
Abstract
Mucosal barriers constitute major body surfaces that are in constant contact with the external environment. Mucosal sites are densely populated by a myriad of distinct neurons and immune cell types that sense, integrate and respond to multiple environmental cues. In the recent past, neuro-immune interactions have been reported to play central roles in mucosal health and disease, including chronic inflammatory conditions, allergy and infectious diseases. Discrete neuro-immune cell units act as building blocks of this bidirectional multi-tissue cross-talk, ensuring mucosal tissue health and integrity. Herein, we will focus on reciprocal neuro-immune interactions in the airways and intestine. Such neuro-immune cross-talk maximizes sensing and integration of environmental aggressions, which can be considered an important paradigm shift in our current views of mucosal physiology and immune regulation.
Similar content being viewed by others

Introduction
Mucosal barriers are major interfaces with the external environment, playing central roles in organismal physiology and immune surveillance. At these sites, an integrated network of distinct tissues and cell types ensures mucosal homeostasis and host protection from environmental insults and infectious agents.1 A major structural component of mucosal surfaces is the epithelial cell layer, which forms a physical barrier with the external environment and exerts dynamic responses that contribute to host defence. These processes encompass the production of mucus and inflammatory mediators, including chemokines and cytokines that recruit and regulate innate and adaptive immune cells.2,3 As such, a compromised barrier function is a prominent feature of mucosal inflammatory disorders, including those affecting the gastrointestinal (GI) tract (inflammatory bowel disease) and the respiratory system (asthma, allergy and chronic obstructive pulmonary disease (COPD)). Importantly, the worldwide incidence and prevalence of these diseases have increased drastically over the past 50 years,4 and understanding the onset and pathophysiology of such pathologies is an undisputed unmet need.
In the past few years, pioneer studies highlighted the importance of neuronal signals in regulating mucosal immune cells and mucosal barrier integrity.5,6,7 Therefore, understanding the communication strategies linking the nervous and immune systems may lead, in the near future, to novel therapies for multiple conditions, including chronic inflammation, cancer and metabolic disorders, amongst others. Herein, we review recent findings on how neuro-immune interactions shape the homeostasis of the GI and respiratory systems and will discuss how these interactions might be harnessed in health and disease.
Mucosal innervation
Mucosal tissues are highly innervated, notably in the intestine, lung and nasal mucosae, where the nervous system exerts a profound influence on digestive and respiratory processes, respectively. Importantly, these barriers also harbour a large set of lymphoid tissues and immune cell subsets, indicating that neuronal and immune cells may share similar anatomical compartments at mucosal sites.
Enteric innervation
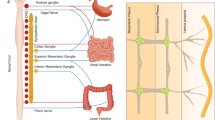
The GI tract may be seen as a dynamic barrier that discriminates between beneficial commensals and pathogens, and that screens ingested material allowing for nutrients to be absorbed while excluding toxic substances.8 In addition, the digestive tract exerts a large range of functions, including digestion, absorption, motility, excretion, secretion and defence. These functions are partly regulated by signals arising from parasympathetic and sympathetic extrinsic neurons, which cell bodies are located within the brainstem and spinal cord, and by the intrinsic enteric nervous system (ENS), characterized by ganglionated plexus embedded in the lining of the GI wall5,7,9,10,11,12 (Fig. 1a). The ENS constitutes an extensive neuronal network with as many neurons as the spinal cord and it is therefore commonly called as the 'second brain'.5 The intrinsic innervation of the gut is arranged in two main ganglionated layers that contain numerous enteric neurons and enteric glial cells (ECGs): the myenteric plexus (or Auerbach’s plexus), which is located between the circular and longitudinal muscle layers, and the submucosal plexus (or Meissner’s plexus), which is placed in the gut submucosa.5,11,13,14 The myenteric plexus forms a continuous entity that extends from the upper oesophagus to the internal anal sphincter and receives inputs mainly from the vagus nerve to coordinate the contractions and relaxations of the gut smooth muscle and ensure the digestive tract motility and peristalsis.15 The submucosal plexus is found in the small and large intestine and contains two distinguishable ganglionated plexus in humans and large mammals. The inner submucosal plexus (closer to the intestinal lumen) mainly innervates the mucosa and the muscularis mucosae and is mostly involved in the sensing and control of the luminal environment, regulating the GI blood flow and epithelial cell functions.15 The outer submucosal plexus (closer to the circular muscle layer) plays a significant role in intestinal motility.15 In addition, scarce and small ganglia can be found in the mucosa, close to the muscularis mucosae.16 The ENS also contains abundant EGCs and neuronal terminal varicosities that are in close proximity with lymphoid tissues and resident immune cells in the lamina propria.5,17 Enteric neurons can be divided into three main classes: (i) intrinsic primary afferent neurons, which are sensitive to chemical and mechanical stimuli; (ii) interneurons that connect to other interneurons and to motor neurons; (iii) motor neurons that act on different effector cells, notably muscle cells, blood vessels, epithelial cells and haematopoietic cells in the intestinal lamina propria.5,9,12,18,19 While the ENS activity is largely autonomous, its functions can be affected and modulated by external neural inputs provided by the Central nervous system (CNS) via vagal, spinal, pelvic and sympathetic innervation.5 Extrinsic neuronal inputs do not control discrete gut motility patterns, but they can enhance or inhibit ENS activities.8,9 Reciprocally, the ENS can also project axons outwards to the periphery through vagal, spinal, pelvic and sympathetic pathways.9,19,20 Notably, 90% of the vagal fibres that transit between the gut and the brain are of afferent origin, suggesting that the brain may be more of a receiver than a transmitter in the so-called gut–brain axis.19

Innervation of mucosal surfaces. a Neuronal network in the intestine. The gastrointestinal tract is innervated by intrinsic neurons (enteric) and by the axons of extrinsic sympathetic and parasympathetic neurons that project to the gut wall. IPANs intrinsic primary afferent sensory neurons, ENS enteric nervous system, CNS central nervous system. b The nervous system in the lungs. Vagal nerves supply all parasympathetic and most of the sensory neurons to the airways. Some sensory innervation originates from the dorsal root ganglia and run through spinal sympathetic nerves. ASM airway smooth muscle
Pulmonary innervation
The nervous system in the respiratory tract plays an important role in the maintenance of lung homeostasis, via regulation of mucus secretion, vascular permeability, airway smooth muscle tone and blood flow.21,22 The pulmonary intrinsic ganglia, mostly comprised of neurons and glia cells, are a recognized component of the developing lung. The lung buds develop as an outgrowth of the foregut, which contains migrating neural crest cells that ultimately give rise to the ENS.23,24 The innervation of the lung consists of afferent and efferent neurons whose axons reach the airways by both vagal and sympathetic nerves22 (Fig. 1b). Vagal nerves supply all parasympathetic and most of the sensory neuronal fibres in the airways. Pulmonary afferent signals are transmitted by vagal sensory axons originating in the lamina propria, which cell bodies reside in the jugular and nodose ganglia and then project into the medulla25 (Fig. 1b). Some additional afferent sensory signals originate in the dorsal root ganglia that connect with thoracic ganglia to supply sympathetic innervation to blood vessels.21 The afferent innervation of the lungs encompasses sensory receptors, mechano-sensors and chemosensors, which have been extensively studied and reviewed elsewhere.22 Nociceptive C-fibres represent 80% of the vagal afferents nerves, and the former are promptly activated by noxious chemical and inflammatory molecules. Mechano-sensory nerves are activated by respiratory mechanical forces and are normally less sensitive to inflammatory stimuli.22,26,27 Lung afferent and efferent neurons connect via the brainstem. Efferent neuronal activation can be classified into sympathetic nerves, cholinergic parasympathetic nerves, non-adrenergic non-cholinergic parasympathetic nerves and the efferent sensory nerves.28 The efferent nerves arising from the thoracic and superior sympathetic ganglia are mainly involved in the relaxation of airway smooth muscles via activation of β-adrenoceptors.28 Mounting evidence indicates that activation of peripheral neurons results in neurotransmitter and neuropeptide release that can lead to neurogenic inflammation and enhanced central reflex activity through the CNS; notably, it was reported that altered innervation can contribute to respiratory diseases such as asthma, rhinitis and COPD.22
Enteric neuro-immune interactions
The expression of neurotransmitter and neuropeptide receptors by distinct immune cell subsets has been suggesting that neuronal-derived signals may regulate immune cell function.5,7,29 Reciprocally, neurons can also express cytokine receptors to detect and respond to inflammation.30 This shared molecular repertoire supports an integrated neuro-immune interaction that is likely to contribute to organ homeostasis. Neuro-immune communications are of particular significance in the intestine, where the diversity and complexity of neuronal and immune cell types is required to sense, react and adapt to ever changing conditions.31
Neuronal–mast cell cross-talk
Mast cells were shown to lie in close juxtaposition with intestinal substance P (SP-) and/or calcitonin gene-related peptide (CGRP-) producing neurons, both in rats and humans.32,33 Activation by external stimuli induces mast cell degranulation with consequent release of inflammatory granule components, such as histamine and mast cell proteases.34 Importantly, mast cell-derived mediators can act on enteric neuron receptors.35 Notably, histamine can activate neurons in the myenteric plexus of the small intestine, inducing neuronal hyperexcitability via neuronal H2 receptors.36 Additionally, mast cell-derived tryptase can trigger proteinase-activated receptor 2 (PAR2) on submucosal neurons to causes acute and long-term neuronal hyperexcitability37 and release of CGRP and SP neuromodulators.38 Interestingly, PAR2 activation was reported to induce neurogenic colitis, and treatment with SP and CGRP receptor antagonists ameliorates PAR2-induced colitis in mice.39 Reciprocally, peptidergic neurons were also shown to modulate mast cell function via secretion of SP, CGRP and vasoactive intestinal peptide (VIP).35 Human mast cells respond to SP and VIP stimulation, which leads to mast cell degranulation and cytokine production.40 In rodents, enteric mast cells isolated from rats infected with Nippostrongylus brasiliensis secrete histamine after stimulation with SP.41 Notably, mast cell proteases were described to enzymatically cleave neuron-derived SP and VIP,42 suggesting that mast cells may limit neuronal activation.35 While there is no clear evidence of a functional mast cell–neuron interaction regulating enteric pathophysiology, some studies have correlated mast cell activation with irritable bowel syndrome (IBS) pathogenesis. Notably, supernatants from colonic biopsies of IBS patients display higher levels of histamine and tryptase, when compared with healthy controls,43,44,45 and increased mast cell numbers are found in the vicinity of enteric neurons from IBS patients.43,44 Interestingly, supernatants of colonic biopsies from IBS patients were shown to increase the firing rates of mesenteric afferent nerves and dorsal root ganglia nociceptive neurons in rats44 and human submucosal neurons.45 Altogether, these findings suggest that mast cell-derived molecules activate adjacent peptidergic neurons, which, in turn, may regulate mast cell function in a bidirectional positive feedback-loop that might be significant in the context of neurogenic inflammatory disorders.
Neuronal-macrophage cross-talk
The intestine also contains a vast reservoir of macrophages, most of which locate in the lamina propria and in the muscularis externa.46 Recently, two independent studies reported that muscularis macrophages (MMs) can establish a bidirectional interplay with mucosal neurons47,48 (Fig. 2a). Muller et al.48 demonstrated that MMs can regulate homeostatic GI motility. Blockage of colony stimulatory factor 1 receptor (CSF1R) signalling, an essential molecule for macrophage development, induces MM depletion and subsequent GI dysfunction.48 In addition, MMs in close proximity to enteric neurons secrete bone morphogenetic protein 2 (BMP2) that control peristalsis.48 In agreement, treatment with a BMP receptor inhibitor phenocopies MM-depleted mice, while ex vivo administration of BMP2 restores colonic peristalsis.48 Interestingly, enteric neurons in the muscularis are the main source of CSF1, suggesting a reciprocal neuronal regulation of MMs.48 Remarkably, the production of CSF1 and BMP2 rely on luminal microbes, demonstrating that commensal signals can shape neuronal–MM interaction and GI activity.48 Thus, an intricate bidirectional interplay between MMs and enteric neurons is regulated by microbial cues to control intestinal peristalsis. More recently, Gabanyi et al.47 demonstrated that intestinal lamina propria macrophages display a pro-inflammatory phenotype, while MMs show a tissue-protective profile.47 Remarkably, MMs differentially express β2 adrenergic receptor (β2AR), which is required for norepinephrine signalling. The expression of β2AR together with a close proximity to neurons suggested a molecular and anatomic basis for a neuron–MM interaction. In agreement, infection with Salmonella typhimurium activates extrinsic tyrosine hydroxylase-expressing neurons in the sympathetic ganglia, with consequent norepinephrine release in the intestinal muscularis.47 In turn, norepinephrine activation of β2AR-expressing MMs drives their polarization towards a tissue-protective phenotype47 (Fig. 2a).

Intestinal neuro-immune interactions. a Microbial commensals tune intestinal peristalsis by shaping the bidirectional communication among CSF1-producing enteric neurons and BMP2-producing MMs. Activation of extrinsic sympathetic ganglia by bacteria drives the polarization of β2AR-expressing MMs into a tissue-protective phenotype. b EGC sense microbiota-derived cues to control GFLs expression. ECG-derived GFLs activate RET-expressing ILC3s, which induces IL-22 that ultimately controls intestinal homeostasis. c Cholinergic enteric neurons sense and respond to worm products by producing the neuropeptide NMU. Neuronal-derived NMU induces an immediate activation of NMUR1-expressing ILC2s that contributes to a protective immune response against parasitic infections. CSF1 colony-stimulating factor 1, Mo monocyte, BMP2, bone morphogenetic protein 2, NE norepinephrine, Arg1 encodes the arginase 1 protein, Chi3l3 encodes the chitinase-like protein 3, GFLs, glial cell line-derived neurotrophic factor family ligands, IL interleukin, ILC innate lymphoid cell, NMU neuromedin U
Neuronal-innate lymphoid cell cross-talk
Group 2 innate lymphoid cells (ILC2s) are abundant at mucosal barriers.49 ILC2s contribute to anti-helminth immunity, regulation of allergic responses and tissue repair.49 Akin to other ILC subsets, ILC2s do not recognize specific antigen or pathogen-associated molecular patterns (PAMPs).49,50 ILC2s rather sense barrier disruption by integrating locally produced host-derived signals, including alarmins, lipid mediators and neuropeptides.49,50 Three recent independent studies reported that mucosal neurons can directly regulate innate type 2 immunity via neuromedin U (NMU)51,52,53 (Fig. 2c). Analysis of the ILC transcriptome revealed that ILC2s selectively express NMU receptor 1 (Nmur1), which encodes the peripheral receptor of the neuropeptide NMU.51,52,53 NMU is a highly conserved neuropeptide, mostly expressed in the GI tract and the CNS.54 Intestinal ILC2s were found in close proximity to NMU-expressing cholinergic neurons51,52 (Fig. 2c). Remarkably, activation of ILC2s with NMU leads to a uniquely fast and potent production of type 2 cytokines, including interleukin-5 (IL-5), IL-13 and amphiregulin in a Nmur1-dependent manner.51,52 Accordingly, in vivo NMU treatment induces a selective increase of innate type 2 cytokine production, while their adaptive counterparts are unaffected. Interestingly, infection with N. brasiliensis induces a strong increase of Nmu expression in infected tissues, suggesting that NMU–NMUR1 axis may orchestrate barrier defence.51,52 In agreement, in vivo administration of NMU to N. brasiliensis-infected mice results in a selective increase of innate type 2 responses, reduced tissue damage and reduced infection burden.51,52 Sensing molecules, such as toll-like receptors (TLRs), contribute for the initiation of type 2 immunity.50 Using neurosphere-derived neuronal organoids, Cardoso et al.51 revealed that cholinergic neurons upregulate Nmu expression in response to N. brasiliensis excretory/secretory products in a myeloid differentiation primary response 88 (MYD88)-dependent manner.51 Taken together, these studies uncover a novel layer of homeostatic regulation whereby mucosal cholinergic neurons sense pathogen-derived molecules that induce NMU production and consequent activation of ILC2s (Fig. 2c).
Adding to the neuronal regulation of ILC2 responses, catecholaminergic neurons, a sympathetic nervous system component, were shown to impair innate type 2 responses.55 Catecholaminergic neurons are a source of norepinephrine that binds the β2AR.5 β2AR deficiency leads to increased ILC2 responses, while β2AR agonist treatment led to deficient ILC2 responses and impaired inflammation, indicating that neuronal-derived signals can limit type 2 inflammation55 (Fig. 2c).
Interestingly, while norepinephrine regulates enteric MM polarization towards an anti-inflammatory protective M2 phenotype,47 adipose tissue sympathetic neuron-associated macrophages, which also sense norepinephrine, exhibit a pro-inflammatory profile.56 In addition, IL-13-dependent and IL-4-dependent mechanisms are essential to maintain M2 macrophage homeostasis.57 These observations suggest that tissue-resident macrophages by differently integrating neuronal-derived and microenvironment-derived cues might be able to rapidly adapt to local settings. Since ILC2s, that also share β2AR expression, are an important source of IL-13, it would be interesting to dissect how norepinephrine levels differently regulate mucosal ILC2 and macrophage pools and how the balance between these two immune subsets may modulate immunity in the context of helminth infections, allergic inflammation and metabolic disorders.
Enteric glial–immune cell interactions
In addition to the cues provided by intrinsic and extrinsic enteric neurons, EGCs were also shown to be important players in the neuro-immune cross-talk.7,58 EGCs represent a key cellular element of the ENS and they have been historically described as providers of neuronal structural support.58 Nevertheless, it is becoming clear that EGCs are more broadly important to GI homeostasis.58 As an example, EGCs were shown to promote neuronal survival to oxidative stress damage.59,60 Earlier studies employing two independent models (Gfap-HSVtk and Gfap-HA.CL4-TCR)61,62 indicated that ablation of glial fibrillary acidic protein (GFAP)-expressing EGCs led to fatal intestinal inflammation, accompanied by disrupted intestinal epithelial barrier.61,62 Nevertheless, recent studies revisited this issue to demonstrate that EGCs are a morphologically and functionally heterogenous populations, and that GFAP expression is limited to a particular EGC subset.63 Notably, the proteolipid protein 1 (PLP1) was found to be broadly expressed by EGCs.63 In contrast to earlier observations, inducible ablation of EGCs using Plp1CreER.Rosa26DTA fail to induce intestinal inflammation and disrupted intestinal barrier permeability.64 The distinct outcomes of these studies may rely on non-cell-autonomous expression of different transgenic lines or to insufficient efficiency in deleting glial cell. Nevertheless, it has been established that during acute intestinal inflammation EGCs can cause intestinal neurodegeneration through secretion of inflammatory mediators that drive neuronal cell death and intestinal dysmotility.65,66 Thus, deficits of glial neuroprotective functions or activation of neurotoxic glial properties can lead to GI derangements. Nonetheless, additional studies with EGC-autonomous genetic models are still required to clearly elucidate the role of EGCs in intestinal homeostasis.
EGCs are the main producers of neurotrophic factors including glial cell line-derived neurotrophic factor (GDNF) family of ligands (GFL), including GDNF, neurturin (NRTN), artemin (ARTN) and persephin (PSPN), all of which can modulate enteric neuronal function.31 As an example, Nrtn-deficient mice exhibit reduced myenteric plexus innervation density and altered GI motility, while Gdnf-knockout mice display intestinal aganglionosis.67,68,69 GFL signalling involves the shared signalling-transducing element RET (rearranged during transfection) and one of four GDNF family receptor-α (GFRα) members.70 GFLs form a binary complex with a specific co-receptor GFRα that in turn binds to and activates the tyrosine kinase RET. RET signals have been reported to have major roles in development and maintenance of neurons, kidney development and spermatogonial stem cells.70 In the ENS, RET expression drives proliferation, survival and migration of the neural crest-derived precursors of enteric neurons and EGCs in the foetal gut.71,72 In agreement, humans with hypomorphic RET mutations develop Hirschsprung’s disease, which is characterized by intestinal aganglionosis.70 Interestingly, RET signals were also shown to modulate haematopoietic cell functions. RET signals are required for intestinal Peyer’s patches development through control of lymphoid tissue initiator cells,73 and RET activation also controls T cell-derived cytokines in vitro74 and haematopoietic stem cell survival and function.75 Strikingly, intestinal group 3 ILC3s in the adult intestine were shown to express high levels of the neuroregulator RET.76 RET-deficient ILC3s have an impaired capacity to produce IL-22, a critical cytokine for epithelial barrier integrity. In agreement, mice with RET-deficient ILC3s displayed a poor epithelial reactivity that was associated with exacerbated dextran sulphate sodium-induced intestinal inflammation and increased susceptibility to Citrobacter rodentium infection.76 GFL-producing EGCs were found in close association with ILC3s, suggesting a paracrine EGC–ILC3 interaction76 (Fig. 2b). Astonishingly, microbial products and host-derived alarmins regulate glial-derived GFL expression in an MYD88-dependent manner.76 Notably, glial cell-specific deletion of MYD88 signalling impaired GFL and innate IL-22 expression and increased susceptibility to intestinal inflammation and infection.76 In conclusion, glial cells translate microbial and host-derived alarmin cues into GFL production, which then target neuronal and ILC3 function to promote intestinal homeostasis (Fig. 2b).
Pulmonary neuro-immune interactions
The airway epithelium is the first line of contact with inhaled agents, including allergens and pathogens. Allergic lung diseases are accompanied by bronchial hyperreactivity, cough, spasm and inflammation.77,78,79 These features can be explained by an impaired airway innervation and/or dysregulated immune responses,80 putting forward a pivotal role of neuro-immune interactions in lung homeostasis and defence. While multiple studies described spatial interaction between airway nerves and infiltrating immune cells, few reports adequately defined the nerve fibres and/or the immune cell populations that may underlie neuro-immune interactions. Nevertheless, it is now established that inflammatory cells and airway nerves may be found in close proximity, notably during airway inflammation.22,81
Neuronal-granulocyte cross-talk
Mast cells and eosinophils, which are key players in allergic inflammation, appear to cluster around airway nerves in asthmatic patients and allergen-challenged animals.82,83,84,85,86 In agreement, parasympathetic and postganglionic neurons express intercellular adhesion molecule-1 and/or eotaxin that mediate eosinophil adhesion and recruitment, respectively.87,88,89 Similarly, eosinophil-derived molecules activate vagal pulmonary neurons to secrete VIP, SP and CGRP that induces eosinophil chemotaxis.80,90,91,92 Interestingly, VIP and SP promotes the release of mast cell-derived allergic mediators such as histamine, leukotriene C4, prostaglandin D2, IL-6 and tumour necrosis factor (TNF).40,85,93
Neuronal-dendritic cell cross-talk
Airway dendritic cells (DCs) can be found in close proximity to CGRP-expressing nociceptors in the airways of allergic mice.94,95,96,97 Interestingly, upon allergic airway inflammation, T cell activation and proliferation preferentially occurs close to DC–neuron clusters, suggesting that neurons may influence DC–T cell interactions.97 Nevertheless, this topic remains a matter of controversy. While some reports showed increased T-helper type 2 (Th2) responses in response to CGRP-exposed DCs upon allergen stimulation,98,99,100 other works revealed CGRP as a negative regulator of DC function. Notably, CGRP-treated DCs decrease antigen-specific T cell responses and increase regulatory T cells numbers, while adoptive transfer of CGRP-treated DCs reduces allergic airway inflammation.80,101 Interestingly, animals subjected to C-fibre denervation display reduced DC numbers and immune cell influx in the lung.102 In line with these findings, stimulation of pulmonary nociceptor neurons with capsaicin increases both neuropeptide release and immune cell infiltration.103
Neuronal-ILC2 cross-talk
Recent studies interrogated the role of the neuropeptide VIP on the regulation of adaptive and innate type 2 responses.80,104 Intestinal and lung ILC2s express VIP receptor 2 (VIPR2) and their activation with VIP-specific or VIPR2-specific agonist promotes IL-5 production and eosinophilia105 (Fig. 3a). These findings suggested a role of VIP/VIPR2 axis in mucosal homeostasis and allergic inflammation. In agreement, Talbot et al.103 elegantly demonstrated that IL-5 activates lung nociceptors to release VIP, which, in turn, stimulates lung-resident ILC2 and Th2 cells leading to bronchial hyper-responsiveness (Fig. 3a). Importantly, the authors further showed that pharmacological silencing of local sensory neurons abolished the pro-inflammatory signalling loop leading to the resolution of allergic inflammation.103

Pulmonary neuro-immune interactions. a Sensory neurons and pulmonary neuroendocrine cells sense and respond to allergens by secreting VIP and CGRP, respectively. VIP activates ILC2s and differentiated Th2 cells, via VIPR2 signalling, to release IL-5. ILC2-derived and Th2-derived IL-5 stimulates IL-5Rα-expressing sensory neurons inducing further VIP production that supports a positive feedforward loop; CGRP activates ILC2s to produce IL-5, which in turn promotes eosinophil recruitment. b Sensory neurons modulate lung immunity by producing CGRP that suppresses protective neutrophil and γδ T cell responses. c Sensory neurons sense and respond to worm products and allergens by producing NMU. Neuronal-derived NMU induces an immediate activation of NMUR1-expressing ILC2s that shape host immunity and allergic inflammation. VIP vasoactive intestinal peptide, CGRP calcitonin gene-related peptide, IL interleukin, NMU neuromedin U
Akin to intestinal ILC2s, lung ILC2s also express Nmur1 and these innate immune cells are in close contact with NMU-cholinergic pulmonary neurons51,52,53 (Fig. 3c). Noteworthy, Wallrapp et al.53 also observed that in vitro treatment with IL-13 increased Nmu expression in dorsal root ganglia neurons, thus suggesting a putative reciprocal neuron–ILC2 regulation. In vitro and in vivo NMU treatment induces lung ILC2 proliferation and production of type 2 inflammatory and tissue repair cytokines.51,52,53 Consistently, in vivo activation of the NMU/NMUR1 axis potentiates type 2 innate responses in the context of parasite infection and allergic reactions. Notably, Nmur1-deficient mice or chimeric mice reconstituted with Nmur1-deficient ILC2s displayed reduced ILC2 cytokine production and increased susceptibility to N. brasiliensis pulmonary infection.51 Moreover, loss of NMU–NMUR1 signalling altered ILC2 transcriptional programs following allergen challenge in vivo.53
Finally, another independent study further revealed the involvement of cholinergic pathways on innate immune responses. Galle-Treger et al.106 showed that ILC2s express α7-nicotinic acetylcholine receptor (α7nAChR), which is known to downregulate airway inflammation. Using a translational approach, the authors observed that engagement of α7nAChR results in impaired human ILC2 cytokine production and reduced airway hyper-responsiveness (AHR) in a humanized ILC2 mouse model.106
Pulmonary neuroendocrine cells are often found in association with peptidergic nerves and are increased in asthmatic patients and in close proximity with ILC2s.107 Pulmonary neuroendocrine cells were shown to regulate goblet cell hyperplasia and ILC2 function via γ-aminobutyric acid (GABA) and CGRP production, respectively.107 Notably, CGRP and GABA co-administration to ovalbumin-challenged pulmonary neuroendocrine cell-deficient mice restored asthma-associated inflammation characterized by goblet cell hyperplasia, type 2 cytokine production and inflammatory cell infiltrates107 (Fig. 3a). Therefore, it is conceivable that pulmonary neuroendocrine–ILC2 interactions cooperate to amplify allergic asthma responses.107 Additionally, it has been found that pulmonary nociceptor neurons repress γδ T cell and neutrophil responses via CGRP release, impairing protective immunity against lethal Staphylococcus aureus pneumonia108 (Fig. 3b). Similarly, Streptococcus pyogenes skin infection activates nociceptors to produce CGRP, which in turn supresses protective neutrophil responses.109 Notably, botulinum neurotoxin A or CGRP antagonists abrogate neuron-mediated immune suppression and improve defence to S. pyogenes infection.109
Altogether, these studies shed light on the central role of neuro-immune signalling in the pathogenesis of lung diseases. Nevertheless, despite major recent advances in this topic, a better understanding of the cellular and molecular mechanisms underlying lung neuro-immune cross-talk is still required.
Mucosal neurons as sensors of environmental signals
Host defence comprises avoidance behaviour of harmful challenges driven by neuronal activity, recognition and response to an invading pathogen, toxin or allergen, and induction of tolerance mechanisms coordinated by the immune system.110 These strategies are particularly important at mucosal sites, where a tightly balanced response needs to be coordinated to induce tolerance to commensals and dietary elements, but also to mount an effective protective response against foreign agents and toxic products.111,112
Mounting evidence indicates that the nervous system can sense environmental cues to instruct immune cells ensuring an integrated neuro-immune response that promotes host homeostasis.7,80 For example, the recognition of immunoglobulin E-mediated mast cell degranulation by adjacent neurons may act as a primordial inflammatory cue that drives avoidance behaviour to discrete antigens.113 An additional example may be provided by food poisoning, where vomit and diarrhoea illustrate the coordinated action of mucosal neurons to eliminate deleterious antigen from the GI tract.114 Thus, neurons are equipped to sense local environmental cues, including noxious stimuli, microbial products, immune-derived mediators and alarmins, and respond to those changes through the release of neurotransmitters and neuropeptides, or by triggering or altering their action potential firing patterns.
Neuronal sensing of noxious stimuli
Sensory neurons detect noxious stimuli from mechanical, thermal and chemical origin via engagement of sensor receptors such as the transient receptor potential (TRP) ion channels.115 The TRP vanilloid 1 and ankyrin 1 (TRPV1 and TRPA1, respectively) channels are the most studied members of the TRP family. For instance, capsazepine, a TRPV1 antagonist, ameliorates acid-evoked hyperaemia, suggesting that sensory neurons in the oesophageal and duodenal mucosa are activated by gastric acid.116 Furthermore, genetic ablation of TRPA1 or treatment with TRPA1 antagonists reduces allergic-driven immune cell infiltration, cytokine and mucus production and ameliorates AHR.117 Similarly, TRPV1-expressing pulmonary vagal sensory neurons can also mediate AHR. In fact, genetic ablation or silencing of TRPV1 neurons abrogate bronchial hyperreactivity, while chemical or opto-genetic stimulation of TRPV1 neurons aggravates AHR.118 Thus, TRPV1-expressing and TRPA1-expressing pulmonary vagal neurons can integrate chemical and mechanical stimuli that are transmitted to the CNS to coordinate a neuronal response that leads to protective airway reflexes such as coughing, sneezing and bronchoconstriction.119
Neuronal and neuroendocrine sensing of microbial products
Immune responses can be initiated by the activation of pattern recognition receptors (PRRs), including TLRs, which recognize highly conserved microbial structures, known as PAMPs.120 Microbial cues were shown to be recognized by PRRs expressed by the ENS.121 Human and murine enteric neurons and EGCs express TLR2, -3, -4 and -7122,123,124,125 and TLR9 expression was also reported for enteric neurons in the rat colon.126 While the importance of microbial cues to neuronal function remains largely unknown, increasing evidence suggest that an interplay between mucosal neurons and the microbiome influences mucosal homeostasis.127 Tlr2-deficient mice display ENS structural alterations, decreased levels of GDNF, GI dysmotility and increased susceptibility to colitis.124 Interestingly, germ-free (GF) mice phenocopy TLR2 deficiency and administration of a TLR2 ligand to these mice partially restores GI alterations.124 Similarly, TLR4-deficient and MYD88-deficient mice or mice harbouring MYD88 signalling disruption on neural crest cells displayed decreased number of enteric neurons and GI motility.128 In line with these findings, mice treated with antibiotics or GF mice show lower enteric neuronal density, GI motility and neuronal excitability.128,129,130 In addition, such mice were also reported to exhibit altered TLR expression in the ENS, decreased Peyer's patches counts and mild intestinal inflammation.131 Microbiota signals were also shown to modulate EGC homeostasis. EGC differentiation and migration from the intestinal muscularis to the lamina propria is determined by microbial cues.132 Furthermore, invading or symbiotic bacteria differently regulate TLR, S100 calcium-binding protein-β expression and nitric oxide production by human EGCs.125 Finally, it has been suggested that maternal microbiota signals might modulate the nervous and immune systems of their progeny.133,134 Altogether, these reports indicate that commensal microbial sensing by the ENS controls GI activity and potentially modulates neuro-immune interactions.
In addition to the recognition of commensal microbiota by neuronal cells, the latter are also able to integrate cues from pathogenic microbes.135 Notably, while the capacity of nociceptor neurons to sense fungal and bacterial products has been mostly established in skin,136,137 this sensing ability is likely to be conserved in mucosal neurons. In the context of Staphylococcus aureus infection in the skin, bacterial components, such as N-formylated peptides and the pore-forming toxin α-hemolysin, can directly activate calcium mobilization and CGRP release in dorsal root ganglia sensory neurons, thus modulating infection-induced hyperalgesia.136 Similarly, during Candida albicans infection, sensory neurons in the dorsal root ganglia are activated upon stimulation with heat-killed C. albicans or zymosan, as revealed by increased neuronal calcium flux and release of CGRP.137 Interestingly, ablation of sensory neurons leads to an impaired control of C. albicans in the skin.137 Multiple studies have now demonstrated that microbial pathogen product are sensed by the ENS. Stimulation of EGCs with lipopolysaccharide (LPS) induces IL-1β production,138 while TNF is induced after LPS activation of enteric neurons.139 The enteric pathogen Campylobacter jejuni was shown to stimulate afferent neurons that innervate the GI tract and spread information to the CNS.140 Furthermore, toxin B of Clostridium difficile activates VIP-positive submucosal neurons.141 An unresolved question relates to the mechanisms by which neuronal cells sense microbial aggressions. It was recently demonstrated that enteric neurons and EGC sense microbial products and parasites, via MYD88 signalling, to produce neuroregulatory molecules that, in turn, activate innate lymphocytes.51,76 Nevertheless, whether this sensing property is broadly used by neuronal cells throughout the body, and whether other sensing molecules might be employed by neurons to instruct immune cells remains elusive.
Microbial cues also modify mucosal neuronal activity in an indirect manner. For example, intestinal enteroendocrine cells can sense the luminal environment and convey information to neurons via release of hormones.142 Commensal spore-forming bacteria induce serotonin synthesis in colonic enteroendocrine cells that in turn act on myenteric neurons to control GI motility.143 More recently, it was also suggested that a direct cell-to-cell contact through cytoplasmic processes (neuropods) might mediate enteroendocrine–neuron interactions.144 Chemosensory serotonin-expressing enteroendocrine cells were found to establish synaptic connections with serotonin-responding neurons, allowing for direct recognition of luminal changes and immediate neuromodulation of intestinal physiologic responses.145 Noteworthy, pulmonary neuroendocrine cells, which are found in the vicinity of pulmonary neurons, were also shown to promptly induce lung immune responses by releasing neuropeptides.107,146
Enteric neuronal and glial cell sensing of inflammation
Sensory vagus neurons express receptors for pro-inflammatory molecules, such as TNF and IL-1β, and administration of TNF and IL-1β activates firing in the mouse cervical vagus nerve. In turn, vagus stimulation can also regulate the production of immunomodulators by immune cells.147 In the airway mucosa, release of pro-inflammatory mediators from resident immune cells, including histamine, nerve growth factor, brain-derived neurotrophic factor, IL-1β, TNF and IL-5, directly affects pulmonary neuronal activity.86,89,103,119 In turn, activated neurons release neuropeptides (SP, VIP and CGRP) that promote further activation and recruitment of immune cells.80,81 Similarly, enteric neurons and EGC were shown to sense host-derived alarmins, in an MYD88-dependent manner, to produce neuroregulatory molecules that, in turn, activate ILCs.51,76 Altogether, these studies indicate that mucosal innervation is able to sense inflammatory conditions and alter its neuronal activity, providing a mechanism for reciprocal neuro-immune regulation.
Conclusion and perspectives
The cross-talk between the nervous and immune systems at mucosal barrier has seen major breakthroughs in the recent past. These pioneering studies uncovered cellular and molecular missing links between the two major sensing systems in the body, and explored how these neuro-immune interactions contribute to tissue homeostasis. As an example, there is solid mounting evidence that mucosal-protective immunity can be regulated by neuronal cells,47,48,51,52,76 while neuro-immune interactions can be also detrimental in defined mucosal pathological scenarios.53,103 Nevertheless, our understanding of how neuronal and immune cells interact and respond to ever changing physiological and pathological conditions is still very limited and has been hampered by conceptual and technical limitations.
Using genetic mouse models or in vitro studies, it has been shown that distinct immune cells can integrate signals from a wide range of neuromodulatory molecules.5,7 However, since multiple non-neuronal cells can also secrete molecules historically ascribed to be of neuronal origin, whether the observed effects are underlined by neuro-immune interactions remains unclear. Another major challenge in interrogating and deciphering novel neuro-immune cell units (NICUs) relates to our poor understanding of mucosal neuronal circuit architecture and their molecular fingerprints. Nevertheless, the use of combined recent technological advances might allow to address these unmet needs in the future. A clear example may be provided by the use of mouse models with RiboTag targeted mutation of the ribosomal protein L22 locus associated with a Cre driver mouse line, which allows for immunoprecipitation of actively translated ribosome-associated mRNAs from specific neuronal cells.47,148 In addition, technologies that interrogate the neuronal anatomical mapping and its functional connectivity, such as noninvasive gene delivery using engineered viral tracing,149 may also help shed light into mucosal neuronal circuits. Classically established methods to functionally address neuronal activities, such as physical lesions, pharmacological or chemical inhibitors and electrical stimulation, can provided useful information on how neuronal circuits shape immunity. Nevertheless, most of these methods are not tractable, and fail to provide tissue-autonomous information at cellular and molecular level. The development of opto- and chemo-genetics allows to manipulate neuronal activity in a tractable manner and, therefore, these approaches will possibly establish causal relationships between defined neuronal activity and discrete immune response.5 As an example, the use of opto-genetic approaches was recently used to reveal macrophage-neuron cell units in the adipose tissue.56 Nevertheless, while these novel experimental approaches are likely to advance our understanding of mucosal neural activity, there are some pitfalls that should be taken in consideration. Notably, despite the existence of a wide range of transgenic mouse lines driving Cre recombinase expression, none of these lines specifically target mucosal neurons since the latter share development and molecular pathways with other central and peripheral neurons. Other limitations can also occur when using chemo- and opto-genetic approaches. For example, when using designer receptors exclusively activated by designer drugs (DREADDs), spatial specificity is only possible with high-resolution delivery of the modulatory drug, which can be an obstacle in the GI tract.150 The implementation of opto-genetic techniques at mucosal sites may also be hampered by the further need to develop a portable and stable micro device that allow normal behaviour in mice.150 Nevertheless, the more recent development of a transparent graphene sensor with a customized abdominal window may allow, in the future, to harness opto-genetics to study neuro-immune interactions at mucosal sites.151
A deeper understanding of how neuro-immune interactions regulate tissue and organ physiology is a major challenge in the future. The interrogation of cellular and molecular players of mucosal NICUs will shed light into the neuro-immune scaffolds that underlie tissue homeostasis. In addition, these efforts will likely lead to the identification of novel targets that may be used in the therapy of mucosal inflammatory, infectious and cancer conditions. In conclusion, the mucosal nervous system partners with immune cells to ensure local tissue health. Since the pressure of pathogens and other environmental cues is an important evolutionary force, it is possible that the establishment of functional NICUs may have occurred very early in evolution. Finally, this new paradigm of coordinated multi-tissue physiology may also exist beyond mucosal tissues, raising the possibility that neuro-immune sensory units may also regulate physiology and homeostasis at an organismal level.
References
Tulic, M. K., Piche, T. & Verhasselt, V. Lung-gut cross-talk: evidence, mechanisms and implications for the mucosal inflammatory diseases. Clin. Exp. Allergy 46, 519–528 (2016).
Onyiah, J. C. & Colgan, S. P. Cytokine responses and epithelial function in the intestinal mucosa. Cell. Mol. Life Sci. 73, 4203–4212 (2016).
Vareille, M., Kieninger, E., Edwards, M. R. & Regamey, N. The airway epithelium: soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 24, 210–229 (2011).
Keely, S., Talley, N. J. & Hansbro, P. M. Pulmonary-intestinal cross-talk in mucosal inflammatory disease. Mucosal Immunol. 5, 7–18 (2012).
Veiga-Fernandes, H. & Mucida, D. Neuro-immune interactions at barrier surfaces. Cell 165, 801–811 (2016).
Powell, N., Walker, M. M. & Talley, N. J. The mucosal immune system: master regulator of bidirectional gut–brain communications. Nat. Rev. Gastroenterol. Hepatol. 14, 143–159 (2017).
Veiga-Fernandes, H. & Pachnis, V. Neuroimmune regulation during intestinal development and homeostasis. Nat. Immunol. 18, 116–122 (2017).
Margolis, K. G., Gershon, M. D. & Bogunovic, M. Cellular organization of neuroimmune interactions in the gastrointestinal tract. Trends Immunol. 37, 487–501 (2016).
Furness, J. B., Callaghan, B. P., Rivera, L. R. & Cho, H. J. The entered nervous system and gastrointestinal innervation: integrated local and central control. Adv. Exp. Med. Biol. 817, 39–71 (2014).
Furness, J. B., Rivera, L. R., Cho, H. J., Bravo, D. M. & Callaghan, B. The gut as a sensory organ. Nat. Rev. Gastroenterol. Hepatol. 10, 729–740 (2013).
Uesaka, T., Young, H. M., Pachnis, V. & Enomoto, H. Development of the intrinsic and extrinsic innervation of the gut. Dev. Biol. 417, 158–167 (2016).
Chow, A. K. & Gulbransen, B. D. Potential roles of enteric glia in bridging neuroimmune communication in the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G145–G152 (2017).
Grubisic, V., Verkhratsky, A., Zorec, R. & Parpura, V. Enteric glia regulate gut motility in health and disease. Brain Res. Bull. 136, 109–117 (2018).
Furness, J. B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 9, 286–294 (2012).
Poole, D. P. & Furness, J. B. in Physiology of the Gastrointestinal Tract (ed Johnson, L. H.) Ch. 19, 557–581 (Academic Press, New York, 2012).
Furness, J. B. The Enteric Nervous System (Blackwell Publishing, Malden, 2006).
Van Landeghem, L. et al. Enteric glia promote intestinal mucosal healing via activation of focal adhesion kinase and release of proEGF. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G976–G987 (2011).
Costa, M., Brookes, S. J. & Hennig, G. W. Anat. Physiol. Enteric Nerv. Syst. Gut 47(Suppl. 4), iv15–iv19 (2000). discussion iv26.
Rao, M. & Gershon, M. D. The bowel and beyond: the enteric nervous system in neurological disorders. Nat. Rev. Gastroenterol. Hepatol. 13, 517–528 (2016).
Furness, J. B. & Stebbing, M. J. The first brain: species comparisons and evolutionary implications for the enteric and central nervous systems. Neurogastroenterol Motil 30, e13234 (2018).
Belvisi, M. G. Overview of the innervation of the lung. Curr. Opin. Pharmacol. 2, 211–215 (2002).
Lee, L. Y. & Yu, J. Sensory nerves in lung and airways. Compr. Physiol. 4, 287–324 (2014).
Burns, A. J. & Delalande, J. M. Neural crest cell origin for intrinsic ganglia of the developing chicken lung. Dev. Biol. 277, 63–79 (2005).
Freem, L. J. et al. The intrinsic innervation of the lung is derived from neural crest cells as shown by optical projection tomography in Wnt1-Cre;YFP reporter mice. J. Anat. 217, 651–664 (2010).
Wang, Y. F. & Yu, J. Structural survey of airway sensory receptors in the rabbit using confocal microscopy. Sheng Li Xue Bao 56, 119–129 (2004).
Zaccone, E. J. & Undem, B. J. Airway vagal neuroplasticity associated with respiratory viral infections. Lung 194, 25–29 (2016).
Undem, B. J. & Kollarik, M. The role of vagal afferent nerves in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2, 355–360 (2005). discussion 371–352.
Otmishi, P. et al. Neuroimmune interaction in inflammatory diseases. Clin. Med. Circ. Respir. Pulm. Med. 2, 35–44 (2008).
Kioussis, D. & Pachnis, V. Immune and nervous systems: more than just a superficial similarity? Immunity 31, 705–710 (2009).
Kipnis, J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 353, 766–771 (2016).
Yoo, B. B. & Mazmanian, S. K. The enteric network: interactions between the immune and nervous systems of the gut. Immunity 46, 910–926 (2017).
Stead, R. H., Dixon, M. F., Bramwell, N. H., Riddell, R. H. & Bienenstock, J. Mast cells are closely apposed to nerves in the human gastrointestinal mucosa. Gastroenterology 97, 575–585 (1989).
Stead, R. H. et al. Intestinal mucosal mast cells in normal and nematode-infected rat intestines are in intimate contact with peptidergic nerves. Proc. Natl. Acad. Sci. USA 84, 2975–2979 (1987).
Urb, M. & Sheppard, D. C. The role of mast cells in the defence against pathogens. PLoS Pathog. 8, e1002619 (2012).
van Diest, S. A., Stanisor, O.I., Boeckxstaens, G.E., de Jonge, W.J. & van den Wijngaard, R.M. et al. Relevance of mast cell–nerve interactions in intestinal nociception. Biochim. Biophys. Acta 1822, 74–84 (2012).
Tamura, K. & Wood, J. D. Effects of prolonged exposure to histamine on guinea pig intestinal neurons. Dig. Dis. Sci. 37, 1084–1088 (1992).
Reed, D. E. et al. Mast cell tryptase and proteinase-activated receptor 2 induce hyperexcitability of guinea-pig submucosal neurons. J. Physiol. 547, 531–542 (2003).
Steinhoff, M. et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat. Med. 6, 151–158 (2000).
Nguyen, C. et al. Colitis induced by proteinase-activated receptor-2 agonists is mediated by a neurogenic mechanism. Can. J. Physiol. Pharmacol. 81, 920–927 (2003).
Kulka, M., Sheen, C.H., Tancowny, B.P., Grammer, L.C. & Schleimer, R.P. et al. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology 123, 398–410 (2008).
Shanahan, F., Denburg, J. A., Fox, J., Bienenstock, J. & Befus, D. et al. Mast cell heterogeneity: effects of neuroenteric peptides on histamine release. J. Immunol. 135, 1331–1337 (1985).
Caughey, G. H., Leidig, F., Viro, N. F. & Nadel, J. A. Substance P and vasoactive intestinal peptide degradation by mast cell tryptase and chymase. J. Pharmacol. Exp. Ther. 244, 133–137 (1988).
Barbara, G. et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology 126, 693–702 (2004).
Barbara, G. et al. Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology 132, 26–37 (2007).
Buhner, S. et al. Activation of human enteric neurons by supernatants of colonic biopsy specimens from patients with irritable bowel syndrome. Gastroenterology 137, 1425–1434 (2009).
Bain, C. C. & Mowat, A. M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 260, 102–117 (2014).
Gabanyi, I. et al. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 164, 378–391 (2016).
Muller, P. A. et al. Cross-talk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 158, 300–313 (2014).
Klose, C. S. & Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 17, 765–774 (2016).
von Moltke, J. & Pepper, M. Sentinels of the type 2 immune response. Trends Immunol. 39, 99–111 (2018).
Cardoso, V. et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281 (2017).
Klose, C. S. N. et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 (2017).
Wallrapp, A. et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 (2017).
Martinez, V. G. & O’Driscoll, L. Neuromedin U: a multifunctional neuropeptide with pleiotropic roles. Clin. Chem. 61, 471–482 (2015).
Moriyama, S. et al. Beta2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061 (2018).
Pirzgalska, R. M. et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 23, 1309–1318 (2017).
Van Dyken, S. J. & Locksley, R. M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu. Rev. Immunol. 31, 317–343 (2013).
Sharkey, K. A. Emerging roles for enteric glia in gastrointestinal disorders. J. Clin. Invest. 125, 918–925 (2015).
Abdo, H. et al. Enteric glial cells protect neurons from oxidative stress in part via reduced glutathione. FASEB J. 24, 1082–1094 (2010).
Abdo, H. et al. The omega-6 fatty acid derivative 15-deoxy-Delta(1)(2),(1)(4)-prostaglandin J2 is involved in neuroprotection by enteric glial cells against oxidative stress. J. Physiol. 590, 2739–2750 (2012).
Bush, T. G. et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 93, 189–201 (1998).
Cornet, A. et al. Enterocolitis induced by autoimmune targeting of enteric glial cells: a possible mechanism in Crohn’s disease? Proc. Natl. Acad. Sci. USA 98, 13306–13311 (2001).
Rao, M. et al. Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 63, 2040–2057 (2015).
Rao, M. et al. Enteric glia regulate gastrointestinal motility but are not required for maintenance of the epithelium in mice. Gastroenterology 153, 1068–1081 e1067 (2017).
Brown, I. A., McClain, J.L., Watson, R.E., Patel, B.A. & Gulbransen, B.D. et al. Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell. Mol. Gastroenterol. Hepatol. 2, 77–91 (2016).
Gulbransen, B. D. & Sharkey, K. A. Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 9, 625–632 (2012).
Moore, M. W. et al. Renal and neuronal abnormalities in mice lacking GDNF. Nature 382, 76–79 (1996).
Pichel, J. G. et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 382, 73–76 (1996).
Sanchez, M. P. et al. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 382, 70–73 (1996).
Mulligan, L. M. RET revisited: expanding the oncogenic portfolio. Nat. Rev. Cancer 14, 173–186 (2014).
Gianino, S., Grider, J. R., Cresswell, J., Enomoto, H. & Heuckeroth, R. O. et al. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development 130, 2187–2198 (2003).
Sasselli, V., Pachnis, V. & Burns, A. J. The enteric nervous system. Dev. Biol. 366, 64–73 (2012).
Veiga-Fernandes, H. et al. Tyrosine kinase receptor RET is a key regulator of Peyer’s Patch organogenesis. Nature 446, 547–551 (2007).
Almeida, A. R. et al. The neurotrophic factor receptor RET regulates IL-10 production by in vitro polarised T helper 2 cells. Eur. J. Immunol. 44, 3605–3613 (2014).
Fonseca-Pereira, D. et al. The neurotrophic factor receptor RET drives haematopoietic stem cell survival and function. Nature 514, 98–101 (2014).
Ibiza, S. et al. Glial cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 535, 440–443 (2016).
Audrit, K. J., Delventhal, L., Aydin, O. & Nassenstein, C. The nervous system of airways and its remodeling in inflammatory lung diseases. Cell Tissue Res. 367, 571–590 (2017).
Nassenstein, C., Kutschker, J., Tumes, D. & Braun, A. Neuro-immune interaction in allergic asthma: role of neurotrophins. Biochem. Soc. Trans. 34, 591–593 (2006).
Chesne, J. et al. IL-17 in severe asthma. Where do we Stand?. Am. J. Respir. Crit. Care Med. 190, 1094–1101 (2014).
Foster, S. L., Seehus, C. R., Woolf, C. J. & Talbot, S. Sense and immunity: context-dependent neuro-immune interplay. Front. Immunol. 8, 1463 (2017).
McGovern, A. E. & Mazzone, S. B. Neural regulation of inflammation in the airways and lungs. Auton. Neurosci. 182, 95–101 (2014).
Costello, R. W. et al. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am. J. Physiol. 273, L93–L103 (1997).
Nilsson, G., Alving, K., Ahlstedt, S., Hokfelt, T. & Lundberg, J. M. Peptidergic innervation of rat lymphoid tissue and lung: relation to mast cells and sensitivity to capsaicin and immunization. Cell Tissue Res. 262, 125–133 (1990).
Jacoby, D. B., Costello, R. M. & Fryer, A. D. Eosinophil recruitment to the airway nerves. J. Allergy Clin. Immunol. 107, 211–218 (2001).
Cyphert, J. M. et al. Cooperation between mast cells and neurons is essential for antigen-mediated bronchoconstriction. J. Immunol. 182, 7430–7439 (2009).
Voisin, T., Bouvier, A. & Chiu, I. M. Neuro-immune interactions in allergic diseases: novel targets for therapeutics. Int. Immunol. 29, 247–261 (2017).
Sawatzky, D. A. et al. Eosinophil adhesion to cholinergic nerves via ICAM-1 and VCAM-1 and associated eosinophil degranulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L1279–L1288 (2002).
Nie, Z., Nelson, C. S., Jacoby, D. B. & Fryer, A. D. Expression and regulation of intercellular adhesion molecule-1 on airway parasympathetic nerves. J. Allergy Clin. Immunol. 119, 1415–1422 (2007).
Fryer, A. D. et al. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J. Clin. Invest. 116, 228–236 (2006).
Gu, Q., Wiggers, M. E., Gleich, G. J. & Lee, L. Y. Sensitization of isolated rat vagal pulmonary sensory neurons by eosinophil-derived cationic proteins. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L544–L552 (2008).
Matsuda, H., Kawakita, K., Kiso, Y., Nakano, T. & Kitamura, Y. Substance P induces granulocyte infiltration through degranulation of mast cells. J. Immunol. 142, 927–931 (1989).
Sagara, H., Yukawa, T., Arima, M. & Makino, S. Effect of capsaicin on the migration of eosinophils into the bronchi of guinea pigs. Arerugi 42, 236–242 (1993).
Ansel, J. C., Brown, J. R., Payan, D. G. & Brown, M. A. Substance P selectively activates TNF-alpha gene expression in murine mast cells. J. Immunol. 150, 4478–4485 (1993).
Assas, B. M., Pennock, J. I. & Miyan, J. A. Calcitonin gene-related peptide is a key neurotransmitter in the neuro-immune axis. Front. Neurosci. 8, 23 (2014).
Veres, T. Z. et al. Spatial interactions between dendritic cells and sensory nerves in allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 37, 553–561 (2007).
Veres, T. Z., Rochlitzer, S. & Braun, A. The role of neuro-immune cross-talk in the regulation of inflammation and remodelling in asthma. Pharmacol. Ther. 122, 203–214 (2009).
Veres, T. Z. et al. Dendritic cell–nerve clusters are sites of T cell proliferation in allergic airway inflammation. Am. J. Pathol. 174, 808–817 (2009).
Levite, M. Nerve-driven immunity. The direct effects of neurotransmitters on T cell function. Ann. NY Acad. Sci. 917, 307–321 (2000).
Levite, M. Neuropeptides, by direct interaction with T cells, induce cytokine secretion and break the commitment to a distinct T helper phenotype. Proc. Natl. Acad. Sci. USA 95, 12544–12549 (1998).
Ding, W., Stohl, L. L., Wagner, J. A. & Granstein, R. D. Calcitonin gene-related peptide biases Langerhans cells toward Th2-type immunity. J. Immunol. 181, 6020–6026 (2008).
Rochlitzer, S. et al. The neuropeptide calcitonin gene-related peptide affects allergic airway inflammation by modulating dendritic cell function. Clin. Exp. Allergy 41, 1609–1621 (2011).
Kradin, R. et al. Pulmonary response to inhaled antigen: neuroimmune interactions promote the recruitment of dendritic cells to the lung and the cellular immune response to inhaled antigen. Am. J. Pathol. 150, 1735–1743 (1997).
Talbot, S. et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87, 341–354 (2015).
Saenz, S. A. et al. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J. Exp. Med. 210, 1823–1837 (2013).
Nussbaum, J. C. et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 (2013).
Galle-Treger, L. et al. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat. Commun. 7, 13202 (2016).
Sui, P. et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 360, eaan8546 (2018).
Baral, P. et al. Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat. Med. 24, 417–426 (2018).
Pinho-Ribeiro, F. A. et al. Blocking neuronal signaling to immune cells treats Streptococcal invasive infection. Cell 173, 1083–1097 e1022 (2018).
Medzhitov, R., Schneider, D. S. & Soares, M. P. Disease tolerance as a defense strategy. Science 335, 936–941 (2012).
Veiga-Fernandes, H. & Freitas, A. A. The S(c)ensory Immune System Theory. Trends Immunol. 38, 777–788 (2017).
Wang, J., Li, F. & Tian, Z. Role of microbiota on lung homeostasis and diseases. Sci. China Life Sci. 60, 1407–1415 (2017).
Costa-Pinto, F. A., Basso, A. S. & Russo, M. Role of mast cell degranulation in the neural correlates of the immediate allergic reaction in a murine model of asthma. Brain Behav. Immun. 21, 783–790 (2007).
Popoff, M. R. & Poulain, B. Bacterial toxins and the nervous system: neurotoxins and multipotential toxins interacting with neuronal cells. Toxins (Basel) 2, 683–737 (2010).
Julius, D. TRP channels and pain. Annu. Rev. Cell. Dev. Biol. 29, 355–384 (2013).
Holzer, P. TRP channels in the digestive system. Curr. Pharm. Biotechnol. 12, 24–34 (2011).
Caceres, A. I. et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc. Natl. Acad. Sci. USA 106, 9099–9104 (2009).
Trankner, D., Hahne, N., Sugino, K., Hoon, M. A. & Zuker, C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc. Natl. Acad. Sci. USA 111, 11515–11520 (2014).
Mazzone, S. B. & Undem, B. J. Vagal afferent innervation of the airways in health and disease. Physiol. Rev. 96, 975–1024 (2016).
Iwasaki, A. & Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 16, 343–353 (2015).
Obata, Y. & Pachnis, V. The effect of microbiota and the immune system on the development and organization of the enteric nervous system. Gastroenterology 151, 836–844 (2016).
Rumio, C. et al. Activation of smooth muscle and myenteric plexus cells of jejunum via Toll-like receptor 4. J. Cell. Physiol. 208, 47–54 (2006).
Barajon, I. et al. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J. Histochem. Cytochem. 57, 1013–1023 (2009).
Brun, P. et al. Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology 145, 1323–1333 (2013).
Turco, F. et al. Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut 63, 105–115 (2014).
Burgueno, J. F. et al. TLR2 and TLR9 modulate enteric nervous system inflammatory responses to lipopolysaccharide. J. Neuroinflamm. 13, 187 (2016).
Shi, N., Li, N., Duan, X. & Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 4, 14 (2017).
Anitha, M., Vijay-Kumar, M., Sitaraman, S. V., Gewirtz, A. T. & Srinivasan, S. Gut microbial products regulate murine gastrointestinal motility via Toll-like receptor 4 signaling. Gastroenterology 143, 1006–1016 e1004 (2012).
Collins, J., Borojevic, R., Verdu, E. F., Huizinga, J. D. & Ratcliffe, E. M. Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol. Motil. 26, 98–107 (2014).
McVey Neufeld, K. A., Mao, Y. K., Bienenstock, J., Foster, J. A. & Kunze, W. A. The microbiome is essential for normal gut intrinsic primary afferent neuron excitability in the mouse. Neurogastroenterol. Motil. 25, 183–e188 (2013).
Grasa, L. et al. Antibiotic-induced depletion of murine microbiota induces mild inflammation and changes in Toll-like receptor patterns and intestinal motility. Microb. Ecol. 70, 835–848 (2015).
Kabouridis, P. S. et al. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron 85, 289–295 (2015).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013).
Gomez de Aguero, M. et al. The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302 (2016).
Yang, N. J. & Chiu, I. M. Bacterial signaling to the nervous system through toxins and metabolites. J. Mol. Biol. 429, 587–605 (2017).
Chiu, I. M. et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57 (2013).
Kashem, S. W. et al. Nociceptive sensory fibers drive interleukin-23 production from CD301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity 43, 515–526 (2015).
Murakami, M., Ohta, T. & Ito, S. Lipopolysaccharides enhance the action of bradykinin in enteric neurons via secretion of interleukin-1beta from enteric glial cells. J. Neurosci. Res. 87, 2095–2104 (2009).
Coquenlorge, S. et al. Modulation of lipopolysaccharide-induced neuronal response by activation of the enteric nervous system. J. Neuroinflamm. 11, 202 (2014).
Goehler, L. E. et al. Activation in vagal afferents and central autonomic pathways: early responses to intestinal infection with Campylobacter jejuni. Brain Behav. Immun. 19, 334–344 (2005).
Neunlist, M. et al. Toxin B of Clostridium difficile activates human VIP submucosal neurons, in part via an IL-1beta-dependent pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G1049–G1055 (2003).
Gribble, F. M. & Reimann, F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu. Rev. Physiol. 78, 277–299 (2016).
Yano, J. M. et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276 (2015).
Bohorquez, D. V. et al. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J. Clin. Invest. 125, 782–786 (2015).
Bellono, N. W. et al. Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 170, 185–198 e116 (2017).
Branchfield, K. et al. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science 351, 707–710 (2016).
Bonaz, B., Sinniger, V., & Pellissier, S. The vagus nerve in the neuro-immune axis: implications in the pathology of the gastrointestinal tract. Front. Immunol. 8, 1452 (2017).
Sanz, E. et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 106, 13939–13944 (2009).
Chan, K. Y. et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179 (2017).
Aviello, G. & D’Agostino, G. Tools for controlling activity of neural circuits can boost gastrointestinal research. Front. Pharmacol. 7, 43 (2016).
Rakhilin, N. et al. Simultaneous optical and electrical in vivo analysis of the enteric nervous system. Nat. Commun. 7, 11800 (2016).
Acknowledgements
V.C. is supported by the LisbonBioMed PhD program funded by Fundação para a Ciência e Tecnologia (FCT), Portugal. J.C. is supported by Marie Skłodowska-Curie fellowship (750030), EU. H.V.-F. is supported by ERC (647274), EU; and FCT, Portugal.
Author information
Authors and Affiliations
Contributions
V.C., J.C. and H.V.-F. contributed to the planning, writing and editing of this review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Chesné, J., Cardoso, V. & Veiga-Fernandes, H. Neuro-immune regulation of mucosal physiology. Mucosal Immunol 12, 10–20 (2019). https://doi.org/10.1038/s41385-018-0063-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-018-0063-y
This article is cited by
-
Interorgan communication networks in the kidney–lung axis
Nature Reviews Nephrology (2024)
-
CCL5/CCR5-mediated peripheral inflammation exacerbates blood‒brain barrier disruption after intracerebral hemorrhage in mice
Journal of Translational Medicine (2023)
-
Enteric neuroimmune interactions coordinate intestinal responses in health and disease
Mucosal Immunology (2022)
-
Inflammation in the gut is encoded by neurons in the brain
Nature (2022)
-
The intestinal neuro-immune axis: crosstalk between neurons, immune cells, and microbes
Mucosal Immunology (2021)
