cre+;Jam-afl/fl mice (Fig. 3e, f). Interestingly, under germ-free conditions, PMN infiltration in the peritoneum remained unaltered in response of zymosan or LPS in Villin-Cre+;Jam-afl/fl mice compared to littermate controls (Fig. 3g). Furthermore, the amount of CXCL1 produced by peritoneal macrophages derived from germ-free Villin-Cre+;Jam-afl/fl mice was unchanged compared to control mice after zymosan treatment ex vivo (Fig. 3h, i). Collectively, these observations suggest important contributions of gut permeability and microbiota in regulating macrophage responses to microbial stimuli at distant sites such as the peritoneum.

Loss of JAM-A on intestinal epithelial cells increases intestinal permeability and results in decreased recruitment of PMN in models of acute peritonitis as well as diminished production of CXCL1 by peritoneal macrophages that is dependent on the presence of gut microbiota. a JAM-A expression (green) in the ileum of Jam-afl/fl versus Villin-Cre+;Jam-afl/fl mice. Shown are representative images of immunostained tissue sections. Nuclei are stained with Dapi (blue). Asterisks represent crypt lumen. Arrows show vessels and immune cells in the sub-epithelial space (lamina propria). Scale bars are 100 μm. b Expression of JAM-A mRNA in intestinal epithelial cells and lamina propria-enriched fractions from ileal tissue of Jam-a fl/fl versus Villin-Cre+;Jam-afl/fl mice. Symbols represent individual mice from two independent experiments. Data are means ± SEM. ns; not significant. ***P < 0.001 by two-way ANOVA with a Bonferroni multiple comparison post hoc test. c Permeability to FITC-dextran (4 kDa) using an ileal loop in vivo model in Jam-afl/fl versus Villin-Cre+;Jam-afl/fl mice. Symbols represent individual mice and data are pooled from four independent experiments. Data are means ± SEM. **P < 0.01 by Student’s t-test. d Number of PMN in the peritoneal cavity of Villin-Cre+;Jam-afl/fl and control mice 2 h post-injection with zymosan or LPS by flow cytometry. Each data point represents individual mice from two independent experiments. Data represent means ± SEM. **P < 0.01, *P < 0.05 by Student’s t-test. Expression of CXCL1 by ELISA (e) or mRNA level (f) in peritoneal macrophages from Jam-afl/fl versus Villin-Cre+;Jam-afl/fl mice either treated or untreated ex vivo with zymosan for 1 h. Symbols represent individual mice and data are pooled from three independent experiments (e) or two independent experiments (f). Data are means ± SEM. ns; not significant. ***P < 0.001 by two-way ANOVA with a Bonferroni multiple comparison post hoc test. g Number of PMN in the peritoneal cavity of germ-free (GF) Villin-Cre+;Jam-afl/fl and control mice 2 h post-injection with zymosan or LPS by flow cytometry. Each data point represents an individual mouse from two independent experiments. Data are means ± SEM. ns; not significant by Student’s t-test. Expression of CXCL1 by ELISA (h) or mRNA level (i) in peritoneal macrophages from germ-free (GF) Jam-afl/fl versus Villin-Cre+;Jam-afl/fl mice, either treated or untreated ex vivo with zymosan for 1 h. Each data point represents an individual mouse from two independent experiments. Data are means ± SEM. ***P < 0.001 by two-way ANOVA with a Bonferroni multiple comparison post hoc test. ns; not significant. ANOVA analysis of variance, LPS lipopolysaccharide
Altered NF-kB signaling in peritoneal macrophages from Jam-a KO mice and Villin-Cre+; Jam-a fl/fl mice
To further understand the basis of decreased CXCL1 generation in JAM-A-deficient peritoneal macrophages, we tested whether phagocytosis of zymosan particles was altered. As seen in Supplemental Fig. 7, phagocytosis of zymosan particles was similar in peritoneal macrophages from Jam-aWT and Jam-aKO mice. We next evaluated the effect of zymosan or LPS on activation of signaling pathways in peritoneal macrophages and with a focus on NF-kB-dependent signaling, which is known to regulate gene expression for multiple cytokines and chemokines including CXCL1.28,29 As shown in Fig. 4a, d, a time course analysis of NF-kB activation in peritoneal macrophages after zymosan or LPS stimulation revealed enhanced Serine 536 phosphorylation of the NF-kB subunit p65 (p65 pSer536) in WT animals that was reduced in macrophages derived from Jam-aKO mice. Similarly, targeted loss of JAM-A on IECs resulted in significant reduction in p65 pSer536 in peritoneal macrophages derived from Villin-Cre+;Jam-afl/fl compared to Jam-afl/fl controls in response to zymosan or LPS (Fig. 4b, e). Conversely, NF-kB activation was not altered in LysM-Cre+;Jam-afl/fl compared to Jam-afl/fl controls (Fig. 4c, f). Taken together, these findings strongly suggest that peritoneal macrophages from Jam-aKO as well as Villin-Cre+;Jam-afl/fl mice have reduced NF-kB activity resulting in dampened CXCL1 production and PMN recruitment into the peritoneal cavity in response to zymosan.

Impaired NF-kB activation in peritoneal macrophages from Jam-aKO and Villin-Cre+;Jam-afl/fl mice in response to zymosan and LPS. Peritoneal macrophages from Jam-aKO, Villin-Cre+;Jam-afl/fl, LysM-Cre+;Jam-afl/fl and controls were either untreated or stimulated with zymosan for 5 and 10 min (a–c) or with LPS for 15 and 30 min (d–f). Protein cell lysates were collected, separated by SDS-PAGE and immunoblotted for NF-kB p65 pSer536, NF-kB p65 total, and GAPDH as a loading control. Data are a representative immunoblots from three independent experiments. Graphs represent densiometric analysis for values of NF-kB p65 pSer536 normalized to p65 total. Data are means ± SEM of three independent experiments (3–5 mice per group). *P < 0.05, **P < 0.01 by two-way ANOVA with a Bonferroni multiple comparison post hoc test. LPS lipopolysaccharide, NF-kB nuclear factor- kB, SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
Discussion
Here, we report new observations on the role of JAM-A in PMN recruitment in vivo in response to multiple inflammatory stimuli using tissue-targeted and total knockout mice. Surprisingly, selective loss of JAM-A on myeloid cells failed to show any defect in PMN infiltration in contrast to significantly reduced PMN infiltration in the peritoneum of Jam-aKO mice. We conclude that expression of JAM-A on PMN does not contribute to recruitment in the peritoneum following zymosan, LPS, and TNFα. This conclusion is supported by observations that loss of JAM-A on PMN from either Jam-aKO or LysM-Cre;Jam-afl/fl animals did not prevent migration in response to CXCL1 or LTB4 in a transwell-based chemotaxis assay. These findings are consistent with another study using dendritic cells (DCs) isolated from Jam-aKO mice that showed normal chemotactic responsiveness to CCL19 and CCL3 in a three-dimensional transwell system; however, JAM-A-null DCs were shown to exhibit increased random motility in another 2-D-based assay.19 Similarly, there are two previous studies that have implicated expression of JAM-A on PMN as being necessary for polarized migration.9,20 A plausible explanation for the lack of effect of JAM-A on PMN migration in this report is that we used a three-dimensional transmigration assay while the others used linear two-dimensional assays on solid surfaces. In addition, in Jam-aKO animals, it has been reported that the overall numbers of transmigrated PMN in response to LTB4 is unchanged compared to controls yet interstitial migration of PMN was impaired20 suggesting contributions of the extracellular substrate in JAM-A-dependent PMN migration. Noteworthy, it has been reported that JAM-A-dependent PMN migration is stimulus-specific.13 Although, the mechanisms that control stimulus-dependent ability of JAM-A to mediate leukocyte transmigration are unknown, it has been suggested that stimuli that activate directly endothelial cells (such as IL-1β) may involve JAM-A and other endothelial cell adhesion molecules like PECAM-1, while stimuli that directly activate neutrophils (such as LTB4 or TNFα) are independent of JAM-A or PECAM-1, and may use other mechanisms.13,30 In addition, JAM-A has been reported to control integrin functions in platelets and PMN.17,18,20 Since integrins are well-known to play an important role in cell adhesion and migration on extracellular substrates, presumably, stimulus-dependent modulation of the interaction between JAM-A and integrins (or integrin binding partners) might also contribute to the ability of JAM-A to mediate leukocyte migration.
Another important observation in this study is reduced peritoneal macrophage production of CXCL1 in response to zymosan in JAM-A-null mice in vivo and ex vivo. These findings suggest that impaired CXCL1 production is responsible for the decreased PMN infiltration in the peritoneum. Surprisingly, JAM-A-null peritoneal macrophages from LysM-Cre;Jam-afl/fl mice did not exhibit a defect in the ability to produce CXCL1 in response to zymosan implying that macrophage-expressed JAM-A does not contribute to the findings in this inflammatory model. Thus, we concluded that tissue microenvironment or exogenous factors are responsible for the observed reduction of PMN infiltration in Jam-aKO mice. These results highlight the importance of using tissue-targeted knockout mice in future studies on the characterization of the specific contributions of JAM-A in leukocyte subsets on observed phenotypes in animal models of inflammation.
JAM-A is also abundantly expressed on epithelia as well as endothelia, and there are multiple reports of a non-redundant role for JAM-A in the maintenance of vascular permeability and epithelial barrier function.1,6 Presumably, total loss of JAM-A in Jam-aKO animals may generate various defects that are restrained by compensatory mechanisms to maintain homeostasis and prevent pathophysiologic outcomes. This idea is supported by our previous studies that demonstrated compensatory mechanisms in the intestine of Jam-aKO mice. In Jam-a-deficient animals, the colonic mucosa has normal architecture yet there is increased intestinal permeability, increased numbers of lymphoid aggregates, enhanced PMN infiltration, and increased bacterial translocation.21,22 Noteworthy, Jam-aKO animals are protected from spontaneous intestinal inflammation due to compensatory mechanisms driven by mucosal T and B lymphocytes, increased TGF-β production and enhanced IgA secretion that presumably results in neutralization of the harmful effects of the exposure to luminal antigens and bacterial products secondary to the intestinal leaky barrier.21 While there is mounting evidence that increased permeability of the gut barrier contributes to many inflammatory disorders, there are several studies concluding that a leaky intestine alone is insufficient to induce chronic inflammatory conditions because of compensatory changes and safety mechanisms that lead to protective responses.31,32,33,34
In order to determine if the tissue microenvironment in Jam-aKO animals might modulate peritoneal macrophage responses to zymosan and LPS we produced Villin-Cre;Jam-afl/fl mice that harbor selective depletion of JAM-A on intestinal epithelial cells (IECs) while maintaining normal expression on other cell types, notably endothelia, and leukocytes. Selective loss of JAM-A on IECs resulted in impaired intestinal barrier function as evident by increased paracellular permeability to 4 kDa dextran. We also observed that depletion of JAM-A on IECs was sufficient to diminish peritoneal PMN migration in vivo in response to zymosan or LPS and dampen the production of CXCL1 by peritoneal macrophages. Interestingly, peritoneal macrophages isolated from germ-free Villin-Cre;Jam-afl/fl animals did not exhibit any defect in PMN infiltration into the peritoneal cavity following stimulation with zymosan or LPS as well as no defects in production of CXCL1. These observations suggest a role for gut microbiota in repressing peritoneal macrophage response to inflammatory stimuli and, therefore, in modulation of immune responses. However, further investigations are needed to determine mechanisms by which gut microbiota-derived factors may regulate peritoneal innate responses in Jam-aKO and Villin-Cre;Jam-afl/fl animals.
Nevertheless, our findings are consistent with several reports that have demonstrated that enteric bacteria are linked to development of colitis35,36,37,38 as well as a previous study demonstrating that antibiotic-treated Jam-aKO Rag1-/- mice are less susceptible to dextran sulfate sodium (DSS)-induced colitis when compared to mice not treated with antibiotics.21 It is likely that bacterial derived products such as LPS are important in controlling the macrophage responses observed as it was recently shown that Jam-aKO mice develop severe steatohepatitis when placed on a diet high in saturated fat, fructose, and cholesterol (HFCD). This effect was shown dependent on activated hepatic macrophages, elevated circulating LPS, and gut microbes.39 In addition, bacterial derived products are linked to release of pro-inflammatory immune mediators such as interferon (IFN)-γ that potently influence innate immune responses. In a recent study, priming of peritoneal macrophages by exogenous IFN-γ treatment prior to LPS administration resulted in impaired NF-kB transcriptional activity, selective decrease in production of CXCL1 and reduced LPS-induced PMN recruitment.40 The authors also demonstrated that IFN-γ priming of macrophages lead to epigenetic modifications that causes changes in the inflammatory repertoire of the macrophages.40 Similarly, our observations showed that in response to zymosan, peritoneal macrophages from Jam-aKO and Villin-Cre+;Jam-afl/fl exhibited reduced production of CXCL1 and impaired NF-kB signaling compared to controls. Of note, in response to zymosan, we observed no consistent and statistically significant decrease in secretion of other pro-inflammatory factors that are driven by NF-kB activity such as CXCL2, IL-6, and TNFα in vivo and ex vivo in Jam-aKO mice compared to control (Supplemental Fig. 3). These observations suggest that the phenotype in Jam-aKO mice might be secondary to priming of peritoneal macrophages and involve epigenetic modifications that cause a selective downregulation of specific genes and not a global shutdown of macrophage responses to zymosan.
The functional consequences of IFN-γ priming of macrophages in inflammatory diseases are complex and incompletely understood. Some studies have reported deleterious effects of type I interferons (IFNs) produced during viral infections that suppress innate protection against subsequent bacterial pathogens and sensitize hosts to secondary bacterial superinfections. Type I IFNs have also been shown to induce decreased production of CXCL1 and CXCL2 leading to impaired neutrophil responses.41,42 By contrast, bacterial endotoxin has been reported to support systemic immunity. Preconditioning with LPS results in an endotoxin tolerance phenotype that has been reported to enhance bacterial clearance and improve survival in murine models of polymicrobial sepsis.43,44,45 Although the molecular mechanisms have not been fully elucidated, there is increasing evidence that pretreatment/priming with LPS results in attenuation of inflammation by specific reprogramming of immune cells and inhibition of NF-kB activation.44
Whether loss of JAM-A in Jam-aKO or Villin-Cre;Jam-afl/fl animals might be protective or detrimental for the immune response to virus and bacteria remains to be investigated in future studies. Analogous mechanisms may be at play in immune mediated tumor surveillance as it has been reported that total loss of JAM-A supports stronger immunologic responses against pancreatic tumors: Rip1Tag2 mice prone to pancreatic beta-cell carcinogenesis crossed with JAM-A-deficient mice developed less tumor growth than Jam-aWT mice. The phenotype was attributed to an enhanced infiltration of DCs and T cells as well as reduced angiogenesis.46 These observations strongly imply that Jam-a-deficient animals are useful tools to unravel mechanisms that protect against inflammatory burden and tumorigenesis.
In conclusion, we provide evidence that impaired intestinal barrier function secondary to selective loss of intestinal epithelial JAM-A downregulates neutrophil recruitment into the peritoneum in response to pro-inflammatory stimuli by repressing NF-kB activity and reducing production of inflammatory mediators by peritoneal macrophages. These findings highlight a critical functional link between the intestinal barrier and regulation of systemic innate immune responses.
Methods
Animals
Experimental studies were conducted in C57BL/6 wild-type mice (Jam-aWT), JAM-A total knockout mice (Jam-aKO),22 tissue-targeted JAM-A-deficient mice under the control of the murine intestinal epithelial villin promoter (Villin-Cre; Jam-afl/fl) or in the myeloid cell lineage (LysM-Cre;Jam-afl/fl), and respective littermate controls (Jam-afl/fl). Villin-Cre; Jam-afl/fl mice were generated by crossing Jam-afl/fl mice with Villin-Cre transgenic mice.47 LysM-Cre;Jam-afl/fl mice were generated by crossing Jam-afl/fl mice with LysM-Cre transgenic mice purchased from Jackson Laboratory (Bar Harbor, ME). Either LysM-Cre;Jam-afl/fl or Villin-Cre; Jam-afl/fl were backcrossed with Jam-afl/fl mice for at least ten generations. Animals (8–12 weeks, both genders) were used in this study and maintained under standard conditions with 12 h day/night cycle with access to food and water ad libitum. LysM-Cre;Jam-afl/fl or Villin-Cre; Jam-afl/fl and respective littermate controls (Jam-afl/fl mice) were co-housed together. Bedding containing feces was mixed between Jam-aWT and Jam-aKO mice. All experimental procedures were performed in accordance with the protocols approved by the University Committee on Use and Care of Animals at University of Michigan.
In vivo models of peritonitis
Zymosan-induced peritonitis was induced as described previously with modifications.48 Briefly, a solution of 20 μg/mL zymosan A from Saccharomyces cerevisiae (Sigma, St. Louis, MO) in sterile phosphate-buffered saline (PBS- Corning, NY) was prepared and autoclaved at 121 °C for 20 min. Two-hundred microliters of zymosan suspension (5 μg of zymosan) was administered by intraperitoneal (i.p.) injection. Mice were divided in two groups, untreated (injected with PBS only) or 2 h post-injection with zymosan. Mice were euthanized by exposure to isoflurane and peritoneal exudate was harvested by lavage with 5 mL of sterile cold PBS supplemented with 2 mM EDTA (Lonza, Rockland, ME) followed by cervical dislocation. Peritoneal exudates were centrifuged at 300 × g for 10 min at 4 °C: pellet-containing cells was processed for flow cytometric analysis while the supernatant was collected for chemokine/cytokine production measurement by enzyme-linked immunosorbent assay (ELISA). For lipopolysaccharide (LPS)-induced peritonitis, 250 μL of LPS suspension (from Escherichia coli O111:B4; Sigma) at 10 ng/mL was administered by i.p. injection. Mice were treated for 2 h with LPS. For TNFα-induced peritonitis, 250 μL of animal-free recombinant murine TNF-α suspension at 10 ng/mL (PeproTech, Rocky Hill, NJ) was administered by i.p. injection. Mice were treated for 4 h with TNF-α.
Naive peritoneal macrophage cell culture and stimulation with zymosan
Peritoneal lavage was collected with 5 mL of sterile PBS/ 2 mM EDTA on untreated mice (naive) euthanized by isoflurane and prior cervical dislocation. After centrifugation at 300 × g for 10 min (4 °C), pellet was resuspended in PBS/ 2 mM EDTA and cells were counted using a handled automated cell counter (Scepter TM, EMD Millipore, Billerica, MA). In all, 2 × 106 cells were collected and after centrifugation were resuspended in 1 mL of RPMI medium containing RPMI medium 1640 (Corning) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, GA) and 1% Penicillin/Streptomycin (Corning). Cell suspension (106 cells) was added in 12-well flat-bottom plates and transferred for 45 min to a 5% CO2 incubator at 37 °C. Adherent cells (~85–90% of peritoneal macrophages) were washed twice with RPMI medium then untreated or treated with 20 μg/mL of zymosan in RPMI medium for 1 h at 37 °C. After incubation with zymosan, cell media were harvested, spun down at 300 × g for 10 min at 4 °C and supernatants were collected to quantify the production of chemokines/cytokines by ELISA. Adherent peritoneal macrophages were washed twice with cold PBS/ 2 mM EDTA and processed for RNA isolation. Results show data from the enrichment of peritoneal macrophages from each individual mouse. However, same results were obtained by pooling peritoneal exudates from 3–5 mice followed by specific isolation of unlabeled macrophages by using Macrophage Isolation Kit (peritoneum) from MACs/Miltenyi Biotec (Bergisch Gladbach, Germany).
Chemotaxis assay of mouse bone marrow neutrophils
Neutrophils were isolated from bone marrow (BMN) by using a density gradient centrifugation technique. Briefly, bone marrow cells from femurs and tibias were flushed with RPMI supplemented with 5% FBS and 1 mM EDTA, and passed through a 70 μm strainer (Falcon, Corning, NY). Cell suspension was centrifuged at 1500 rpm for 5 min and red blood cells were lysed with ACK lysis buffer (Lonza). Cells were resuspended in 1 mL of ice-cold PBS without Ca2+ or Mg2+ and layered over a separation media formed by 3 mL of Histopaque 1077 (1.077 g/mL; Sigma) overlaid on 3 mL of Histopaque 1119 (1.119 g/mL) in a 15-mL tube-column. The column was centrifuged at 2000 rpm for 20 min at 25 °C and BMN at the interphase of the two Histopaques were collected, washed in PBS and counted. Then, BMN chemotaxis assay was performed using collagen-coated (10 μg/mL rat tail collagen), permeable 0.33-cm2 polycarbonate filters (5-μm pore size; Costar Corp., Corning, NY). A total of 106 BMNs were added to the upper chambers of transwell filter and migration to the lower chamber was stimulated for 2 h at 37 °C in the presence of a chemotactic gradient of 10 nmol/L of LTB4 (Cayman, Ann Arbor,MI) or 2 μg/mL of CXCL1/KC (R&D systems). Migrated BMNs were quantified by detecting the PMN azurophilic marker MPO, as published previously49 and color development was assayed at 405 nm on a microtiter plate reader. The rate of migration was calculated as the percentage of the total BMN added to the upper chamber of the transwell filter that have migrated into the bottom.
Flow cytometry and data analysis
Cellular composition of peritoneal exudates was determined by incubating the cells in FACS buffer (PBS, 2% FBS) with fluorescent conjugated antibodies against well-accepted surface markers for PMN (Ly6G + CD11b), monocytes (Ly6C + CD11b), T lymphocytes (CD3 + TCRβ), B lymphocytes (CD19), macrophages (F4/80 + CD11b), or Mast cells (c-Kit and Fc Receptor ε). Antibodies used were purchased from eBioscience (San diego. CA): CD11b–eFluor 450, F4/80-PE, TCRβ-FITC, CD19- PE, c-kit-APC, CD45- PerCP and Fc Recptor ε- PerCP-eFluor710, Rat anti-mouse isotype controls (IgG), and Ly6G- APC (1A8; BD Bioscience, San Jose, CA). Cells were mixed with cell counting beads (30,000 beads per sample, CountBrignt Absolute Counting Beads, Invitrogen). Sample were analyzed by NovoCyte Flow Cytometer (ACEA Bioscience, San Diego, CA) and data processed with FlowJo (FLOWJO, LLC). The absolute number of cells was determined by using fluorescent counting beads as recommended by the manufacturer (CountBright, Life Technologies, Carlsbad, CA). For assessment of the depletion of JAM-A on bone marrow-derived neutrophils and monocytes or T cells from spleen, cells were pooled (3–5 mice per group) and specifically enriched using magnetic-activated cell sorting (Miltenyi Biotec, Germany). Cells were stained with JAM-A-PE (BD Bioscience) in combination with Ly6G-APC + CD11b-FITC (PMN), Ly6C-APC + CD11b-FITC (monocytes), CD3-FITC (T lymphocytes), then analyzed by flow cytometry.
Enzyme-linked immunosorbent assay (ELISA)
ELISA were performed following manufacturer’s instruction (R&D systems) to quantify the production of murine CXCL1/KC (DY453), CXCL2 (DY452), IL-6 (DY406), and TNFα (DY410). As indicated, the expression was measured either in cell free exudate supernatants of peritoneal lavage from mice untreated or treated with zymosan in vivo, or in culture media from enriched peritoneal macrophages untreated or treated with zymosan ex vivo.
RNA isolation and real-time quantitative PCR
Total RNA was isolated from 500,000 enriched peritoneal macrophages using the Qiagen RNeasy Micro Plus kit (Hilden, Germany) then followed by cDNA synthesis using 4 μL of total RNA with the BioRAD iScript™ Reverse Transcription supermix (Hercules, CA). Semi-quantitative real-time PCR was performed using 1 μL of the generated cDNA pool using iQ SYBR Green Master Mix (BioRAD) on a BioRAD CFX Connect Real-time System using the following intron spanning primers following amplification program: 95 °C for 2 min, 95 °C for 5 s, 60 °C for 30 s (40 cycles) followed by a melt curve program. Cq values were determed using a BioRAD CFX Manager 3.1 software. Relative mRNA expression was computed using the 2(-ΔΔCT) method using Rps18 as reference gene.
(Cxcl1-F) 5′-TGCACCCAAACCGAAGTCATAG-3′;
(Cxcl1-R) 5′-CAGAGCAGTCTGTCTTCTTTCTCC-3′;
(F11r-F) 5′-TCTGTGACCCGGAAGGACAATG-3′;
(F11r-R) 5′-TCGGCTTGGATGGAGGTACAAG-3′;
(Rps18-F) 5′-ACTTTTGGGGCCTTCGTGTC-3′;
Rps18-R) 5′-GCCCAGAGACTCATTTCTTCTTG-3′:
In vivo permeability assay
Intestinal permeability was assessed by injection of fluorescein isothiocyanate (FITC) conjugated dextran (FD4; Sigma) using an ileal loop model as described previously.50 Briefly, animals were anesthetized with isoflurane (Fluriso™, VETONE®, Boise, Idaho) at constant rate using a rodent anesthesia vaporizer machine (E-Z anesthesia® 7000, Palmer, PA) and placed on a controlled temperature heat pad. A midline abdominal incision was made to expose the ileum, and a 4-cm loop was exteriorized, cleaned, and cut-ends were closed by ligations using non-absorbable silk suture 3.0 (Braintree Scientific, Braintree, MA). In all, 1 mg/mL FITC-dextran (FD4) in 200 μL Hank’s Buffered Salt Solution (HBSS) was injected into the ileal loop. The loop was re-inserted into the abdominal cavity then peritoneum and skin were closed with sutures. After 2 h, whole blood was collected by cardiac puncture and fluorescence intensity was measured in the serum using a fluorescence microtiter plate reader (FLUOstar; BMG Labtech, Cary, NC).
Immunofluorescence microscopy
In all, 8-μm OCT-embedded tissue sections of mouse ileum were fixed and permeabilized in absolute ethanol at −20 °C for 20 min, blocked with 3% donkey serum-PBS for 1 h at room temperature (RT) and incubated with primary antibody goat anti-mouse JAM-A (R&D systems) diluted in blocking buffer overnight at 4 °C. Alexa Fluor 488 donkey anti-goat antibody (Life technologies) diluted in blocking buffer was added for 1 h at RT. To visualize cell nuclei, tissue sections were incubated with Dapi (Life technologies) for 10 min at RT then mounted in Prolong Antifade Agent (Life technologies). Immunofluorescence images were captured on a confocal microscope (Leica SP5 Inverted 2-Photon FLIM Confocal). ImageJ (National Institutes of Health, Bethesda, MD) was used for image processing.
Enrichment of intestinal epithelial and lamina propria cells
A section of Ileum (7 cm-long above the cecum) was collected, opened longitudinally, and washed with PBS minus supplemented with 5 mM 1,4-dithiothreitol solution (DTT; Fisher BioReagents™) and 2% FBS for 20 min at 37 °C under mechanical agitation at 200 rpm. Intestinal epithelial cells (IECs)-enriched fraction was obtained by mechanical agitation (200 rpm) for 10 min at 37 °C in 10 mL solution of PBS minus supplemented with 5 mM EDTA and 2% FBS. This step was repeated three times and fractions were pooled (30 mL) prior centrifugation at 400 × g for 10 min at 4 °C. For the lamina propria-enriched fraction, the remaining tissue after IECs removal was cut into small pieces (1–2 mm) and enzymatically digested using RPMI solution supplemented with Liberase TM (2.5 mg/mL; Roche Diagnostics, Basel, Switzerland) and DNAse I (2 × 104 Kuntz units/mL; EMD Millipore) for 30 min at 37 °C under mechanical agitation at 200 rpm. Cell suspension was filtered through a 70 μm cell strainer (Falcon) and washed with PBS minus supplemented with 10% FBS, and 2 mM EDTA then with ice-cold PBS minus. Cells from both, epithelial and lamina propria-enriched fractions were counted using a Scepter 2.0 Cell Counter (EMD Millipore). In all, 2 × 106 cells for each fraction were prepared for mRNA isolation.
Western blotting
Naive peritoneal macrophages were obtained by pooling peritoneal exudates from 3–5 mice followed by specific isolation of unlabeled macrophages by using Macrophage Isolation Kit (peritoneum) from MACs/Miltenyi Biotec. Cell suspension (5 × 105 cells) in RPMI medium supplemented with 10% FBS were added in 12-well flat-bottom plates and transferred for 45 min to a 5% CO2 incubator at 37 °C. Adherent cells were washed twice with RPMI medium then untreated or treated with 20 μg/mL of zymosan in RPMI medium for 5 min or 10 min at 37 °C. For LPS stimulation, cells were treated with 10 ng/mL of LPS in RPMI medium for 15 min or 30 min at 37 °C. After treatment, cells were washed twice with cold PBS and directly lysed in Laemmli sample buffer complemented with 2-mercaptoethanol and benzonase (Sigma). Lysates were sonicated and boiled for 5 min at 100 °C. Western blot was performed using antibodies against NFκB p65 pSer536, NFκB p65 (Cell Signaling Technology, Danvers, MA) or GAPDH (Sigma) followed by horseradish peroxidase-conjugated secondary antibodies and detection by enhanced chemiluminescence (GE Healthcare Bio-Sciences, Pittsburgh, PA). Band intensity was quantified by densitometry using ImageJ (NIH) software and values of NFκB p65 pSer536 were normalized to p65 total.
Phagocytosis assay
Peritoneal macrophages were enriched by adherence as described above. Adherent peritoneal macrophages were untreated (time 0) or incubated in RPMI-1640 medium containing 5% FBS with pHrodo-red conjugated zymosan (Life Technologies) at 37 °C for 30 min and 60 min. After incubation, cells were washed three times with ice-cold PBS, detached in PBS-2mM EDTA, centrifuged and resuspended in FACS buffer (PBS, 2% FBS). Cells were incubated with DAPI and labeled with anti-CD11b-e450 and anti-F4/80-APC. Zymosan was analyzed using Imagestream X Mark II.51 Positive zymosan uptake was defined as intracellular uptake pHrodo-red conjugated zymosan. Data is represented as percent phagocytosis of the total population.
Statistical analysis
All experiments were performed independently at least twice. Values were pooled from multiple experiments. Data represent means and error bars are standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism© 7.00. The differences were assessed with t-test or two-way analysis of variance (ANOVA) followed by a Bonferroni multiple comparison post hoc test. P-values ≤ 0.05 were considered statistically significant.
References
Ebnet, K. Junctional adhesion molecules (JAMs): Cell adhesion receptors with pleiotropic functions in cell physiology and development. Physiol. Rev. 97, 1529–1554 (2017).
Liu, Y. et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J. Cell Sci. 113(Pt 13), 2363–2374 (2000).
Martin-Padura, I. et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142, 117–127 (1998).
Williams, L. A., Martin-Padura, I., Dejana, E., Hogg, N. & Simmons, D. L. Identification and characterisation of human Junctional Adhesion Molecule (JAM). Mol. Immunol. 36, 1175–1188 (1999).
Bazzoni, G. et al. Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J. Cell Sci. 118(Pt 3), 623–632 (2005).
Luissint, A. C., Nusrat, A. & Parkos, C. A. JAM-related proteins in mucosal homeostasis and inflammation. Semin. Immunopathol. 36, 211–226 (2014).
Monteiro, A. C. & Parkos, C. A. Intracellular mediators of JAM-A-dependent epithelial barrier function. Ann. N. Y. Acad. Sci. 1257, 115–124 (2012).
Naik, M. U., Vuppalanchi, D. & Naik, U. P. Essential role of junctional adhesion molecule-1 in basic fibroblast growth factor-induced endothelial cell migration. Arterioscler. Thromb. Vasc. Biol. 23, 2165–2171 (2003).
Corada, M. et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA 102, 10634–10639 (2005).
Del Maschio, A. et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J. Exp. Med. 190, 1351–1356 (1999).
Khandoga, A. et al. Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood 106, 725–733 (2005).
Lechner, F. et al. Antibodies to the junctional adhesion molecule cause disruption of endothelial cells and do not prevent leukocyte influx into the meninges after viral or bacterial infection. J. Infect. Dis. 182, 978–982 (2000).
Woodfin, A. et al. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood 110, 1848–1856 (2007).
Ostermann, G., Weber, K. S., Zernecke, A., Schroder, A. & Weber, C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 3, 151–158 (2002).
Shaw, S. K. et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J. Exp. Med. 200, 1571–1580 (2004).
Karshovska, E. et al. Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ. Res. 116, 587–599 (2015).
Naik, M. U., Caplan, J. L. & Naik, U. P. Junctional adhesion molecule-A suppresses platelet integrin alphaIIbbeta3 signaling by recruiting Csk to the integrin-c-Src complex. Blood 123, 1393–1402 (2014).
Sobocka, M. B. et al. Signaling pathways of the F11 receptor (F11R; a.k.a. JAM-1, JAM-A) in human platelets: F11R dimerization, phosphorylation and complex formation with the integrin GPIIIa. J. Recept. Signal. Transduct. Res. 24, 85–105 (2004).
Cera, M. R. et al. Increased DC trafficking to lymph nodes and contact hypersensitivity in junctional adhesion molecule-A-deficient mice. J. Clin. Invest. 114, 729–738 (2004).
Cera, M. R. et al. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J. Cell Sci. 122(Pt 2), 268–277 (2009).
Khounlotham, M. et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity 37, 563–573 (2012).
Laukoetter, M. G. et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 204, 3067–3076 (2007).
Koch, A. E. et al. Growth-related gene product alpha. A chemotactic cytokine for neutrophils in rheumatoid arthritis. J. Immunol. 155, 3660–3666 (1995).
Goetzl, E. J. & Pickett, W. C. Novel structural determinants of the human neutrophil chemotactic activity of leukotriene B. J. Exp. Med. 153, 482–487 (1981).
Ajuebor, M. N. et al. Endogenous monocyte chemoattractant protein-1 recruits monocytes in the zymosan peritonitis model. J. Leukoc. Biol. 63, 108–116 (1998).
Sung, S. S., Nelson, R. S. & Silverstein, S. C. Yeast mannans inhibit binding and phagocytosis of zymosan by mouse peritoneal macrophages. J. Cell Biol. 96, 160–166 (1983).
Flemming S., Luissint A. C., Nusrat A. & Parkos C. A. Analysis of leukocyte transepithelial migration using an in vivo murine colonic loop model. JCI Insight. 3, pii: 99722 (2018).
Amiri, K. I. & Richmond, A. Fine tuning the transcriptional regulation of the CXCL1 chemokine. Prog. Nucleic Acid. Res. Mol. Biol. 74, 1–36 (2003).
Liu T., Zhang L., Joo D. & Sun S.C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2, e17023 (2017).
Thompson, R. D. et al. Platelet-endothelial cell adhesion molecule-1 (PECAM-1)-deficient mice demonstrate a transient and cytokine-specific role for PECAM-1 in leukocyte migration through the perivascular basement membrane. Blood 97, 1854–1860 (2001).
Edelblum, K. L. et al. The microbiome activates CD4 T-cell-mediated immunity to compensate for increased intestinal permeability. Cell. Mol. Gastroenterol. Hepatol. 4, 285–297 (2017).
Hollander, D. et al. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann. Intern. Med. 105, 883–885 (1986).
Lechuga, S. & Ivanov, A. I. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim. Biophys. Acta 1864, 1183–1194 (2017).
Wyatt, J., Vogelsang, H., Hubl, W., Waldhoer, T. & Lochs, H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 341, 1437–1439 (1993).
Gkouskou, K. K., Deligianni, C., Tsatsanis, C. & Eliopoulos, A. G. The gut microbiota in mouse models of inflammatory bowel disease. Front. Cell. Infect. Microbiol. 4, 28 (2014).
Madsen, K. L. et al. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm. Bowel Dis. 5, 262–270 (1999).
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323 (2009).
Sellon, R. K. et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224–5231 (1998).
Rahman, K. et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 151, 733–746 e12 (2016).
Hoeksema, M. A. et al. IFN-gamma priming of macrophages represses a part of the inflammatory program and attenuates neutrophil recruitment. J. Immunol. 194, 3909–3916 (2015).
Didierlaurent, A. et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 205, 323–329 (2008).
Shahangian, A. et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119, 1910–1920 (2009).
Murphey, E. D., Fang, G. & Sherwood, E. R. Endotoxin pretreatment improves bacterial clearance and decreases mortality in mice challenged with Staphylococcus aureus. Shock 29, 512–518 (2008).
Shi, D. W. et al. LPS pretreatment ameliorates multiple organ injuries and improves survival in a murine model of polymicrobial sepsis. Inflamm. Res. 60, 841–849 (2011).
Wheeler, D. S. et al. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock 30, 267–273 (2008).
Murakami, M. et al. Inactivation of junctional adhesion molecule-A enhances antitumoral immune response by promoting dendritic cell and T lymphocyte infiltration. Cancer Res. 70, 1759–1765 (2010).
el Marjou, F. et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 (2004).
Cash, J. L., White, G. E. & Greaves, D. R. Zymosan-induced peritonitis as a simple experimental system for the study of inflammation. Methods Enzymol. 461, 379–396 (2009).
Colgan, S. P., Parkos, C. A., Delp, C., Arnaout, M. A. & Madara, J. L. Neutrophil migration across cultured intestinal epithelial monolayers is modulated by epithelial exposure to IFN-gamma in a highly polarized fashion. J. Cell Biol. 120, 785–798 (1993).
Monteiro, A. C. et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol. Biol. Cell. 24, 2849–2860 (2013).
Ploppa, A., George, T. C., Unertl, K. E., Nohe, B. & Durieux, M. E. ImageStream cytometry extends the analysis of phagocytosis and oxidative burst. Scand. J. Clin. Lab. Invest. 71, 362–369 (2011).
Acknowledgements
We thank Nicholas Potochick and Meenal Mhaskar for technical support in study. This work was supported by NIH grants 1R01DK097256 to T.L.D., DK59888, DK55679 to A.N., and DK072564, DK061379, and DK079392 to C.A.P.
Author information
Authors and Affiliations
Contributions
A.C.L. designed and performed experiments, interpreted the data, and wrote the manuscript. H.C.W., W.K., S.F., V.A., and R.H. performed experiments. M.N.O. helped in generating germ-free Villin-Cre;Jam-afl/fl mice. T.L.D., A.N., and C.A.P. designed and supervised the study, edited the manuscript, and provided financial support.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Luissint, AC., Williams, H.C., Kim, W. et al. Macrophage-dependent neutrophil recruitment is impaired under conditions of increased intestinal permeability in JAM-A-deficient mice. Mucosal Immunol 12, 668–678 (2019). https://doi.org/10.1038/s41385-019-0143-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-019-0143-7
This article is cited by
-
The F11 Receptor (F11R)/Junctional Adhesion Molecule-A (JAM-A) (F11R/JAM-A) in cancer progression
Molecular and Cellular Biochemistry (2022)
-
Neutrophil expressed CD47 regulates CD11b/CD18-dependent neutrophil transepithelial migration in the intestine in vivo
Mucosal Immunology (2021)
-
Regulatory mechanisms of neutrophil migration from the circulation to the airspace
Cellular and Molecular Life Sciences (2021)
