
Abstract
The small intestinal and colonic lamina propria are populated with forkhead box P3 (FOXP3)+CD4+ regulatory T cells (Tregs) and interleukin-10-producing T cells that orchestrate intestinal tolerance to harmless microbial and food antigens. Expression of co-inhibitory receptors such as CTLA-4 and PD-1 serve as checkpoints to these cells controlling their T-cell receptor (TCR)-mediated and CD28-mediated activation and modulating the phenotype of neighboring antigen presenting cells. Recent discoveries on the diversity of co-inhibitory receptors and their selective cellular expression has shed new light on their tissue-dependent function. In this review, we provide an overview of the co-inhibitory pathways and checkpoints of Treg and effector T cells and their mechanisms of action in intestinal homeostasis. Better understanding of these inhibitory checkpoints is desired as their blockade harbors clinical potential for the treatment of cancer and their stimulation may offer new opportunities to treat chronic intestinal inflammation such as inflammatory bowel disease.
Similar content being viewed by others


Introduction
The intestinal mucosa is continuously exposed to exogenous antigens derived from food proteins and microbiota. Upon encounter of foreign antigens, antigen presenting cells (APCs) migrate to draining lymph nodes to present antigens to naive T cells. The interactions between APCs and responding T cells are modulated by additional signals from costimulatory and co-inhibitory receptors. The balance between these signals determines the functional outcome of T-cell receptor (TCR)-mediated activation, including the strength, nature and duration of the T-cell response.1 In addition to controlling APC-T-cell interactions during the initial activation of naive T-cells, costimulatory and co-inhibitory signals also control effector, memory and regulatory T-cell responses.2 Under homeostatic circumstances, encounter of innocuous antigens such as food proteins and molecular components of commensal bacteria in mucosa draining lymph nodes result in a preferential tolerogenic T-cell response. Inflammatory T-cell responses to food and microbial antigens can result in allergy and chronic inflammation, such as seen in patients with inflammatory bowel disease (IBD) and celiac disease,and might contribute to autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA).3,4,5,6,7
Over the years, studies in both mice and humans have established a central role for Forkhead box P3 (Foxp3)+CD4+ regulatory T cells (Tregs) and interleukin 10-producing CD4+Foxp3neg T cells in maintaining intestinal tolerance to harmless microbial and food antigens.8 Foxp3+ Tregs are classified into thymus-derived Treg (tTreg) and peripherally-derived Treg (pTreg). tTreg arise during CD4+ T-cell differentiation in the thymus, whereas pTreg differentiate from naive CD4+ T cells exposed to antigens under tolerogenic conditions in lymphoid tissue.9 Special environmental control in intestinal lymphoid tissues favors de novo pTreg development in response to TCR-specific recognition of food-derived and microbiota-derived antigens.8,10,11,12 In particular, soluble factors including transforming growth factor-β (TGF-β) and retinoic acid promote intestinal pTreg differentiation.13,14,15,16 Besides Foxp3-expressing Tregs, the intestine contains IL-10-secreting CD4+Foxp3neg type 1 regulatory T cells (Tr1 cells), differentiation of which is facilitated by the cytokine IL-27.17,18 A common feature of both Foxp3+ and Foxp3neg regulatory CD4+ T-cell populations is the expression of inhibitory receptors.19 Recently, immunotherapies directed against co-inhibitory receptors such as cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), aiming to enhance antitumor T-cell responses in cancer, show that co-inhibitory receptor blockade can lead to the development of immune-mediated intestinal inflammation.20 These findings provide new insight into the critical role of co-inhibitory receptors in the maintenance of intestinal homeostasis, but their involvement in the regulation of mucosal immune responses in the intestine remains poorly understood. In this review, we will discuss the role of co-inhibitory receptors in the maintenance of intestinal homeostasis and how the timing of inhibitory receptor upregulation, ligand distributions and specific effects on different cell-types contribute to this regulation. Better mechanistic understanding of how co-inhibitory receptor pathway modulation leads to intestinal inflammation is desired as stimulating co-inhibitory receptor pathways may offer new opportunities to treat chronic intestinal inflammation as observed in IBD.
Inhibitory receptors in the Ig superfamily that engage B7 ligands: CTLA-4 and PD-1
Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4; CD152)
CTLA-4: expression, ligands, and function
CTLA-4, a member of the Ig superfamily, is a structural homolog of the costimulatory receptor CD28 and is a ligand for CD80 (B7-1) and CD86 (B7-2) expressed on the surface of APCs.21,22 Unlike CD28, CTLA-4 undergoes constitutive clathrin-mediated endocytosis such that it is rapidly internalized after reaching the cell surface.23,24,25 CTLA-4 has a higher affinity for CD80 and CD86 than CD28 and can prevent the costimulatory signal normally provided by CD28/B7 interactions (Fig. 1).26 As CD28/B7 costimulation is essential to prime naive T cells, the CTLA-4 pathway is thought to inhibit T-cell proliferation early in the immune response, preferentially in the lymph node.27,28 Since CTLA-4 is a target gene of Foxp3,29,30,31 Tregs have high intracellular stores of CTLA-4 and constitutively traffic high levels of CTLA-4 to their cell surface.32,33 Resting naive T cells can be induced to express CTLA-4 in response to TCR ligation, in particular together with CD28 costimulation, reaching a maximum level after 48–72 h.34,35,36

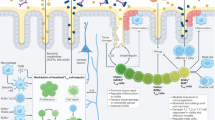
Interactions between co-inhibitory receptors of the immunoglobulin superfamily and their ligands. The majority of T-cell co-inhibitory receptors belong to the immunoglobulin (Ig) superfamily. Many co-inhibitory receptors of the Ig superfamily are expressed on activated T cells, and their specific ligands are expressed in professional antigen presenting cells (APCs), neutrophils, macrophages, or stromal cells. The inhibitory receptor cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) is a structural homolog of the costimulatory receptor CD28, but binds B7 ligands CD80 and CD86 with much higher binding affinity. Programmed cell death 1 (PD-1) is a member of the B7/CD28 family that has two ligands, PD-L1 and PD-L2. B- and T-lymphocyte attenuator (BTLA) is part of a shared receptor-ligand network and binds to the TNF receptor family member herpesvirus entry mediator (HVEM). Lymphocyte activation gene-3 (Lag-3) structurally resembles CD4 and binds to MHC-II with higher affinity. T-cell immunoglobulin and mucin domain-containing protein (Tim-3) has many ligands, including galectin-9 and Ceacam-1. T-cell immunoreceptor with Ig and ITIM domain (TIGIT) and the costimulatory receptor CD226 share the ligands CD112 and CD155. Together they form a pathway reminiscent of the CTLA-4/CD28/B7 pathway, in which TIGIT delivers an inhibitory signal
The critical role of CTLA-4 in controlling T-cell activation and tolerance is well-established. Ctla4−/− mice develop a lymphoproliferative disorder within 3 weeks of age that is characterized by the infiltration of CD4+ T cells into multiple non-lymphoid tissues, leading to organ destruction and death.37,38 Specific deletion of CTLA-4 in CD4+Foxp3+ Tregs results in a similar lymphoproliferative disease and multi-organ failure as seen in Ctla4−/− mice, but with a delayed onset, demonstrating that expression of CTLA-4 by Foxp3+ Tregs is essential to prevent autoimmunity in vivo.39 Mice lacking both CTLA-4 and CD28 or their ligands CD80/CD86 show no signs of disease,40,41,42 demonstrating that the main function of CTLA-4 is to control CD28-dependent T-cell activation by competing for CD80 and CD86. This so-called cell extrinsic regulation, in which CTLA-4+ cells inhibit the activation of neighboring CTLA-4neg cells, is exerted by conventional T cells as well as by Tregs.43,44,45,46 In addition to competing for CD80/CD86, CTLA-4 can downregulate expression of these ligands on APCs.39,47,48 This can occur via transendocytosis, whereby CTLA-4 captures CD80 and CD86 from the surface of APCs and internalizes them for degradation, impairing the capacity of these APCs to provide CD28 costimulation.49 The role of transendocytosis in CTLA-4 function offers a plausible biological explanation for the unusual endocytic and recycling behavior of the CTLA-4 molecule. Although it has been suggested that the cytoplasmic domain of CTLA-4 transmits an inhibitory signal leading to cell-intrinsic regulation within CTLA-4 expressing cells, this is not supported by experiments with mixed bone marrow chimeric mice containing a mixture of wildtype and CTLA-4-deficient cells.50,51,52 Thus while cell intrinsic competition between CTLA-4 and CD28 expressed on the same cell must surely occur, this does not appear to be a major mechanism of CTLA-4 action, at least in conventional T cells.32,52 In sum, CTLA-4 regulates T-cell function primarily by restricting T-cell activation early in the immune response through cell extrinsic ligand competition.
In the intestine, approximately 90% of all Foxp3+ Tregs express CTLA-4.60 Several murine studies have addressed the role of CTLA-4 in the induction of intestinal Foxp3+ cells, its effect on suppressive capacity and its role in maintaining the Foxp3+ Tregs pool. CTLA-4 is not required for pTreg induction, as in culture with TGF-β, IL-2, and TCR signals, naive Ctla4−/− T cells and Ctla4+/+ T cells are equally efficient in converting into Foxp3+ pTregs.53 However, CTLA-4 does appear to play a role in maintaining the intestinal Treg pool. The intestinal lamina propria of Rag2−/− mice reconstituted with a mixture of Ctla4−/− and Ctla4-sufficient BALB/c bone marrow cells contains reduced percentages of Foxp3+ T cells derived from Ctla4−/− bone marrow when compared to Foxp3+ T cells derived from BALB/c bone marrow.53 In contrast, in the spleen and mesenteric lymph nodes, CTLA-4-deficient (Ctla4−/−) and -sufficient (BALB/c) T cells contributed equally to the Foxp3+ and Foxp3neg compartments. This is not because of a general inability of CTLA-4−/− cells to migrate to the gut, as CTLA-4−/− cells in the intestine contribute to the CD4+Foxp3neg cell pool to a similar percentage as in other organs.53 These observations thus argue that, in vivo, CTLA-4 plays a role in regulating pTreg frequencies in the intestine in particular.53 In sum, CTLA-4 is not essential for intestinal pTreg induction but does regulate the accumulation of intestinal pTreg in the intestine.
CTLA-4 plays a fundamental role in Treg suppressive function during intestinal inflammation.33,40,55,56,61 The majority of data on the role of CTLA-4 in intestinal inflammation has been generated using the T-cell transfer model of colitis. In this model, naive CD4+CD45RBhigh T cells are adoptively transferred into lymphopenic Scid−/− or Rag−/− mice and cause colitis upon activation in response to intestinal antigens.62,63 Co-transfer of CD4+CD25+ T cells, enriched in Tregs, with naive CD4+CD45RBhigh T cells prevents development of colitis.64 In most studies, CTLA-4 expression on co-transferred CD4+CD25+ T cells is essential to prevent colitis induced by naive T-cell transfer into Rag−/− recipients (Table 1).32,45,54 Similarly, blocking the interaction of CTLA-4 and its ligands through use of anti-CTLA-4 antibodies or Fab fragments, abrogates CD4+CD25+ cell mediated suppression of colitis.40,55,56 This argues that CTLA-4 expression on Tregs is required for colitis suppression (Fig. 2(2)). It should be noted that genetic deficiency of CTLA-4 in CD4+CD25+ Tregs has been reported to result in a compensatory IL-10 production in some settings, allowing the prevention of transfer colitis in an IL-10 dependent fashion (Table 1). It is unclear whether CTLA-4 expression by colitogenic naive CD4+CD45RBhigh T cells plays a role in transfer colitis, as anti-CTLA-4 antibody treatment exacerbated colitis in some experimental settings but not others.40,56 An overview of the role of CTLA-4 in colitis suppression in the different transfer colitis studies is provided in Table 1. Altogether, data demonstrate a key role for CTLA-4 in Foxp3+ Tregs to prevent T-cell transfer induced colitis.

Functional effects of co-inhibitory receptors in the intestine. 1. PD-1 promotes peripheral Foxp3 induction in naive CD4+ T cells, especially in TGF-β rich environments. 2. CTLA-4 enhances the suppressive function of Tregs to suppress colitogenic CD4+ effector T cells. In addition, the CTLA-4 pathway is important in establishing oral tolerance through suppression of Th1 and/or Th2 responses. 3. PD-1 expression on CD8+ effector T cells is involved in preventing responses to intestinal self-antigens. 4. PD-1 expression on Tfh cells regulates Tfh-cell numbers in Peyer’s patches, promoting intestinal tolerance to microbiota through secretory IgA. 5. HVEM-CD160 interactions at the mucosal surface enhance IL-22R mediated STAT-3 activation in epithelial cells. 6. IL-27 is produced by APCs upon activation by microbial products. IL-27 induces Lag-3 expression on Tregs and inhibiting effector T-cell responses through enhancing Treg-suppressive function. Additionally, Lag-3 can prevent T-cell activation through inhibition of DC maturation. 7. Frequencies of CD4+CD25negLag-3+ T cells are high in Peyer’s patches, but their role in regulating intestinal humoral immune responses remains unknown. 8. Tim-3 inhibits polarization of pathogenic pro-inflammatory M1 macrophages. 9. IL-27 enhances TCR-mediated induction of Tim-3 on naïve CD4+ T cells and can directly suppress Th1 cell-mediated colitis via the induction of Tim-3. 10. TIGIT identifies a Treg subset that specifically suppresses pro-inflammatory Th1 and Th17 but not Th2 responses
In addition to their crucial role in preventing intestinal inflammation, Foxp3+ pTregs are required for the induction of oral tolerance.10,65,66 Feeding of soluble antigen induces systemic immunological hyporesponsiveness that is characterized by a suppressed delayed type hypersensitivity (DTH) reaction after antigen injection in the footpad or the auricle.67,68 At the cellular level, tolerance is a consequence of Foxp3+ pTreg mediated suppression of the Th1 and Th2 response to the particular antigen. Several studies have demonstrated a role for CTLA-4 function in the development of oral tolerance, but effects on cytokine secretion varied on the dose of the antigen fed while the DTH response was not monitored.69,70 Administration of anti-CTLA-4 antibodies during 50 mg ovalbumin (OVA) feeding completely abrogated the suppression of cytokine production by Th1 and Th2 cells.70 By contrast, the administration of anti-CTLA-4 antibodies during 1 mg OVA feeding only abrogated suppression of Th2 type immune responses but not Th1.69 Co-administration of IL-12 along with anti-CTLA-4 antibodies was required to abrogate the suppression of Th1 cytokine secretion during tolerance induction 1 mg OVA feed.69 These results indicate that the CTLA-4 pathway is important for establishing oral tolerance, but suggests that its role is most important in high dose oral tolerance. Whether this relates to different mechanisms mediating oral tolerance to low and high antigen doses, i.e., low doses favoring the induction of pTregs and higher doses the induction of anergy or clonal deletion remains to be established. In particular, whether OVA-specific pTregs require CTLA-4 for full suppression of Th1 and Th2 immune responses in models for low dose oral tolerance remains to be further investigated (Fig. 2(2)).
In some settings, CTLA-4 deficiency may allow for induction of other subsets of CD4+ T cells with the capacity to suppress colitis. Cre-recombinase based deletion of CTLA-4 in adult mice results in significantly increased frequencies of Foxp3neg T cells, that exhibit high levels of IL-10 and increased expression of coinhibitory receptors Lag-3 and PD-1.71 High IL-10-producing capacity and the expression of co-inhibitory receptors such as Lag-3 and PD-1 are characteristic of IL-10-secreting CD4+Foxp3neg Tr1, a cell type that has been shown to inhibit T-cell responses and colitis in an IL-10-dependent manner.72,73,74 Along the same lines, it was recently demonstrated that anti-CTLA-4 treatment can induce IL-10-producing ICOS+ regulatory CD4+ T cells with anti-inflammatory properties that inhibit 2,4,6-Trinitrobenzenesulfonic acid solution (TNBS) induced colitis.75 This is in contrast with the finding that anti-CTLA-4 antibodies abrogated Treg control of colitis in the setting of T-cell transfer experiments (described above), but fits with the observation that IL-10 can compensate for a genetic deficiency of CTLA-4 in some settings (Table 1).40 The precise circumstances in which blockade or lack of CTLA-4 allows for the differentiation of immunosuppressive IL-10 secreting CD4+Foxp3neg T cells with the ability suppress colitis requires further investigation.
Quantitative deficiencies in CTLA-4 expression in humans
In humans, heterozygous mutations in CTLA-4 resulting in CTLA-4 haploinsufficiency are associated with lymphoproliferation reminiscent of the mouse model, resulting in severe clinical manifestations of autoimmunity and early-onset Crohn’s disease.57,58,59 The organs of affected CTLA4+/− individuals, including the intestine and lungs, show extensive CD4+ T-cell infiltration.59 Frequencies of Tregs within the CD4+ T-cell compartment of CTLA4+/− individuals are significantly increased compared to healthy CTLA4+/+ controls and their suppressive function is impaired.57,58,59 In addition to impaired Treg function, it has been suggested that the CTLA4+/− genotype alters naive T-cell responses, as naive T cells obtained from a clinically affected CTLA4+/− individual showed hyperproliferation after in vitro activation in the presence of autologous T-cell depleted feeder cells.57 However, other studies did not observe hyperproliferation in naive T-cell responses isolated from CTLA4+/− individuals suggesting that the variations in experimental set-up may be key.59,76 Interestingly, the clinical penetrance of CTLA-4 haploinsufficiency is incomplete, suggesting that additional genetic or environmental factors influence disease susceptibility in individuals with impaired CTLA-4 function. It is currently unknown whether clinically unaffected CTLA4+/− individuals are protected from disease by CD4+ T cells with suppressive capacity independent of CTLA-4.
Genetic variants in the CTLA4 gene locus are associated with intestinal inflammation and autoimmune diseases.77,78,79 The single nucleotide polymorphism (SNP) CT60 (rs3087243; A/G) located in the 3′ UTR of the CTLA4 gene has been associated with IBD in a Slovenian cohort of adult IBD patients.80 This variant of the CTLA4 gene is associated with a functionally altered TCR signaling in CD4+ T cells and decreased production of the soluble CTLA-4 isoform.81,82 However, the association between the CT60 CTLA4 allele and IBD was not confirmed in a separate cohort of Spanish patients.77 Several studies have shown that the CT60 CTLA4 allele is also weakly associated with celiac disease.83,84 Taken together, these data indicate that quantitative deficiencies in CTLA-4 protein expression can predispose selected subgroups of individuals to autoimmunity, including CD4+ T-cell mediated inflammation in the gastrointestinal tract.
Programmed death 1 (PD-1; CD279)
PD-1: expression, ligands, and function
PD-1 is a cell-surface molecule belonging to the B7 receptor family that contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) in its cytoplasmic tail.85,86 PD-1 has two ligands, PD-L1 (B7-H1; also termed CD274) and PD-L2 (B7-DC; also termed CD273). Most of the inhibitory roles of PD-1 have been attributed to its interaction with PD-L1 (Fig. 1).87 Upon PD-L1 or PD-L2 binding, the ITIM or ITSM tyrosine motifs in the cytoplasmic tail of PD-1 are phosphorylated.88 This leads to recruitment of Src-homology 2 domain-containing phosphatase 2 (SHP-2) and augments phosphatase and tensin homolog (PTEN) expression, inhibiting phosphatidylinositol 3-kinase (PI3K) and Akt activation.89,90,91 CD28, not the TCR or its associated components, has been reported to be the most sensitive target of PD-1, as shown by the strong degree of CD28-dephosphorylation compared to dephosphorylation of TCR-signaling components after PD-1 activation.90,92 In line with its inhibitory function, PD-1 deficient mice (Pdcd1−/−) develop spontaneous autoimmune disease although the incidence of disease depends on the genetic background and symptoms only manifest later in life.93,94,95 Moreover, the disease is often tissue-specific which is in contrast to the rapid multi-organ autoimmune disease observed in CTLA-4−/− mice in the first few weeks of life.37,38
PD-1 is expressed on diverse hematopoietic cells, including T and B cells,96 natural killer (NK) cells and other innate immune cells.97 PD-1 expression is absent on naive T cells but is rapidly upregulated after antigen encounter and can be detected after only 6 h, with a peak in expression at 48 h after antigen encounter.96,98,99,100 Of the two PD-1 ligands, PD-L2 is mainly expressed by DCs and macrophages, whereas PD-L1 is more widely expressed by both hematopoietic and non-hematopoietic cells101,102,103 and can be induced by inflammatory cytokines such as interferons.104,105 The broad expression of PD-1 and PD-L1 is in contrast to CTLA-4 and its ligands that are mainly expressed by hematopoietic cells present in the lymph node environment. Therefore, it is often hypothesized that while CTLA-4 impacts T-cell activation primarily during the priming phase in secondary lymphoid organs, PD-1 plays a dominant role during the maintenance of tolerance in peripheral tissues.27,28 Many murine models for autoimmune disease and cancer have established a role for PD-1/PD-L1 interactions in maintaining tissue tolerance by controlling the effector T-cell responses in non-lymphoid tissues.94,106,107,108,109,110 However, the rapid upregulation of PD-1 after activation suggests that PD-1 can control T-cell activation at the time of initial antigen encounter in the lymph node in addition to modulating effector responses in target tissues.
Although Foxp3+ Tregs highly express PD-1,111 the role of PD-1 on Tregs is only beginning to be fully understood. PD-1 regulates the generation of pTreg and PD-L1 synergizes with TGF-β to promote pTreg induction,111,112 suggesting that PD-1/PD-L1-mediated pTreg induction might play a role in TGF-β-rich environments such as the intestinal mucosa-draining lymphoid tissue (Fig. 2(1)). A role for PD-1 in the maintenance of Tregs has been suggested by several studies. PD-1 maintains the suppressive phenotype of Tregs through inducing PTEN expression.120 In line with this finding, PD-1 prevents the conversion of Foxp3+ Tregs into pro-inflammatory effector/memory CD4+ T cells .113 Recently, it was shown that low dose IL-2 therapy, used as a therapy to induce expansion of pTregs, increases PD-1 expression on Tregs of patients treated for graft-versus-host disease.114 Examination of Tregs from IL-2-treated mice demonstrated that PD-1 reduced IL-2-induced Treg proliferation but prevented their terminal differentiation, rendering Tregs less susceptible for apoptosis and promoting their survival and subsequent regulation.114 IL-2-induced Tregs isolated from PD-1−/− mice exhibited normal levels of suppressive activity, indicating that PD-1 does not directly affect Treg function.114 In addition to Foxp3+ Tregs, PD-1 expression has recently been described on intestinal Tr1 cells (defined as CD3+CD4+CD25−IL-7R+ cells) in both mice and humans.115 Expression of CCR5 and PD-1 allowed enrichment for IL-10+ Tr1 cells in the human intestine. In mice, co-transfer of IL-10-producing CCR5+PD-1+ Tr1 cells strongly inhibited colitis induced by transfer of Th17 cells, whereas IL-10-producing control T cells that lacked CCR5 and PD-1 were less efficient.115 Taken together, PD-1 is expressed on both Foxp3+ Treg and CD4+Foxp3neg Tr1 cells, and can take part in regulating the induction, survival and IL-10 production of these cells in the intestinal environment.
In addition to PD-1 on regulatory CD4+ T-cell populations, PD-1 expression on CD8+ effector T cells is involved in preventing responses to intestinal self-antigens (Fig. 2(3)).116,117 In a transfer model of OVA-specific OT-I CD8+ T cells into iFABP-OVA mice that express OVA as a self-antigen on intestinal epithelial cells, blocking PD-1/PD-L1 interaction at the time of OT-I cell transfer prevents tolerance induction and resulted in small intestinal inflammtion.116,117 The intestinal pathology was characterized by apoptosis of epithelial cells, villous atrophy and leukocytic infiltration, reminiscent of the pathology in human celiac disease. Crucially, when PD-L1 is blocked 30 days after OT-I cell transfer, mice do not develop intestinal pathology, indicating that PD-1/PD-L1 interaction is crucial for the induction, but not the maintenance of mucosal CD8+ T-cell tolerance.117 PD-1 upregulation on CD8+ effector T cells upon antigen encounter in vivo is highly dependent on the type of antigen recognized. Transfer of CD8+ effector T cells specific for influenza hemagglutinin (HA) into mice expressing HA as a self-antigen induces robust PD-1 expression on HA-specific CD8+ T cells.87 In contrast, when HA-specific CD8+ T cells were transferred into mice infected with an HA-expressing Listeria monocytogenes, there was virtually no PD-1 expression on HA-specific CD8+ T cells, demonstrating that PD-1 is differentially induced on CD8+ effector T cells responding to self-antigen versus microbial antigen.87 Of note, the recipient mice in this model expressed HA under the control of the C3 promoter, which directs HA expression on a variety of parenchymal tissues. Whether HA expressed as a self-antigen specifically on intestinal cells induces PD-1 expression on HA-specific T cells remains unknown. Overall, these data demonstrate that the role of PD-1/PD-L1 interactions in CD8+ T-cell tolerance to self-antigens and microbial antigens is likely determined by multiple factors, including the timing of the immune response, type of antigen and signals from the tissue microenvironment.
Besides CD8+ effector T cells, PD-1-expressing Tfh cells are involved in regulating microbial-host interactions through secretory IgA (Fig. 2(4)). PD-1 is highly expressed on cells in the germinal centers of Peyer’s patches,121 the major sites for induction of mucosal secretory IgA antibody responses.122 Secretory IgA plays a key role in regulating microbial-host interactions at the mucosal surface by maintaining a balanced and highly diverse microbial communities in the gut.123 In humans, the protective role of IgA is illustrated by the fact that IgA-deficiency is associated with increased susceptibility to autoimmunity and gastrointestinal infections.124,125,126 It has been demonstrated that PD-1 deficient mice (Pdcd1−/−) have significantly more Tfh cells in Peyer’s patches compared to wildtype mice.118 The production of IL-21, the cytokine that promotes proliferation and differentiation of IgA+ B cells into plasma cells,127,128 is reduced in Tfh cells from Pdcd1−/− mice causing an impaired ability to support the generation of IgA plasma cells in gut.118,119 As a result, Pdcd1−/− mice have lower proportions of bacteria coated with IgA and altered microbial communities in the intestine,118 which resemble alterations of the microbiome observed in several pathological conditions including human IBD.129 Serum from Pdcd1−/− mice contains increased titers of commensal-specific IgG,118 indicating a breach of normal host-microbe interaction in the absence of PD-L1/PD-1 interactions.
Besides a role for the PD-1/PD-L1 pathway in adaptive immune cells, PD-L1 serves as an essential ligand for innate immune cells in the intestine to prevent intestinal inflammation.130 Experiments with bone marrow chimeras have demonstrated that PD-L1 expression on intestinal epithelial cells reduces dextran sulfate sodium (DSS)-induced intestinal inflammation.130 This protective effect was mediated through inhibition of TNFα secretion of CD11c+CD11b+ lamina propria cells and was independent of adaptive immunity, as PD-L1-deficient Rag1−/− mice exhibited a significantly higher morbidity and mortality than Rag1−/− mice after DSS administration.130 In humans, PD-L1 is expressed by intestinal epithelial cells of IBD patients but not of healthy controls,131 and PD-1 expression is increased on lamina propria mononuclear cells,132 which might reflect an effort to promote protective intestinal immune responses through PD-1.
Lessons learned from CTLA-4 and PD-1 blockade in cancer
Over the past decade, a role for co-inhibitory receptors in the maintenance of intestinal homeostasis in humans has been widely appreciated following the implementation of anti-CTLA-4 and anti-PD-1/PD-L1 immunotherapies to promote anti-tumor T-cell responses and tumor regression in cancer patients. Blockade of CTLA-4 and PD-1/PD-L1 is thought to activate a wide repertoire of T cells, not only tumor-specific T cells. In consequence, these therapies have a broad range of adverse effects, of which diarrhea and colitis are very frequently observed.133,134 The incidence of diarrhea and colitis is 35.4 and 8.8%, respectively with CTLA-4 inhibitors and 13.7 and 1.6%, respectively for PD-1 inhibitors.135,136 Combining CTLA-4 and PD-1 inhibitors may increase the risk of diarrhea but not colitis.135,136 Colonic bowel perforation is the most common cause of fatal immune-related adverse events in patients who develop immunotherapy-induced colitis, but the incidence of life-threatening colon perforation is low (<1% of patients).20 Thus, although blockade of co-inhibitory receptors is a promising new approach to improve tumor control, it can cause severe life-threatening immune-related adverse events, most often through a dysregulation of intestinal homeostasis.
In contrast to the chronic colitis-associated histological alterations observed in IBD, anti-CTLA-4 and anti-PD-1 colitis are characterized by neutrophilic inflammation or a mononuclear expansion in the lamina propria and increased intraepithelial lymphocytes.137,138,139 In most patients with immunotherapy-induced colitis, disease remission is achieved by discontinuing the drug,134 but patients are likely to relapse when restarting the same drug.137,140 Development of recurrent colitis has been observed in patients without a past medical history of intestinal inflammation who had stopped anti-CTLA-4 or anti-PD-1 therapy for multiple months.137,140 The incidence of recurrent colitis is unknown. Colonic biopsies in these patients often demonstrated features of chronicity such as crypt architectural irregularity, suggesting that immunotherapy-induced colitis may progress to chronic intestinal inflammation as seen in IBD. Further studies are needed to understand how short-term co-inhibitory receptor blockade can result in long-term effects.
The exact mechanisms of immunotherapy-induced colitis are still elusive. There is evidence that Treg-cell depletion contributes to the pathogenesis of CTLA-4 induced colitis in some studies,141,142 but not in others.143,144 More investigation is required to establish a role for Tregs in immunotherapy-induced colitis. Other possible mechanisms of immunotherapy-induced colitis include the priming of naïve lymphocytes with reactivity to intestinal bacteria, self-antigens or cross-reactivity with tumor antigens, and the worsening of a pre-existing colitogenic immune response upon co-inhibitory receptor blockade. Interestingly, the risk of immunotherapy-related colitis is increased in patients with lower abundance of intestinal bacteria belonging to the Bacteroidetes phylum before start of anti-CTLA-4 therapy, which is reminiscent of the dysbiosis observed in IBD patients.145,146,147 It has been suggested that the risk of immunotherapy-related colitis may be depend on a patient’s co-inhibitory receptor allele polymorphisms,148 although in general SNPs associated with autoimmune disease do not appear useful in predicting the side-effects of immunotherapy.149 Moreover, the majority of individuals experience no immunotherapy-related adverse events on anti-CTLA-4 or anti-PD-(L)1 therapy. Therefore, it is likely that multiple factors contribute to intestinal disease development during co-inhibitory receptor blockade.
Other members of the Ig superfamily: BTLA, Lag-3, Tim-3, and TIGIT
The success of immunotherapies directed against CTLA-4 and PD-1 in enhancing anti-tumor activity has prompted intense investigation into the targeting of other co-inhibitory receptors in order to broaden the therapeutic repertoire. A next generation immune checkpoint inhibitors directed against co-inhibitory receptors BTLA, Lag-3, Tim-3, and TIGIT, are currently being explored in clinical trials and may emerge soon.150,151 It is expected that many of these new checkpoint inhibitors will be used in combination with the already approved checkpoint inhibitors against CTLA-4 and PD-1.152 In order to maximize success of novel combination therapies while minimizing gastro-intestinal adverse effects, it is crucial to increase our current knowledge on the basic molecular mechanisms of these co-inhibitory molecules and their tissue-specific functions. In the paragraphs below, we highlight the unique tissue-specific functions in the intestine of four newly emerging immune checkpoints.
HVEM ligands: B- and T-lymphocyte attenuator (BTLA; also known as CD272) and CD160
BTLA is a co-inhibitory receptor that is expressed on a wide range of hematopoietic cells, including CD4+ and CD8+ T cells, γδ T cells, B cells, innate lymphoid cells (ILCs), NK cells and DCs.153,154 In contrast to other co-inhibitory receptors that are induced upon TCR ligation, BTLA is constitutively expressed on naive CD4+ T cells and its expression increases upon T-cell activation, with the highest level of BTLA observed 2–3 days after TCR stimulation.155 Unlike CTLA-4 and PD-1, BTLA expression is not enriched on Tregs compared to naïve CD4+ T cells.156 However, BTLA is expressed most highly on CD4+ T cells after antigen-specific anergy induction in vivo.156 In vitro ligation of BTLA with an agonistic antibody during CD4+ T-cell stimulation reduces IL-2 and CD25 induction. BTLA is part of a shared receptor-ligand network and binds to the TNF receptor family member Herpesvirus entry mediator (HVEM), a receptor that is constitutively expressed on the surface of various cell types, including hematopoietic cells and non-hematopoietic cells.154 HVEM also binds to the co-inhibitory receptor CD160, an Ig superfamily member that is co-expressed by a small percentage of unstimulated BTLA-expressing CD4+ T cells and can be upregulated during CD4+ T-cell activation (Fig. 1).157 In BTLA and CD160 co-expressing CD4+ T cells both co-inhibitory receptors may act coordinately in HVEM-mediated inhibition and use different intracellular pathways to exert their suppression.157 Additionally, HVEM also triggers costimulatory signals by ligating tumor necrosis factor (TNF) superfamily members, including the TNF superfamily members LIGHT and LTα.158,159 As a result, the functional outcome of HVEM engagement can be opposing with negative regulation of T-cell responses through BTLA and CD160-derived inhibitory signals but stimulatory signals when binding LIGHT and LTα.1,154 Overall, in vivo the net outcome of HVEM-signaling via its stimulatory and inhibitory ligands on CD4+ T cells appears a more dominant inhibitory function as HVEM-knockout mice have higher T-cell activation,160 indicating that inhibition via HVEM is the essential, non-redundant function of HVEM. This dominant inhibitory function of HVEM-ligand interaction in T cells is also seen in mouse and human CD4+ T cells stimulated with APCs transfected with mouse or human HVEM, respectively.157,161
Antagonistic anti-human BTLA antibodies are currently in clinical development for cancer treatment and urge us to further understand the dual effects of HVEM-LIGHT-LTα-BTLA-CD160 interactions in intestinal immune function. In line with the pro-inflammatory capacities of HVEM-ligand interaction, Hvem−/− mice are resistant to DSS colitis.162,163 As LIGHT deficiency only partially reduces DSS colitis, both LIGHT and other stimulatory ligands may convey HVEM signaling, a process attributed to innate immune cells.162 Deletion of HVEM expression in transferred naive CD4+CD45RBhigh T cells was reported to contribute to the induction of T-cell mediated colitis in recipient Rag−/− mice in some studies but not others, suggesting that the role of HVEM in T cells is not crucial in intestinal homeostasis.155,162 Conversely, absence of HVEM expression on irradiation resistant structural cells in the recipient Rag−/− mice (Hvem−/−Rag−/− mice) results in a dramatic acceleration of colitis development compared to Rag−/− recipients. This suggested that recipient HVEM expression on structural cells binds inhibitory ligands leading to an anti-inflammatory HVEM-mediated regulation of T-cell transfer colitis.155 As treatment of Hvem−/−Rag−/− recipients with an agonistic anti-BTLA antibody during T-cell transfer rescued disease, it was postulated that inhibitory BTLA signaling on T cells mediated by HVEM is crucial to prevent colitis acceleration in the transfer colitis model. However, a possible role for ligation of HVEM to the other co-inhibitory ligand CD160 was not experimentally addressed in this study.155 Using the Citrobacter rodentium-induced murine colitis model, the Kronenberg group demonstrated that CD160 is the non-redundant ligand engaging HVEM in the mucosa during intestinal anti-bacterial responses.164 HVEM-CD160 interactions at the mucosal surface enhanced IL-22R mediated STAT-3 activation in epithelial cells and promoted their innate response to acute bacterial infection (Fig. 2(5)).164 In both uninfected and infected mice, CD160 was expressed by several intraepithelial lymphocyte (IEL) subpopulations, particularly CD8αα-expressing IEL. These CD160+CD8αα+ IEL rapidly increased during the early stages of C. Rodentium infection.164 Given their close contact with intestinal epithelial cells, CD160+CD8αα+ IEL seem likely candidates to interact with HVEM, which is known to be highly expressed on intestinal epithelial cells.155,164 Whether, reciprocally, epithelial ligation of HVEM to CD160 expressed on IEL elicits functional changes in the IEL is not clear. Human CD160 expression on intestinal IEL has been described,165 but further studies are needed to decipher whether it functions to prevent intestinal inflammation as shown in mice. Taken together, BTLA-HVEM and CD160-HVEM interactions help maintain mucosal immune homeostasis and protect against mucosal infections. Caution is warranted when blocking BTLA-HVEM interactions due to the potential interaction of HVEM with costimulatory ligands present in the intestine.
Lymphocyte activation gene-3 (Lag-3; also known as CD223)
Lag-3 is a co-inhibitory receptor that is expressed on activated CD4+ and CD8+ T cells and subsets of NK cells.166,167 Lag-3 structurally resembles CD4 and binds to MHC-II with higher affinity than CD4 (Fig. 1),168 resulting in downregulation of antigen-specific CD4+ T-cell responses.169,170,171 Lag-3 also interferes with CD8+ T-cell function,172 suggesting that Lag-3 has other unidentified ligands in addition to MHC-II.173 In addition to activated T cells,174 Lag-3 is expressed on Tregs and is often used as a marker for IL-10-secreting CD4+Foxp3neg Tr1 cells.175
Multiple companies have developed Lag-3 specific antagonist antibodies that are currently being tested in phase I clinical trials, but it remains to be seen what the effects on gastro-intestinal homeostasis will be. Lag-3 expression in Tregs is critical for Treg-mediated suppression of colitogenic T-cell responses.176 Lag-3 expression on Tregs is induced by IL-27, a cytokine that is mainly produced by APCs upon activation by microbial products.177 IL-27 stimulation has been shown to enhance Treg-suppressive function through a Lag-3-dependent mechanism (Fig. 2(6)).176 In contrast to wildtype Tregs, Lag3−/− Tregs failed to suppress effector T-cell expansion and cytokine expression in mesenteric lymph nodes, even after IL-27 stimulation.176 In addition to inhibiting effector T-cell responses through enhancing Treg-suppressive function, Lag-3 can prevent T-cell activation through inhibition of DC maturation (Fig. 2(6)).176,178
Lag-3 on CD4+Foxp3neg T cells also has a role in regulating or suppressing other cells in the intestine. CD4+CD25negLag-3+ T cells prevent colitis induced in Rag1−/− recipients by the transfer of naive CD4+CD25negCD45RBhigh T cells in a Foxp3-independent, IL-10-dependent manner.179 In a murine model for SLE, CD4+CD25negLag-3+ T cells suppressed B-cell activation and antibody production through TGF-β3.180 As frequencies of CD4+CD25negLag-3+ T cells are high in Peyer’s patches,179 it can be expected that intestinal Lag-3+ T-cells regulate intestinal humoral immune responses, but further studies are needed to prove this hypothesis (Fig. 2(7)). In sum, Lag-3-dependent mechanisms contribute to intestinal homeostasis influencing the function of both intestinal Tregs and suppressive CD4+Foxp3neg T-cell subsets. However, whether Lag-3-driven regulation also modulates effector T cells or B-cell responses in the intestine has not yet been determined.
T-cell immunoglobulin and mucin domain-containing protein-3 (Tim-3)
Tim-3 is a co-inhibitory receptor initially identified on interferon-gamma (IFNγ)-producing CD4+ T cells and CD8+ T cells,181 but is also expressed on innate immune cells including DCs, NK cells, and monocytes.182,183,184 TCR ligation induces Tim-3 expression on conventional CD4+ T cells and Tregs, although with different kinetics.185 Activated human CD4+ T cells that express Tim-3 secrete reduced levels of IFNγ and IL-17A,186 and Tim-3+ Tregs have been shown to specifically suppress Th17 cells.185 Over the past decade, Tim-3 expression on CD4+ and CD8+ T cells has especially been associated with exhausted and dysfunctional T-cell phenotypes,187,188,189 suggesting that Tim-3 negatively regulates T cells that have previously undergone activation. Tim-3 has many ligands, including galectin-9190 and Ceacam-1,191 and can bind to its ligands both in cis and in trans (Fig. 1).186,191 Therefore, Tim-3 has the capacity to function via both autocrine and paracrine mechanisms to inhibit T-cell responses.
Several Tim-3 antagonists are currently being tested in phase I clinical trials. Data available from animal models support a role for Tim-3 in inhibiting intestinal inflammation. Tim-3 inhibits polarization of pathogenic pro-inflammatory M1 macrophages by preventing phosphorylation of IRF3, a transcriptional factor downstream of TLR-4 that regulates macrophage polarization.183 In consequence, blockade of Tim-3 exacerbates DSS-induced colitis in wildtype mice, but not in Tlr4−/− mice (Fig. 2(8)).192 In addition to a role in macrophages, IL-27 greatly enhances TCR-mediated induction of Tim-3 on naïve CD4+ T cells190 but not on Tregs.176 IL-27-conditioned Th1 cells induce less severe colitis in Rag1−/− recipients compared to unconditioned Th1 cells, suggesting that IL-27 can directly suppress Th1 cell-mediated colitis via the induction of Tim-3 (Fig. 2(9)).183,190
In humans, Tim-3 expression is strongly enriched on CD4+ T cells isolated from the intestinal mucosa compared to CD4+ T cells in peripheral blood.193 In contrast, IBD patients had significantly lower frequencies of Tim-3+ cells in CD4+ T-cell populations isolated from peripheral blood and intestinal biopsies when compared to healthy individuals, possibly suggesting that decreased Tim-3 expression or blockade of Tim-3 may contribute to intestinal inflammation.193,194 Whether restoration of Tim-3 expression in patients with IBD harbors potential clinical benefit needs to be further investigated.
T-cell immunoreceptor with Ig and ITIM domain (TIGIT)
TIGIT is a co-inhibitory receptor specifically expressed by immune cells, including NK cells, memory CD4+ T cells and subsets of Tregs.195,196 TIGIT binds to CD155 and CD112, which are expressed on the surface of APCs, T cells and non-hematopoietic cells.195,197 The costimulatory receptor CD226 also binds to the same ligands198 and together with TIGIT forms a pathway in which CD226 enhances the activation of T cells while TIGIT inhibits their activation,199 which is reminiscent of the CTLA-4/CD28/B7 pathway (Fig. 1). On effector T cells, TIGIT directly targets molecules in the TCR signaling pathway, dampening effector T-cell activation, proliferation and inflammatory cytokine secretion.200 Moreover, TIGIT modifies DC function via bi-directional co-signaling through CD155, promoting the generation of immunoregulatory DCs with decreased IL-12 and increased IL-10 production.195
In addition to effector cells, TIGIT promotes the suppressive function of both Foxp3+ Treg and regulatory CD4+Foxp3neg T cells. TIGIT expression on CD4+Foxp3neg T cells discriminates IL-10-secreting CD4+ T cells induced by immunotherapy.201 On Tregs, TIGIT ligation induces the expression of IL-10 and other effector molecules, such as fibrinogen-like protein 2 (Fgl2).202 TIGIT identifies a Treg subset that specifically suppresses pro-inflammatory Th1 and Th17 but not Th2 responses (Fig. 2(10)).202 The TIGIT gene is a direct target of Foxp3 and TIGIT expression on Foxp3+ Tregs results in higher levels of Treg signature genes.202,203 Moreover, TIGIT+ Tregs show enhanced demethylation of Treg-specific demethylated regions (TSDR) ensuring stable Foxp3 expression.202 Combined, these data show that TIGIT can promote the stability and function of various subsets of regulatory T cells.
In line with its inhibitory functions, TIGIT deficiency or blockade exacerbates disease in models for autoimmune disease.200,204 However, little is known regarding the role of TIGIT in intestinal homeostasis. One study has investigated TIGIT expression in human colonic tissue, and demonstrated that TIGIT is expressed by approximately 30% of intestinal CD4+Foxp3neg T cells and virtually all Tregs.60 Recently, our group has identified TIGIT as a key regulatory molecule in circulating CD38+ effector T cells, a population enriched for T-cells with specificity for mucosal antigens.205 Frequencies of TIGIT+CD38+ effector T cells were decreased in a subgroup of pediatric IBD patients before start of treatment and TIGIT percentages below 25% identified patients with a shorter duration of clinical remission.205 Mechanistic studies are needed to establish whether TIGIT contributes to regulatory T-cell function in the intestine and whether reduced functioning of the TIGIT pathway accelerates or exacerbates intestinal inflammation.
Future perspectives: targeting co-inhibitory receptors in intestinal inflammation
Over the past decades, the concepts of T-cell costimulation and co-inhibition have substantially increased our understanding of the mechanisms controlling the development and maintenance of intestinal homeostasis (Table 2; Fig. 2). In autoimmune diseases such as RA and SLE, mechanistic analysis of co-inhibitory pathways has already led to the identification of potential therapeutic targets and initiation of clinical trials.3 As such, co-inhibitory receptors may represent potential novel therapeutic targets to treat chronic intestinal inflammation as seen in patients with IBD or intestinal graft-versus-host disease. Moreover, studies have only now begun to identify altered expression of co-inhibitory receptors in peripheral blood and intestinal tissue of IBD patients, raising the possibility that their expression could serve as predictive biomarkers to identify patients that are most likely benefit from targeted therapies.
Increasing the expression levels of co-inhibitory receptors is one possible mechanism to reduce intestinal inflammation. In peripheral blood of IBD patients, expression of Tim-3 is significantly decreased compared to healthy controls and preliminary data indicate that Tim-3 expression increases after anti-TNFα therapy.206 Similar results have been obtained in patients with multiple sclerosis (MS).207,208 Tim-3 expression is decreased on T cells in peripheral blood of MS patients during active disease and is induced specifically in responders to IFN-β therapy,209,210 a potent inducer of IL-27.211 This suggests that IL-27 administration might be a promising therapy to treat chronic inflammatory disease, possibly by overcoming intrinsic defects in the upregulation co-inhibitory receptor expression during inflammation.207,212 In humans, IL-27 polymorphisms are associated with susceptibility to IBD.213 Individuals homozygous for the IBD risk allele near the IL27 gene express less colonic IL-27 than individuals homozygous for the protective allele.214 Although multiple studies in mice have identified IL-27 as a suppressor of intestinal inflammation,176,215,216,217,218 investigations are ongoing to establish whether IL-27 administration is effective in IBD. In this context, it will be interesting to see whether decreased co-inhibitory receptor expression in intestinal tissue or peripheral blood are predictive biomarkers to identify patients that are most likely benefit from IL-27-directed therapy, or from other cytokines involved in inducing co-inhibitory receptor expression.
In addition to restoration of co-inhibitory receptor expression, co-inhibitory receptor stimulation or mimicking co-inhibitory receptor function could be beneficial to treat intestinal inflammation. An example of such an approach is abatacept, a chimeric CTLA-4 and IgG-Fc fusion protein that mimics the function of CTLA-4 by blocking CD28 costimulation.219 CTLA-4-Fc results in downregulation of T-cell activation and proliferation and has demonstrated efficacy in the treatment of autoimmune diseases such as RA and psoriatic arthritis.220 However, abatacept was not effective treating intestinal inflammation in IBD patients,221 which could reflect the relative lack of dependence of CD28 costimulation in memory T cells in the intestine. Moreover, as costimulation through CD28 is also required for the maintenance of Tregs in the periphery,222 abatacept may impede intestinal Treg survival by inhibiting essential costimulation mediated by CD28.223 Co-inhibitory receptor fusion proteins other than CTLA-4-Fc might be more successful to treat intestinal inflammation. As an example, the therapeutic potential of targeting the TIGIT/CD226/CD155 pathway by using a TIGIT-Fc fusion protein has been demonstrated in vivo in collagen-induced arthritis,204 but its efficacy in models of IBD remains to be tested.
Co-inhibitory molecules often have synergistic effects to dampen effector T-cell responses and enhance regulatory T-cell function. This is illustrated by models for T-cell exhaustion, where co-inhibitory receptors, such as TIGIT and PD-1, are often co-expressed on exhausted virus-specific CD8+ T cells.224 Expression of PD-1, Lag-3, Tim-3, and TIGIT all correlate with IL-10 production by human CD4+ T cells, although none of these inhibitory receptors are exclusively expressed on IL-10-producing T cells.201 This suggests that cooperative function of co-inhibitory receptor molecules is needed to achieve optimal T-cell regulation.201 In consequence, targeting multiple co-inhibitory receptors simultaneously might be needed for effective treatment of intestinal inflammation. Bi-specific antibodies can be used to target more than one co-inhibitory receptor and could co-ligate co-inhibitory receptors and stimulatory receptors simultaneously. As an example of the latter, crosslinking of Lag-3 and CD3 inhibits T-cell proliferation,172 suggesting that Lag-3-CD3 bispecific antibodies could harbor potential clinical efficacy in T-cell mediated inflammatory diseases, including inflammatory disease of the intestine.
Taken together, accumulating evidence suggests that targeting co-inhibitory receptors is a promising approach to treat intestinal inflammation that warrants further investigation. To define co-inhibitory targets for successful treatment of intestinal inflammation, it is essential to acknowledge their tissue-dependent functions and distinct responses based on cell type and associated kinetics.
Conclusion
This review highlights the emerging role of co-inhibitory receptors in intestinal homeostasis and elucidates many potential prospects for translation to human disease, such as IBD. Despite the substantial insights into the cell-specific expression and function of co-inhibitory receptors in the intestine over the past decades, there is still much to be learned. Key issues remaining to be resolved include the mechanisms of co-inhibitory molecule induction in the intestinal environment, how co-inhibitory signaling pathways integrate to achieve intestinal homeostasis, and whether memory T cells that reside in the intestinal mucosa require specific co-inhibitory signals. In addition, there is a need for a better mechanistic understanding of why inhibitory receptor pathway blockade leads to intestinal inflammation in some individuals but not in others. This should help us to develop more effective therapies while guaranteeing their safety profiles and obtain a better understanding of chronic intestinal inflammation.
References
Chen, L. & Flies, D. B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 (2013).
Schildberg, F. A. et al. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity 44, 955–972 (2016).
Zhang, Q. & Vignali, D. A. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 44, 1034–1051 (2016).
Brusca, S. B., Abramson, S. B. & Scher, J. U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 26, 101–107 (2014).
Wu, H. J. et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827 (2010).
Lee, Y. K. et al. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 108(Suppl 1), 4615–4622 (2011).
Pianta, A. et al. Two rheumatoid arthritis-specific autoantigens correlate microbial immunity with autoimmune responses in joints. J. Clin. Invest. 127, 2946–2956 (2017).
Tanoue, T., Atarashi, K. & Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 16, 295–309 (2016).
Abbas, A. K. et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat. Immunol. 14, 307–308 (2013).
Hauet-Broere, F. et al. Functional CD25- and CD25+ mucosal regulatory T cells are induced in gut-draining lymphoid tissue within 48 h after oral antigen application. Eur. J. Immunol. 33, 2801–2810 (2003).
Veenbergen, S. et al. Colonic tolerance develops in the iliac lymph nodes and can be established independent of CD103(+) dendritic cells. Mucosal Immunol. 9, 894–906 (2016).
Mowat, A. M. To respond or not to respond—a personal perspective of intestinal tolerance. Nat. Rev. Immunol. 18, 405–415 (2018).
Chen, W. et al. Conversion of peripheral CD4+ CD25- naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 (2003).
Coombes, J. L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Mucida, D. et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260 (2007).
Sun, C. M. et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785 (2007).
Awasthi, A. et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat. Immunol. 8, 1380–1389 (2007).
Zhao, H., Liao, X. & Kang, Y. Tregs: where we are and what comes next? Front. Immunol. 8, 1578 (2017).
Akdis, M. et al. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J. Exp. Med. 199, 1567–1575 (2004).
De Velasco, G. et al. Comprehensive meta-analysis of key immune-related adverse events from CTLA-4 and PD-1/PD-L1 inhibitors in cancer patients. Cancer Immunol. Res 5, 312–318 (2017).
Carreno, B. M. & Collins, M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 20, 29–53 (2002).
Collins, A. V. et al. The interaction properties of costimulatory molecules revisited. Immunity 17, 201–210 (2002).
Qureshi, O. S. et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J. Biol. Chem. 287, 9429–9440 (2012).
Chuang, E. et al. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J. Immunol. 159, 144–151 (1997).
Leung, H. T. et al. Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. J. Biol. Chem. 270, 25107–25114 (1995).
Sansom, D. M. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology 101, 169–177 (2000).
Fife, B. T. & Bluestone, J. A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 224, 166–182 (2008).
Buchbinder, E. I. & Desai, A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 39, 98–106 (2016).
Zheng, Y. et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940 (2007).
Wu, Y. et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387 (2006).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Tai, X. et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood 119, 5155–5163 (2012).
Takahashi, T. et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192, 303–310 (2000).
Jago, C. B. et al. Differential expression of CTLA-4 among T cell subsets. Clin. Exp. Immunol. 136, 463–471 (2004).
Perez-Garcia, A. et al. Kinetics of the CTLA-4 isoforms expression after T-lymphocyte activation and role of the promoter polymorphisms on CTLA-4 gene transcription. Hum. Immunol. 74, 1219–1224 (2013).
Alegre, M. L. et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 157, 4762–4770 (1996).
Tivol, E. A. et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 (1995).
Waterhouse, P. et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988 (1995).
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008).
Read, S. et al. Blockade of CTLA-4 on CD4+ CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 177, 4376–4383 (2006).
Mandelbrot, D. A., McAdam, A. J. & Sharpe, A. H. B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). J. Exp. Med. 189, 435–440 (1999).
Tai, X. et al. Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc. Natl Acad. Sci. USA 104, 13756–13761 (2007).
Corse, E. & Allison, J. P. Cutting edge: CTLA-4 on effector T cells inhibits in trans. J. Immunol. 189, 1123–1127 (2012).
Wang, C. J. et al. Cutting edge: cell-extrinsic immune regulation by CTLA-4 expressed on conventional T cells. J. Immunol. 189, 1118–1122 (2012).
Jain, N. et al. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl Acad. Sci. USA 107, 1524–1528 (2010).
Walker, L. S. & Sansom, D. M. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 36, 63–70 (2015).
Schmidt, E. M. et al. Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J. Immunol. 182, 274–282 (2009).
Oderup, C. et al. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology 118, 240–249 (2006).
Qureshi, O. S. et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603 (2011).
Walker, L. S. & Sansom, D. M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 11, 852–863 (2011).
Bachmann, M. F. et al. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 163, 1128–1131 (1999).
Homann, D. et al. Lack of intrinsic CTLA-4 expression has minimal effect on regulation of antiviral T-cell immunity. J. Virol. 80, 270–280 (2006).
Barnes, M. J. et al. CTLA-4 promotes Foxp3 induction and regulatory T cell accumulation in the intestinal lamina propria. Mucosal Immunol. 6, 324–334 (2013).
Sojka, D. K., Hughson, A. & Fowell, D. J. CTLA-4 is required by CD4+ CD25+ Treg to control CD4+ T-cell lymphopenia-induced proliferation. Eur. J. Immunol. 39, 1544–1551 (2009).
Read, S., Malmstrom, V. & Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 192, 295–302 (2000).
Liu, Z. et al. B7 interactions with CD28 and CTLA-4 control tolerance or induction of mucosal inflammation in chronic experimental colitis. J. Immunol. 167, 1830–1838 (2001).
Zeissig, S. et al. Early-onset Crohn’s disease and autoimmunity associated with a variant in CTLA-4. Gut 64, 1889–1897 (2015).
Kuehn, H. S. et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345, 1623–1627 (2014).
Schubert, D. et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 20, 1410–1416 (2014).
Lord, J. D. et al. Human blood and mucosal regulatory T cells express activation markers and inhibitory receptors in inflammatory bowel disease. PLoS One 10, e0136485 (2015).
Walker, L. S. Treg and CTLA-4: two intertwining pathways to immune tolerance. J. Autoimmun. 45, 49–57 (2013).
Powrie, F. & Mason, D. OX-22high CD4+ T cells induce wasting disease with multiple organ pathology: prevention by the OX-22low subset. J. Exp. Med. 172, 1701–1708 (1990).
Powrie, F. et al. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5, 1461–1471 (1993).
Mottet, C., Uhlig, H. H. & Powrie, F. Cutting edge: cure of colitis by CD4+ CD25+ regulatory T cells. J. Immunol. 170, 3939–3943 (2003).
Hadis, U. et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246 (2011).
Curotto de Lafaille, M. A. et al. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29, 114–126 (2008).
Husby, S. et al. Oral tolerance in humans. T cell but not B cell tolerance after antigen feeding. J. Immunol. 152, 4663–4670 (1994).
Mowat, A. M. et al. Immunological responses to fed protein antigens in mice. I. Reversal of oral tolerance to ovalbumin by cyclophosphamide. Immunology 45, 105–113 (1982).
Barone, K. S. et al. Effect of in vivo administration of anti-CTLA-4 monoclonal antibody and IL-12 on the induction of low-dose oral tolerance. Clin. Exp. Immunol. 130, 196–203 (2002).
Samoilova, E. B. et al. CTLA-4 is required for the induction of high dose oral tolerance. Int. Immunol. 10, 491–498 (1998).
Paterson, A. M. et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med. 212, 1603–1621 (2015).
Vieira, P. L. et al. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+ CD25+ regulatory T cells. J. Immunol. 172, 5986–5993 (2004).
Groux, H. et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389, 737–742 (1997).
Huber, S. et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(-) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565 (2011).
Coquerelle, C. et al. Anti-CTLA-4 treatment induces IL-10-producing ICOS+ regulatory T cells displaying IDO-dependent anti-inflammatory properties in a mouse model of colitis. Gut 58, 1363–1373 (2009).
Verma, N. et al. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin. Exp. Immunol. 190, 1–7 (2017).
Rueda, B. et al. CTLA4/CT60 polymorphism is not relevant in susceptibility to autoimmune inflammatory intestinal disorders. Hum. Immunol. 66, 321–325 (2005).
Howson, J. M. et al. A type 1 diabetes subgroup with a female bias is characterised by failure in tolerance to thyroid peroxidase at an early age and a strong association with the cytotoxic T-lymphocyte-associated antigen-4 gene. Diabetologia 50, 741–746 (2007).
Blomhoff, A. et al. Polymorphisms in the cytotoxic T lymphocyte antigen-4 gene region confer susceptibility to Addison’s disease. J. Clin. Endocrinol. Metab. 89, 3474–3476 (2004).
Repnik, K. & Potocnik, U. CTLA4 CT60 single-nucleotide polymorphism is associated with Slovenian inflammatory bowel disease patients and regulates expression of CTLA4 isoforms. DNA Cell Biol. 29, 603–610 (2010).
Maier, L. M. et al. Allelic variant in CTLA4 alters T cell phosphorylation patterns. Proc. Natl Acad. Sci. USA 104, 18607–18612 (2007).
Ueda, H. et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423, 506–511 (2003).
Hunt, K. A. et al. A common CTLA4 haplotype associated with coeliac disease. Eur. J. Hum. Genet. 13, 440–444 (2005).
van Belzen, M. J. et al. A genomewide screen in a four-generation Dutch family with celiac disease: evidence for linkage to chromosomes 6 and 9. Am. J. Gastroenterol. 99, 466–471 (2004).
Greenwald, R. J., Freeman, G. J. & Sharpe, A. H. The B7 family revisited. Annu. Rev. Immunol. 23, 515–548 (2005).
Ishida, Y. et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992).
Goldberg, M. V. et al. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood 110, 186–192 (2007).
Okazaki, T. et al. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl Acad. Sci. USA 98, 13866–13871 (2001).
Patsoukis, N. et al. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol. Cell. Biol. 33, 3091–3098 (2013).
Hui, E. et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 (2017).
Parry, R. V. et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553 (2005).
Kamphorst, A. O. et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355, 1423–1427 (2017).
Nishimura, H. et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 (1999).
Ansari, M. J. et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 198, 63–69 (2003).
Nishimura, H. et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291, 319–322 (2001).
Agata, Y. et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8, 765–772 (1996).
Yao, S. et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 113, 5811–5818 (2009).
Vibhakar, R. et al. Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Exp. Cell Res. 232, 25–28 (1997).
Yamazaki, T. et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 169, 5538–5545 (2002).
Terawaki, S. et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol. 186, 2772–2779 (2011).
Dong, H. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800 (2002).
Nomi, T. et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin. Cancer Res. 13, 2151–2157 (2007).
Hamanishi, J. et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl Acad. Sci. USA 104, 3360–3365 (2007).
Freeman, G. J. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000).
Taube, J. M. et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med 4, 127ra37 (2012).
Salama, A. D. et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 198, 71–78 (2003).
Blank, C. et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 64, 1140–1145 (2004).
Keir, M. E. et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203, 883–895 (2006).
Spranger, S. et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med 5, 200ra116 (2013).
Gubin, M. M. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014).
Francisco, L. M. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 (2009).
Wang, L. et al. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+ CD4+regulatory T cells. Proc. Natl Acad. Sci. USA 105, 9331–9336 (2008).
Zhang, B. et al. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc. Natl Acad. Sci. USA 113, 8490–8495 (2016).
Asano, T. et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood 129, 2186–2197 (2017).
Alfen J. S., et al. Intestinal IFN-gamma-producing type 1 regulatory T cells coexpress CCR5 and programmed cell death protein 1 and downregulate IL-10 in the inflamed guts of patients with inflammatory bowel disease. J Allergy Clin Immunol 2018.
Reynoso, E. D. et al. Intestinal tolerance is converted to autoimmune enteritis upon PD-1 ligand blockade. J. Immunol. 182, 2102–2112 (2009).
Pauken, K. E. et al. Cutting edge: identification of autoreactive CD4+ and CD8+ T cell subsets resistant to PD-1 pathway blockade. J. Immunol. 194, 3551–3555 (2015).
Kawamoto, S. et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science 336, 485–489 (2012).
Good-Jacobson, K. L. et al. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 11, 535–542 (2010).
Sharma, M. D. et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv. 1, e1500845 (2015).
Maruya, M. et al. Impaired selection of IgA and intestinal dysbiosis associated with PD-1-deficiency. Gut Microbes 4, 165–171 (2013).
Gutzeit, C., Magri, G. & Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 260, 76–85 (2014).
Johansen, F. E. et al. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J. Exp. Med. 190, 915–922 (1999).
Ludvigsson, J. F., Neovius, M. & Hammarstrom, L. Association between IgA deficiency & other autoimmune conditions: a population-based matched cohort study. J. Clin. Immunol. 34, 444–451 (2014).
Latiff, A. H. & Kerr, M. A. The clinical significance of immunoglobulin A deficiency. Ann. Clin. Biochem. 44, 131–139 (2007).
Zinneman, H. H. & Kaplan, A. P. The association of giardiasis with reduced intestinal secretory immunoglobulin A. Am. J. Dig. Dis. 17, 793–797 (1972).
Kwon, H. et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 31, 941–952 (2009).
Vinuesa, C. G. et al. Follicular B helper T cells in antibody responses and autoimmunity. Nat. Rev. Immunol. 5, 853–865 (2005).
Arthur, J. C. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 (2012).
Scandiuzzi, L. et al. Tissue-expressed B7-H1 critically controls intestinal inflammation. Cell Rep. 6, 625–632 (2014).
Nakazawa, A. et al. The expression and function of costimulatory molecules B7H and B7-H1 on colonic epithelial cells. Gastroenterology 126, 1347–1357 (2004).
Kanai, T. et al. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J. Immunol. 171, 4156–4163 (2003).
Bertrand, A. et al. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 13, 211 (2015).
Gupta, A. et al. Systematic review: colitis associated with anti-CTLA-4 therapy. Aliment Pharmacol. Ther. 42, 406–417 (2015).
Tandon P. et al. The risk of diarrhea and colitis in patients with advanced melanoma undergoing immune checkpoint inhibitor therapy: a systematic review and meta-analysis. J. Immunother. 2018.
Wolchok, J. D. et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 377, 1345–1356 (2017).
Chen, J. H. et al. Histopathologic features of colitis due to immunotherapy with anti-PD-1 antibodies. Am. J. Surg. Pathol. 41, 643–654 (2017).
Oble, D. A. et al. Alpha-CTLA-4 mAb-associated panenteritis: a histologic and immunohistochemical analysis. Am. J. Surg. Pathol. 32, 1130–1137 (2008).
Garcia-Varona, A., Odze, R. D. & Makrauer, F. Lymphocytic colitis secondary to ipilimumab treatment. Inflamm. Bowel Dis. 19, E15–E16 (2013).
Marthey, L. et al. Cancer immunotherapy with anti-CTLA-4 monoclonal asntibodies induces an inflammatory bowel disease. J. Crohns Colitis 10, 395–401 (2016).
Nancey, S. et al. Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab is associated with a profound long-lasting depletion of Foxp3+ regulatory T cells: a mechanistic explanation for ipilimumab-induced severe enterocolitis? Inflamm. Bowel Dis. 18, E1598–E1600 (2012).
Simpson, T. R. et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 210, 1695–1710 (2013).
Maker, A. V., Attia, P. & Rosenberg, S. A. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J. Immunol. 175, 7746–7754 (2005).
Lord, J. D. et al. Refractory colitis following anti-CTLA4 antibody therapy: analysis of mucosal FOXP3+ T cells. Dig. Dis. Sci. 55, 1396–1405 (2010).
Zhou, Y. & Zhi, F. Lower level of bacteroides in the gut microbiota is associated with inflammatory bowel disease: a meta-analysis. Biomed. Res. Int. 2016, 5828959 (2016).
Swidsinski, A. et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 122, 44–54 (2002).
Andoh, A. et al. Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn’s disease. J. Gastroenterol. 47, 1298–1307 (2012).
Sanderson, K. et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J. Clin. Oncol. 23, 741–750 (2005).
June, C. H., Warshauer, J. T. & Bluestone, J. A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 23, 540–547 (2017).
Torphy R. J., Schulick R. D., Zhu Y. Newly emerging immune checkpoints: promises for future cancer therapy. Int. J. Mol. Sci. 18 (2017).
Marin-Acevedo, J. A. et al. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J. Hematol. Oncol. 11, 39 (2018).
Collin, M. Immune checkpoint inhibitors: a patent review (2010–2015). Expert Opin. Ther. Pat. 26, 555–564 (2016).
Bekiaris, V. et al. The inhibitory receptor BTLA controls gammadelta T cell homeostasis and inflammatory responses. Immunity 39, 1082–1094 (2013).
Murphy, T. L. & Murphy, K. M. Slow down and survive: enigmatic immunoregulation by BTLA and HVEM. Annu. Rev. Immunol. 28, 389–411 (2010).
Steinberg, M. W. et al. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J. Exp. Med. 205, 1463–1476 (2008).
Hurchla, M. A. et al. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly Induced in anergic CD4+ T cells. J. Immunol. 174, 3377–3385 (2005).
Cai, G. et al. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat. Immunol. 9, 176–185 (2008).
Mauri, D. N. et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8, 21–30 (1998).
Montgomery, R. I. et al. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87, 427–436 (1996).
Wang, Y. et al. The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J. Clin. Invest. 115, 711–717 (2005).
Sedy, J. R. et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 6, 90–98 (2005).
Schaer, C. et al. HVEM signalling promotes colitis. PLoS ONE 6, e18495 (2011).
Kim, W. K. et al. Role of TNFR-related 2 mediated immune responses in dextran sulfate sodium-induced inflammatory bowel disease. Mol. Cells 31, 99–104 (2011).
Shui, J. W. et al. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature 488, 222–225 (2012).
Anumanthan, A. et al. Cloning of BY55, a novel Ig superfamily member expressed on NK cells, CTL, and intestinal intraepithelial lymphocytes. J. Immunol. 161, 2780–2790 (1998).
Triebel, F. et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405 (1990).
Bruniquel, D. et al. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics 48, 116–124 (1998).
Huard, B. et al. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 25, 2718–2721 (1995).
Huard, B. et al. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur. J. Immunol. 26, 1180–1186 (1996).
Workman, C. J., Dugger, K. J. & Vignali, D. A. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 169, 5392–5395 (2002).
Workman, C. J. & Vignali, D. A. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol. 33, 970–979 (2003).
Hannier, S. et al. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J. Immunol. 161, 4058–4065 (1998).
Anderson, A. C., Joller, N. & Kuchroo, V. K. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004 (2016).
Durham, N. M. et al. Lymphocyte activation gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS One 9, e109080 (2014).
Gagliani, N. et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 19, 739–746 (2013).
Do, J. S. et al. An IL-27/Lag3 axis enhances Foxp3+ regulatory T cell-suppressive function and therapeutic efficacy. Mucosal Immunol. 9, 137–145 (2016).
Hall, A. O., Silver, J. S. & Hunter, C. A. The immunobiology of IL-27. Adv. Immunol. 115, 1–44 (2012).
Liang, B. et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol. 180, 5916–5926 (2008).
Okamura, T. et al. CD4+ CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc. Natl Acad. Sci. USA 106, 13974–13979 (2009).
Okamura, T. et al. TGF-beta3-expressing CD4+ CD25(-)LAG3+ regulatory T cells control humoral immune responses. Nat. Commun. 6, 6329 (2015).
Monney, L. et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002).
Chiba, S. et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 13, 832–842 (2012).
Jiang, X. et al. Tim-3 promotes intestinal homeostasis in DSS colitis by inhibiting M1 polarization of macrophages. Clin. Immunol. 160, 328–335 (2015).
Patel, J., Bozeman, E. N. & Selvaraj, P. Taming dendritic cells with TIM-3: another immunosuppressive strategy used by tumors. Immunotherapy 4, 1795–1798 (2012).
Gautron, A. S. et al. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur. J. Immunol. 44, 2703–2711 (2014).
Hastings, W. D. et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur. J. Immunol. 39, 2492–2501 (2009).
Golden-Mason, L. et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 83, 9122–9130 (2009).
Jones, R. B. et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 205, 2763–2779 (2008).
Jin, H. T. et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl Acad. Sci. USA 107, 14733–14738 (2010).
Zhu, C. et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 6, 6072 (2015).
Huang, Y. H. et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517, 386–390 (2015).
Li, X. et al. Involvement of T cell Ig Mucin-3 (Tim-3) in the negative regulation of inflammatory bowel disease. Clin. Immunol. 134, 169–177 (2010).
Morimoto, K. et al. Dysregulated upregulation of T-cell immunoglobulin and mucin domain-3 on mucosal T helper 1 cells in patients with Crohn’s disease. Scand. J. Gastroenterol. 46, 701–709 (2011).
Shi, F. et al. Dysregulated Tim-3 expression and its correlation with imbalanced CD4 helper T cell function in ulcerative colitis. Clin. Immunol. 145, 230–240 (2012).
Yu, X. et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 10, 48–57 (2009).
Wang, F. et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur. J. Immunol. 45, 2886–2897 (2015).
Casado, J. G. et al. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol. Immunother. 58, 1517–1526 (2009).
Bottino, C. et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 198, 557–567 (2003).
Lozano, E. et al. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 188, 3869–3875 (2012).
Joller, N. et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 186, 1338–1342 (2011).
Burton, B. R. et al. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nat. Commun. 5, 4741 (2014).
Joller, N. et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40, 569–581 (2014).
Fuhrman, C. A. et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J. Immunol. 195, 145–155 (2015).
Levin, S. D. et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 41, 902–915 (2011).
Joosse M. E. et al. Frequencies of circulating regulatory TIGIT(+)CD38(+) effector T cells correlate with the course of inflammatory bowel disease. Mucosal Immunol. https://doi.org/10.1038/s41385-018-0078-4 2018. [Epub ahead of print].
Kim, M. J., Lee, W. Y. & Choe, Y. H. Expression of TIM-3, human beta-defensin-2, and FOXP3 and correlation with disease activity in pediatric Crohn’s disease with infliximab therapy. Gut Liver 9, 370–380 (2015).
Koguchi, K. et al. Dysregulated T cell expression of TIM3 in multiple sclerosis. J. Exp. Med. 203, 1413–1418 (2006).
Yang, L. et al. Lack of TIM-3 immunoregulation in multiple sclerosis. J. Immunol. 180, 4409–4414 (2008).
Ottoboni, L. et al. An RNA profile identifies two subsets of multiple sclerosis patients differing in disease activity. Sci. Transl. Med 4, 153ra131 (2012).
Boivin, N. et al. Interferon-beta suppresses murine Th1 cell function in the absence of antigen-presenting cells. PLoS One 10, e0124802 (2015).
Sweeney, C. M. et al. IL-27 mediates the response to IFN-beta therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav. Immun. 25, 1170–1181 (2011).
Andrews, C., McLean, M. H. & Durum, S. K. Interleukin-27 as a novel therapy for inflammatory bowel disease: a critical review of the literature. Inflamm. Bowel Dis. 22, 2255–2264 (2016).
Li, C. S. et al. Interleukin-27 polymorphisms are associated with inflammatory bowel diseases in a Korean population. J. Gastroenterol. Hepatol. 24, 1692–1696 (2009).
Imielinski, M. et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 41, 1335–1340 (2009).
Dann, S. M. et al. Attenuation of intestinal inflammation in interleukin-10-deficient mice infected with Citrobacter rodentium. Infect. Immun. 82, 1949–1958 (2014).
Hanson, M. L. et al. Oral delivery of IL-27 recombinant bacteria attenuates immune colitis in mice. Gastroenterology 146, 210–221 e13 (2014).
Sasaoka, T. et al. Treatment with IL-27 attenuates experimental colitis through the suppression of the development of IL-17-producing T helper cells. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G568–G576 (2011).
Troy, A. E. et al. IL-27 regulates homeostasis of the intestinal CD4+ effector T cell pool and limits intestinal inflammation in a murine model of colitis. J. Immunol. 183, 2037–2044 (2009).
Dall’Era, M. & Davis, J. CTLA4Ig: a novel inhibitor of costimulation. Lupus 13, 372–376 (2004).
Genovese, M. C. et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N. Engl. J. Med. 353, 1114–1123 (2005).
Sandborn, W. J. et al. Abatacept for Crohn’s disease and ulcerative colitis. Gastroenterology 143, 62–69 e4 (2012).
Bour-Jordan, H. & Bluestone, J. A. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol. Rev. 229, 41–66 (2009).
Mayer, L., Kaser, A. & Blumberg, R. S. Dead on arrival: understanding the failure of CTLA4-immunoglobulin therapy in inflammatory bowel disease. Gastroenterology 143, 13–17 (2012).
Johnston, R. J. et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937 (2014).
White, A. M. & Wraith, D. C. Tr1-like T cells—an enigmatic regulatory T cell lineage. Front. Immunol. 7, 355 (2016).
Acknowledgements
We thank the members of the Pediatric Gastroenterology laboratory for valuable discussions. This work was supported by the Netherlands Organization for Scientific Research grant 2013/09420/BOO and Dutch Sophia Research Foundation S13-19. LSKW is supported by the Medical Research Council.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Joosse, M.E., Nederlof, I., Walker, L.S.K. et al. Tipping the balance: inhibitory checkpoints in intestinal homeostasis. Mucosal Immunol 12, 21–35 (2019). https://doi.org/10.1038/s41385-018-0113-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-018-0113-5
This article is cited by
-
Post-transplant Inflammatory Bowel Disease Associated with Donor-Derived TIM-3 Deficiency
Journal of Clinical Immunology (2024)
-
Reduced immune-regulatory molecule expression on human colonic memory CD4 T cells in older adults
Immunity & Ageing (2021)
-
Mechanisms of activation of innate-like intraepithelial T lymphocytes
Mucosal Immunology (2020)
-
Fine tuning of the DNAM-1/TIGIT/ligand axis in mucosal T cells and its dysregulation in pediatric inflammatory bowel diseases (IBD)
Mucosal Immunology (2019)
