
Abstract
Goblet cells (GCs) are specialized epithelial cells that line multiple mucosal surfaces and have a well-appreciated role in barrier maintenance through the secretion of mucus. Moreover, GCs secrete anti-microbial proteins, chemokines, and cytokines demonstrating functions in innate immunity beyond barrier maintenance. Recently it was appreciated that GCs can form goblet cell-associated antigen passages (GAPs) and deliver luminal substances to underlying lamina propria (LP) antigen-presenting cells (APCs) in a manner capable of inducing adaptive immune responses. GCs at other mucosal surfaces share characteristics with the GAP forming intestinal GCs, suggesting that GAP formation may not be restricted to the gut, and that GCs may perform this gatekeeper function at other mucosal surfaces. Here we review observations of how GCs contribute to immunity at mucosal surfaces through barrier maintenance, the delivery of luminal substances to APCs, interactions with APCs, and secretion of factors modulating immune responses.
Similar content being viewed by others


Introduction
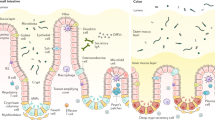
Mucosal surfaces are a diverse environment where microbes live in close proximity to mammalian host cells in mutualistic symbiosis in the steady state. The most prominent example of this symbiotic relationship occurs in the gastrointestinal tract, where the largest number of microbes in the body reside. The gut microbiota functions to increase the bioavailability of nutrients obtained from the diet, and to prevent pathogens from gaining a niche in the lumen. In exchange, these commensal microbes receive a suitable environment in which to reside. Underlying this symbiotic commensal relationship is (1) a strong barrier to prevent encroachment of the microbiota from their luminal compartment, (2) tolerogenic host immune responses to innocuous substances to prevent inflammation, and (3) clearance of, and adaptive immune responses against, pathogens to restore homeostasis. Observations indicate that goblet cells (GCs), specialized epithelial cells that line mucosal surfaces, contribute to each of these functions. This review will highlight new findings on the multiple functions GCs perform that can limit, regulate, and shape mucosal immune responses to environmental substances throughout life (Fig. 1).

Putative functions of GCs in immunity to shape the outcome of immune responses
Limiting encounters with luminal microbes through barrier maintenance
GCs are notable for the secretion of mucin, providing the mucosal surfaces with a thick mucus lining, which acts as a barrier to limit interactions with luminal microbes. Different gel forming mucins are produced and secreted by GCs at various mucosal surfaces. GCs in the intestine predominantly secrete MUC2,1,2 while GCs in the conjunctiva of the ocular epithelium secrete MUC5AC.3 GCs in the upper respiratory tract, including the nasal passages, trachea, and bronchi, secrete MUC5AC and MUC5B.4,5 Airway GCs also express the membrane-bound mucin MUC1,4 but unlike MUC5 and MUC2, this is not a secreted mucin and does not contribute to the gel-forming mucus layer. The mucus layer acts as a physical barrier keeping commensal bacteria, or other environmental organisms, separated from epithelial cells.6,7,8,9,10 The necessity of this physical separation is demonstrated when mucin secretion is disrupted, and commensal bacteria encroaches on the colonic epithelium. Mice lacking the mucin protein MUC2 or mice unable to secrete mucins due to disruptions in the mucin assembly process11,12,13,14,15 quickly develop spontaneous colitis due to the close contact of the commensal bacteria with the epithelium. Similarly, thinner mucus layers can be found in ulcerative colitis patients,16,17 and microbial encroachment can be associated with CD19+ cell recruitment and hyperglycemia in patients with metabolic disorders.18 Traditionally the mucus barrier in the distal colon has been thought to be comprised of two distinct mucus layers, the outer layer serving as the habitat for the microbiota2 and providing a food source for commensal organisms19 and the inner layer being free of bacteria and providing a buffer between the epithelium and the microbiota.20 However, recent work suggests an alternative model where the mucus layer in the distal colon covers the feces and confines the bacteria to the fecal contents. 21 Both models of mucus architecture include a bacteria-free zone near the epithelium, and breach of this buffer by bacteria can place the host at risk of inflammation, even in response to commensal organisms.22 Some bacteria, such as Akkermansia mucinphilia, can digest the dense mucus:23,24 indeed, a mucus-degrading microbiota selected by a low-fiber diet can increase sensitivity to Citrobacter rodentium.25 Additionally, pathogens have been found to propel through the dense mucus.26 To protect against this, GCs secrete RELM-β along with mucins. RELM-β has direct bactericidal properties, killing commensal bacteria and pathogens that penetrate into the mucus layer27,28,29 and targeting helminths for expulsion.10 Other components found in the inner layer of the mucus, like the zymogen granulae protein 16 (ZG16) aggregates bacteria, preventing adherence of the bacteria to the epithelium.30 In the absence of ZG16, spontaneous bacterial translocation and inflammation occurs. These observations demonstrate that GC secrete multiple products with differing mechanisms to limit microbe interaction with the epithelium. In addition to GC-secreted products, the mucus barrier also contains compounds that can control bacteria adherence to the epithelium, including secretory IgA,31 bacteriophages,32 and Paneth cell-derived antimicrobials products. 33
Mucin release is controlled to appropriately maintain the mucus barrier. GCs secrete mucin in response to a number of mediators, including acetylcholine (ACh), histamine, and prostaglandins.34 Having multiple stimuli inducing GC secretion not only provides several ways to induce mucin release to maintain the barrier, they also provide ways to induce mucin release to maintain the barrier without inducing the formation of goblet cell-associated antigen passages (GAPs) and exposing the immune system to luminal substances in disadvantageous situations. Two processes have been described for intestinal GC release of mucins, primary exocytosis, a cAMP signaling process, which induces the release of single vesicles,35 and compound exocytosis, an intracellular Ca2+ signaling process resulting in the fusion and subsequent release of multiple mucin vesicles.36 However, only stimuli inducing compound exocytosis has been associated with the formation of GAPs and delivery of luminal antigens,37 which is discussed in the following section. While most GCs sense mediators and release mucin directly, a subset of GCs can control the release of mucins by neighboring GCs. In response to bacterial components, sentinel GCs located in the upper crypts of the distal colon trigger the rapid release of mucin from neighboring GCs38 quickly restoring the mucus barrier. Thus, through the secretion of mucus and other compounds, GC play a critical role in limiting the immune system’s exposure to luminal microbes in order to avoid inflammatory responses, giving support to the adage “good fences make for good neighbors”.
Controlling the immune system’s exposure to luminal substances through goblet cell-associated antigen passages
How is delivery of luminal substances in the lamina propria controlled?
Despite the presence of a physical barrier due to the mucus layer and epithelium, it has long been appreciated that the immune system underlying the SI villous epithelium is not ignorant of the luminal contents, but instead continuously monitors the contents to promote tolerance to innocuous luminal antigens in the steady state.39 This process, referred to as oral tolerance, involves the induction of peripherally derived induced T regulatory cells (iTregs) specific for luminal antigens, which are then able to suppress and prevent inflammatory responses.40,41,42 The initial step in this process is acquisition of luminal antigens by antigen-presenting cells (APCs) in the LP.43,44 Multiple pathways have been identified by which luminal substances can traverse the epithelium including paracellular leak, transcellular permeability, M cells, and the extension of transepithelial dendrites (TEDs) by LP-APCs.45,46 Paracellular leak is associated with and induced by inflammation,47 suggesting that while luminal substances may traverse the epithelium via paracellular leak, this may not lead to the induction of tolerance. Luminal substances can traverse epithelial cells transcellularly following endocytosis by M cells, though M cells are rare outside of the follicle-associated epithelium in the SI and distal colon where tolerance to luminal substances is induced in the steady state.48,49 Additionally, intestinal epithelial cells have been shown to endocytose luminal substances, though this is thought to mostly contribute to nutrient absorption.50 TED extension is not necessary for oral tolerance, as oral tolerance is induced in mice lacking TEDs,44,51 and oral tolerance is induced to luminal substances in the distal colon where TED formation has not been observed.52,53 In contrast, TEDs are induced by microbial sensing and increase during enteric infection, suggesting TEDs extension as a mechanism for LP-APCs to directly sample luminal bacteria, or alternatively TEDs extension might be a step in the migration of APCs into the lumen to sequester pathogenic bacteria.54,55,56,57,58
In contrast to these pathways, there is evidence suggesting that GC transport of luminal antigen to LP-APCs supports tolerance in the steady state. Multiple studies have observed that GCs are endocytic and able to take up luminal substances;59,60,61,62 however, it was only recently appreciated that this process could result in transfer of luminal substances to LP-APCs in a manner capable of inducing adaptive immune responses.63 This process, referred to as GAP formation, occurs in the SI and distal colon in the steady state, the sites in the GI tract where tolerance to luminal antigens is induced,53,64 but not in the adult proximal colon, where tolerance to luminal substances is not induced in the steady state.64 Moreover, the induction of tolerance to commensal bacterial antigens in the pre-weaning GI tract required GCs and GAPs.65 While currently unexplored, these observations suggest that GCs and GAPs may also be required for the induction of tolerance to dietary antigens post-weaning. Other observations consistent with GAPs as a pathway promoting tolerance to luminal antigens center around the tight regulation of GAP formation. GAPs are induced by ACh acting upon the muscarinic ACh receptor 4 (mAChR4) expressed on GCs, which triggers the release of mucin granules through compound exocytosis.66 GAP formation was not associated with stimuli inducing GC secretion by primary exocytosis.35,37,63,67 The differential ability of secretagogues to induce GAP formation indicates that GCs can secrete mucus to maintain the barrier and not expose the immune system to luminal substances. The association of GAP formation with compound exocytosis, but not with stimuli inducing secretion by primary exocytosis, may be a mechanism that allows GCs to maintain the mucus barrier in settings where exposure of the immune system to the luminal contents would be disadvantageous. This is believed to be the situation in the adult proximal colon, where the mucus barrier is maintained, yet GAP formation is rare and GCs are less responsive to ACh.37
It has been shown that the secretory epithelial cell type enteroendocrine cells can endocytose luminal antigens.61 It remains to be seen if other secretory cells, such as Paneth cells can also endocytose luminal substances, and if these cells can also contribute to the loading of antigens on LP-APCs and induction of mucosal responses.
What is the role of GAPs?
GAP formation has been evaluated throughout the small intestine and colon and throughout the murine life. GAPs were also found in healthy human jejunal resections obtained from patients undergoing bariatric surgery.63 While the role of altered metabolic status on GAP formation remains to be investigated, in aggregate we interpret these observations to indicate that GAPs introduce luminal substances to the mucosal immune system within the intestinal lamina propria (LP) as part of normal physiology in mice and humans.
How are GAPs regulated in the small intestine?
GCs are present in the intestinal epithelium in the early days following birth;68,69 however, GAPs are not formed until later in life, beginning in the SI around day 18. Prior to this, GAP formation is inhibited by phosphorylation of the epidermal growth factor receptor (EGFR) in GCs, which results in activation of p42/p44 MAPK repressing the ability of GCs to respond to ACh to form GAPs (Table 1).37 Prior to weaning direct ligation of EGFR by growth factors, such as EGF, inhibited GAP formation. These growth factors can be secreted in breast milk and found in high concentration in the intestinal lumen during early life and are also necessary for proper intestinal growth during the early days of life and protection from pathogen-induced tissue injury.70,71,72
After day of life (DOL) 18, GAP formation in the SI continues throughout adulthood. GAP formation and antigen delivery in the SI is neither activated nor inhibited by the commensal microbiota,37,53 and there is no change in GAP formation in the SI in germfree mice, or mice on antibiotics.37 However, during pathogenic infections, GAP formation in the SI becomes inhibited,73 ceasing antigen delivery to the SI LP during a time of heightened distress. Inhibition of GAPs during Salmonella infection required Myd88-activation of EGFR pathway, via IL1β acting on the IL1 receptor.74 GAP inhibition during Salmonella infection was rapid, and necessary to prevent increased dissemination of Salmonella to the draining lymph nodes, as Salmonella was able to use GAPs as a portal of entry.73 Listeria monocytogenes has also been shown to associate with GCs to gain entry past the epithelium75 and Citrobacter rodentium directly infect colonic GCs,76 suggesting multiple bacteria species may use GAPs as a portal of entry.
How are GAPs regulated in the proximal colon?
Interestingly, GAP formation occurs in the proximal colon only for a brief window of time, between DOL 10 through weaning (around DOL 21) (Table 1).65 Prior to DOL 10, GAP formation was inhibited in an EGFR-dependent manner, similar to the SI, and activation of Myd88 by TLRs led to EGFR-dependent inhibition of GAP formation post-weaning in response to the microbiota present in specific pathogen-free housed mice.37,65 Such distinct periods of GAP regulation and luminal antigen exposure in the proximal colon may represent the distinct phases of immune development by defining a pre-weaning interval during which the immune system is exposed to luminal antigens for the induction of antigen-specific tolerance to gut bacteria.65
Perhaps as or more important than the stimuli inducing GAP formation, are the pathways that inhibit GAP formation and antigen delivery in unfavorable situations. When present, inappropriately formed GAPs in the proximal colon allowed for the translocation of commensal and pathogenic bacteria.53,73,77 Moreover, overriding the GAP inhibition in these situations results in inflammatory responses.65,73,77
How are GAPs regulated in the distal colon?
GAP formation in the distal colon was not inhibited by the post-weaning microbiota making it distinct from the proximal colon (Table 1).53 However, translocation of bacteria was not associated with the presence of GAPs in the distal colon.53 While not investigated, the presence of GAPs in the distal colon and yet the lack of bacterial translocation might be due to the dense mucus layer in the distal colon, which may reduce the GCs’ exposure to microbes and microbial products allowing GC to respond to ACh to form GAPs and preventing microbes from accessing the GCs. As tolerogenic responses can be induced through the distal colon, it remains to be seen what, if any, regulatory mechanisms may control GAP formation and antigen delivery in the distal colon.
Throughout life, GAP inhibition through EGFR phosphorylation offers an elegant regulatory pathway utilizing a similar inhibitory signal, with separate regulatory ligands defining the time when antigen delivery is a desirable event. Thus, multiple pathways provide tight control of GAP formation and luminal antigen exposure to limit inappropriate inflammatory responses in disadvantageous situations and allow luminal antigen exposure when it is beneficial (Table 1).
GCs interactions with immune cells
The LP contains multiple APC populations78,79 with distinct functions necessary for appropriate immune responses to luminal substances. While initial studies reported that GAPs preferentially delivered luminal antigens to SI CD103+ DCs,63 CX3CR1+ APCs have also been observed to interact with and obtain luminal substances from GAPs.53 When colonic GAPs were formed by bypassing GAP inhibitory pathways in mice post-weaning, CX3CR1+ APCs were seen interacting with colonic GAPs, and were loaded with bacteria that translocated via GAPs.37,53,77 As multiple populations of APCs interact with and obtain antigen from GAPs in the intestine, it remains to be seen if factors exist to recruit a specific population of APCs to GCs to acquire antigen for the induction of oral tolerance as certain populations of APCs, such as CD103+ APCs are thought to be better suited for the induction of tolerogenic responses.80
CD103+ LP-APC expression of retinaldehyde dehydrogenase (ALDH1) is necessary for the production of all-trans retinoic acid (ATRA).81 The production of ATRA plays multiple roles in mucosal immune responses to luminal antigens including promoting IgA responses, imprinting gut homing molecule expression by lymphocytes, and inducing Tregs.82,83,84 In vivo studies revealed that CD103+ LP-APC expression of ALDH was dependent upon recruitment to the epithelium and epithelial expression of the cellular retinol-binding protein II (CRBPII).85,86
During interactions with APCs, GCs transfer GC products along with luminal antigens to APCs.63 This transfer of GC products to APCs might imprint APCs with mucosal properties. One such GC product, MUC2, has been suggested to imprint APCs with an anti-inflammatory gene signature required for oral tolerance,87 suggesting that when APCs acquire luminal antigens from GAPs, they also acquire tolerogenic signals. Disrupting interactions between APCs and the epithelium decreased transfer of GC products to the APCs, and subsequently reduced the induction of mucosal responses by APCs.85 Whether CX3CR1+ APCs, which largely do not express ALDH, are likewise imprinted with specific properties during interactions with GCs remains to be investigated.
The subsequent immune response that follows the acquisition of antigens by APCs is orchestrated through the secretion of cytokines, chemokines, and other proteins, which is based on the signals APCs receives from the antigens88,89 and the tissue environment.90 Secreted cytokines and chemokines recruit cells and drive differentiation of effector cells, regulating the type of immune response that follows antigen exposure, and can come from a variety of cell types, GCs included. During C. rodentium infection, a murine model of enterohemorrhagic Escherichia coli infection, RELM-β is secreted basolaterally by GCs, can be found in the serum, and functions as a chemoattractant recruiting CD4 T cells to the colon LP.91 Additionally, RELM-β supports IL-22 production during infections, which helps promote tissue repair and restoration, indicating RELM-β may perform multiple functions similar to cytokines and chemokines to help resolve pathogen infections. Another GC product, trefoil factor 3, TFF3 is important for the resolution of inflammation, helping in the process of tissue repair and restoration,76,92 and the production of TFF3 can be triggered by microbial sensing, through TLR2 stimulation.93 While it remains to be seen if GCs in the intestine secrete cytokines during immune responses in vivo, GCs express mRNA for a variety of cytokines, such as IL13, IL18, IL15, IL6, IL7, IL17, and IL25, and chemokines eotaxin, CCL6, CCL20, CCL9 (MIP1γ),37,85,94 the latter has been shown to attract APCs to the epithelium.95 Thus, through the secretion of soluble factors GCs help shape and control immune responses.
Can GCs at other mucosal surfaces form GAPs?
Intestinal GCs control immune responses by regulating antigen exposure through mucus secretion to maintain the barrier, luminal antigen delivery, and interactions with APCs underlying the epithelium. Some of these functions have been observed in GCs at other mucosal surfaces; however, it remains to be seen if GCs at other mucosal surfaces have the capacity to form GAPs and deliver antigens to APCs. While unexplored, it is intriguing to speculate, based on characteristic shared between intestinal GCs and GCs at other mucosal surfaces, that the GAP function may exist at other sites. GAP formation in the GI tract is associated with secretion via compound exocytosis,37 a characteristic shared by GCs in the airway epithelium and conjunctiva. Moreover, the stimuli inducing compound exocytosis at these surfaces is similar to those inducing compound exocytosis in intestinal GCs.4,96,97 Of further interest, compound exocytosis and secretion in conjunctival GCs is regulated by EGFR phosphorylation,97,98,99 and TLR activation in airway GCs leads to EGFR transactivation,100 indicating that the pathways regulating GAPs in the intestine are present and functional in GCs at other mucosal surfaces.
The simple columnar intestinal epithelium is well suited for the delivery of luminal antigens as this allows for a single cell to have access to the lumen and the APCs in the LP permitting a GC to directly take up and deliver luminal antigens to APCs. The conjunctiva in the ocular epithelium is also a simple columnar epithelium, and interactions between conjunctival GCs and APCs have been described,101 suggesting that GC-mediated antigen delivery to APCs at this mucosal surface may also occur. Similar to the intestine, a loss of GCs or impaired mucin secretion in the conjunctiva results in inflammatory responses,102,103 96 consistent with a role for conjunctival GCs in maintaining tolerance. Interestingly, ocular GCs secrete TGFβ2 and express CD36, which is necessary to activate TGFβ2.101 Further ocular GCs can imprint a tolerogenic phenotype onto APCs,101 indicating that GCs in the conjunctiva may imprint APCs with a tolerogenic phenotype while delivering antigens. Moreover, the conjunctiva contains a commensal flora,104 and similar to intestinal GCs, conjunctival GCs are responsive to microbial stimulation,105 further suggesting that intestinal GCs and conjunctival GCs may play parallel roles in immune responses.
While the epithelium of the upper airways in not a simple columnar epithelium, it is a pseudostratified epithelium, in which the nuclei of the epithelial cells are not aligned in the same plane but all epithelial cells make contact with the basement membrane and can make contact with the lumen.106 Thus, the pseudostratified epithelium might still allow GCs in the upper airway to have access to both the lumen and immune cells below the basement membrane for the purpose of antigen delivery. Moreover, APCs are in close contact with the upper airway epithelium,107 and increased differentiation of GCs lead to the recruitment of APCs to the airway epithelium,108 suggesting that the presence of GCs might recruit APCs facilitating interactions and antigen transfer. In addition, airway GCs secrete multiple cytokines and chemokines, recruiting immune cells and shaping the immune response to inhaled antigens,108,109,110 suggesting that like intestinal GCs upper airway GCs may deliver antigens to APCs and direct the phenotype of the immune response. Like the intestine and conjunctiva, the lung has its own unique, though limited, microbiota,111 which can become dysbiotic and expanded in disease or harbor pathogens and represent potential danger,112,113 raising the possibility that airway GCs may also respond to microbial signals to modulate immune responses to inhaled antigens.
While this review has focused on the role of GCs in maintaining homeostasis, it is worth noting that GCs can also contribute to disease pathogenesis. Goblet cell hyperplasia is a hallmark of Th2 responses in both the intestine and in the lung and driven by IL-13 in both organs. This process is important for the expulsion of helminths and protection of airway through increased mucus secretion;114,115,116 however, increased mucus production is also a contributing factor to the pathogenesis of asthma and chronic obstructive pulmonary disease.117 It is unknown if the increased GCs that expand during disease have the capacity to form GAPs and deliver antigen and whether these GAPs contribute to homeostatic or pathogenic immune responses. However, observations that IL-13-driven GC expansion resulted in the increased translocation of Listeria monocytogenes75 suggest that these GCs may have GAP function and contribute to disease. GC expansion is often a component of an active infection or resolution to disease during which the system may not be geared for tolerogenic responses. If GAPs do form from GCs during GC expansion, this could result in increased antigen delivery during an inopportune time promoting inflammatory responses against dietary and commensal bacterial antigens, resulting in food allergy or colitis. Moreover, GC dysfunction has been associated with and contributory to multiple diseases including inflammatory bowel disease, cystic fibrosis, asthma, metabolic disorders, Sjögren syndrome, and chronic obstructive pulmonary disease, indicating that GCs are not always innocent bystanders and can be active participants in disease pathogenesis.
Conclusions
A large body of work indicates that GCs have multiple contributions to innate and adaptive immune responses at mucosal surfaces. While the GAP and sentinel GC functions have only recently been appreciated in the intestinal tract, similarities between intestinal GCs and GCs at other mucosal surfaces suggest that these GC functions may exist at other sites. Further investigations will be required to determine if these functions do exist at other surfaces, how they are regulated, their contributions to mucosal immune responses, and whether these functions are a characteristic of all or a subset of GCs at mucosal surfaces. While barrier maintenance remains the stalwart function of GCs, it is becoming increasingly clear that complexity of these cells’ contribution to mucosal immunity extends well beyond mucus secretion.
References
Chang, S.-K. et al. Localization of mucin (MUC2 and MUC3) messenger RNA and peptide expression in human normal intestine and colon cancer. Gastroenterology 107, 28–36 (1994).
Johansson, M. E., Larsson, J. M. & Hansson, G. C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. . Natl. Acad. Sci. USA 108(Suppl 1), 4659–4665 (2011).
Jumblatt, M. M., McKenzie, R. W. & Jumblatt, J. E. MUC5AC mucin is a component of the human precorneal tear film. Invest. Ophthalmol. Vis. Sci. 40, 43–49 (1999).
Rogers, D. F. Airway goblet cells: responsive and adaptable front-line defenders. Eur. Respir. J. 7, 1690 (1994).
Hovenberg, H. W., Davies, J. R. & Carlstedt, I. Different mucins are produced by the surface epithelium and the submucosa in human trachea: identification of MUC5AC as a major mucin from the goblet cells. Biochem. J. 318, 319 (1996).
Birchenough, G. M., Johansson, M. E., Gustafsson, J. K., Bergstrom, J. H. & Hansson, G. C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 8, 712–719 (2015).
Deplancke, B. & Gaskins, H. R. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am. J. Clin. Nutr. 73, 1131S–1141S (2001).
Pelaseyed, T. et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 260, 8–20 (2014).
Button, B. et al. Periciliary Brush Promotes the Lung Health by Separating the Mucus Layer from Airway Epithelia. Science (New York, NY) 337, 937–941 (2012).
Paulsen, F. P. & Berry, M. S. Mucins and TFF peptides of the tear film and lacrimal apparatus. Prog. Histochem. Cytochem. 41, 1–53 (2006).
Van der Sluis, M. et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129 (2006).
Heazlewood, C. K. et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation rsembling ulcerative colitis. PLoS Med. 5, e54 (2008).
Rahman, A. A., Robinson, A. M., Brookes, S. J. H., Eri, R. & Nurgali, K. Rectal prolapse in Winnie mice with spontaneous chronic colitis: changes in intrinsic and extrinsic innervation of the rectum. Cell Tissue Res. 366, 285–299 (2016).
Robinson, A. M. et al. Alterations of colonic function in the Winnie mouse model of spontaneous chronic colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 312, G85–G102 (2016).
Eri, R. D. et al. An intestinal epithelial defect conferring ER stress results in inflammation involving both innate and adaptive immunity. Mucosal Immunol. 4, 354–364 (2011).
Gustafsson, J. K. et al. An ex vivo method for studying mucus formation, properties, and thickness in human colonic biopsies and mouse small and large intestinal explants. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G430–G438 (2012).
Johansson, M. E. et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 63, 281–291 (2014).
Chassaing, B., Raja, S. M., Lewis, J. D., Srinivasan, S. & Gewirtz, A. T. Colonic microbiota encroachment correlates with dysglycemia in humans. Cell. Mol. Gastroenterol. Hepatol. 4, 205–221 (2017).
Martens, E. C., Roth, R., Heuser, J. E. & Gordon, J. I. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent human gut symbiont. J. Biol. Chem. 284, 18445–18457 (2009).
Johansson, M. E. et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. . Natl. Acad. Sci. USA 105, 15064–15069 (2008).
Kamphuis, J. B. J., Mercier-Bonin, M., Eutamene, H. & Theodorou, V. Mucus organisation is shaped by colonic content; a new view. Sci. Rep. 7, 8527 (2017).
Johansson, M. E. et al. Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PLoS ONE 5, e12238 (2010).
Cani, P. D. & de Vos, W. M. Next-generation beneficial microbes: the case of Akkermansia muciniphila. Front. Microbiol. 8, 1765 (2017).
Derrien, M., Vaughan, E. E., Plugge, C. M. & de Vos, W. M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 54, 1469–1476 (2004).
Desai, M. S. et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e1321 (2016).
Stecher, B. et al. Flagella and chemotaxis are required for efficient induction of Salmonella enterica Serovar Typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 72, 4138–4150 (2004).
Propheter, D. C., Chara, A. L., Harris, T. A., Ruhn, K. A. & Hooper, L. V. Resistin-like molecule β is a bactericidal protein that promotes spatial segregation of the microbiota and the colonic epithelium. Proc. Natl. Acad. Sci. USA 114, 11027–11033 (2017).
Herbert, D. et al. Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J. Exp. Med. 206, 2947 (2009).
Morampudi, V. et al. The goblet cell-derived mediator RELM-β drives spontaneous colitis in Muc2-deficient mice by promoting commensal microbial dysbiosis. Mucosal Immunol. 9, 1218 (2016).
Bergström, J. H. et al. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl. Acad. Sci. USA 113, 13833–13838 (2016).
Slack, E., Balmer Maria, L. & Macpherson Andrew, J. B cells as a critical node in the microbiota–host immune system network. Immunol. Rev. 260, 50–66 (2014).
Barr, J. J. et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 110, 10771 (2013).
Meyer-Hoffert, U. et al. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 57, 764–771 (2008).
Halm, D. R. & Halm, S. T. Secretagogue response of goblet cells and columnar cells in human colonic crypts. Am. J. Physiol. 277(3 Pt 1), C501–C522 (1999).
Epple, H. J. et al. Differential stimulation of intestinal mucin secretion by cholera toxin and carbachol. Pflug. Arch.: Eur. J. Physiol. 433, 638–647 (1997).
Specian, R. D. & Neutra, M. R. Mechanism of rapid mucus secretion in goblet cells stimulated by acetylcholine. J. Cell Biol. 85, 626–640 (1980).
Knoop, K. A., McDonald, K. G., McCrate, S., McDole, J. R. & Newberry, R. D. Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol. 8, 198–210 (2015).
Birchenough, G. M., Nystrom, E. E., Johansson, M. E. & Hansson, G. C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542 (2016).
Pabst, O. & Mowat, A. M. Oral tolerance to food protein. Mucosal Immunol. 5, 232 (2012).
Zhang, X., Izikson, L., Liu, L. & Weiner, H. L. Activation of CD25+CD4+ Regulatory T Cells by Oral Antigen Administration. J. Immunol. 167, 4245 (2001).
Mucida, D. et al. Oral tolerance in the absence of naturally occurring Tregs. J. Clin. Invest. 115, 1923–1933 (2005).
Kim, K. S., & Surh, C. D . Induction of immune tolerance to dietary antigens In Crossroads Between Innate and Adaptive Immunity V (eds Schoenberger, S. P., Katsikis, P. D. & Pulendran, B.) 93–118 (Springer International Publishing, Cham, 2015).
Spahn, T. W. & Kucharzik, T. Modulating the intestinal immune system: the role of lymphotoxin and GALT organs. Gut 53, 456–465 (2004).
Worbs, T. et al. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J. Exp. Med. 203, 519–527 (2006).
Knoop, K. A., Miller, M. J. & Newberry, R. D. Transepithelial antigen delivery in the small intestine: different paths, different outcomes. Curr. Opin. Gastroenterol. 29, 112–118 (2013).
Schulz, O. & Pabst, O. Antigen sampling in the small intestine. Trends Immunol. 34, 155–161 (2013).
Turner, J. R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 (2009).
Jacob, E., Baker, S. J. & Swaminathan, S. P. ‘M’ cells in the follicle-associated epithelium of the human colon. Histopathology 11, 941–952 (1987).
Owen, R. L., Piazza, A. J. & Ermak, T. H. Ultrastructural and cytoarchitectural features of lymphoreticular organs in the colon and rectum of adult BALB/c mice. Am. J. Anat. 190, 10–18 (1991).
Ménard, S., Cerf-Bensussan, N. & Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol. 3, 247 (2010).
Vallon-Eberhard, A., Landsman, L., Yogev, N., Verrier, B. & Jung, S. Transepithelial pathogen uptake into the small intestinal lamina propria. J. Immunol. 176, 2465–2469 (2006).
Hapfelmeier, S. et al. Microbe sampling by mucosal dendritic cells is a discrete, MyD88-independent stepin invG S. Typhimurium colitis. J. Exp. Med. 205, 437–450 (2008).
Knoop, K. A. et al. Antibiotics promote the sampling of luminal antigens and bacteria via colonic goblet cell associated antigen passages. Gut Microbes 8, 400–411 (2017).
Cruickshank, S. M. et al. Rapid dendritic cell mobilization to the large intestinal epithelium is associated with resistance to Trichuris muris infection. J. Immunol. 182, 3055–3062 (2009).
Niess, J. H. et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258 (2005).
Chieppa, M., Rescigno, M., Huang, A. Y. & Germain, R. N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 203, 2841–2852 (2006).
Arques, J. L. et al. Salmonella induces flagellin- and MyD88-dependent migration of bacteria-capturing dendritic cells into the gut lumen. Gastroenterology 137, 579–587 (2009).
Nicoletti, C., Arques, J. L. & Bertelli, E. CX(3)CR1 is critical for Salmonella-induced migration of dendritic cells into the intestinal lumen. Gut Microbes 1, 131–134 (2010).
Colony, P. C. & Specian, R. D. Endocytosis and vesicular traffic in fetal and adult colonic goblet cells. Anat. Rec. 218, 365–372 (1987).
Walker, W. A., Cornell, R., Davenport, L. M. & Isselbacher, K. J. Macromolecular absorption. J. Cell Biol. 54, 195 (1972).
Nagatake, T., Fujita, H., Minato, N. & Hamazaki, Y. Enteroendocrine cells are specifically marked by cell surface expression of Claudin-4 in mouse small intestine. PLoS ONE 9, e90638 (2014).
Kang, S. K. et al. Identification of a peptide sequence that improves transport of macromolecules across the intestinal mucosal barrier targeting goblet cells. J. Biotechnol. 135, 210–216 (2008).
McDole, J. R. et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483, 345–349 (2012).
Veenbergen, S. et al. Colonic tolerance develops in the iliac lymph nodes and can be established independent of CD103(+) dendritic cells. Mucosal Immunol. 9, 894–906 (2016).
Knoop, K. A. et al. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci. Immunol. 2, eaao1314 (2017).
Neutra, M. R., O’Malley, L. J. & Specian, R. D. Regulation of intestinal goblet cell secretion. II. A survey of potential secretagogues. Am. J. Physiol. 242, G380–G387 (1982).
Miller, M. J., Knoop, K. A. & Newberry, R. D. Mind the GAPs: insights into intestinal epithelial barrier maintenance and luminal antigen delivery. Mucosal Immunol. 7, 452–454 (2014).
Birchenough, G. M. H. et al. Altered innate defenses in the neonatal gastrointestinal tract in response to colonization by neuropathogenic Escherichia coli . Infect. Immun. 81, 3264–3275 (2013).
Birchenough, G. M. H. et al. Postnatal development of the small intestinal mucosa drives age-dependent, regio-selective susceptibility to Escherichia coli K1 infection. Sci. Rep. 7, 83 (2017).
Kobata, R. et al. High levels of growth factors in human breast milk. Early Hum. Dev. 84, 67–69 (2008).
Wagner, C. L., Taylor, S. N. & Johnson, D. Host factors in amniotic fluid and breast milk that contribute to gut maturation. Clin. Rev. Allergy Immunol. 34, 191–204 (2008).
Good, M. et al. Breast milk protects against the development of necrotizing enterocolitis through inhibition of Toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor receptor. Mucosal Immunol. 8, 1166–1179 (2015).
Kulkarni, D. et al. Goblet cell associated antigen passages are inhibited during Salmonella typhimurium infection to prevent pathogen dissemination and limit responses to dietary antigens. Mucosal Immunol. Feb 14 (2018).
Muzio, M., Ni, J., Feng, P. & Dixit, V. M. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612 (1997).
Nikitas, G. et al. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J. Exp. Med. 208, 2263–2277 (2011).
Bergstrom, K. S. B. et al. Modulation of intestinal goblet cell function during infection by an attaching and effacing bacterial pathogen. Infect. Immun. 76, 796–811 (2008).
Knoop, K. A., McDonald, K. G., Kulkarni, D. H. & Newberry, R. D. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut 65, 1100–1109 (2016).
Bogunovic, M. et al. Origin of the lamina propria dendritic cell network. Immunity 31, 513–525 (2009).
Varol, C. et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity 31, 502–512 (2009).
Schulz, O. et al. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J. Exp. Med. 206, 3101–3114 (2009).
Molenaar, R. et al. Expression of retinaldehyde dehydrogenase enzymes in mucosal dendritic cells and gut-draining lymph node stromal cells is controlled by dietary vitamin A. J. Immunol. 186, 1934–1942 (2011).
Coombes, J. L. et al. A functionally specialized population of mucosal CD103 + DCs induces Foxp3 + regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Sun, C. M. et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785 (2007).
Mora, J. R. et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006).
McDonald, K. G. et al. CCR6 promotes steady-state mononuclear phagocyte association with the intestinal epithelium, imprinting and immune surveillance. Immunology 152, 613–627 (2017).
McDonald, K. G. et al. Epithelial expression of the cytosolic retinoid chaperone cellular retinol binding protein II is essential for in vivo imprinting of local gut dendritic cells by lumenal retinoids. Am. J. Pathol. 180, 984–997 (2012).
Shan, M. et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 342, 447–453 (2013).
Pulendran, B., Tang, H. & Denning, T. Division of labor, plasticity, and crosstalk between dendritic cell subsets. Curr. Opin. Immunol. 20, 61–67 (2008).
Manicassamy, S. & Pulendran, B. Dendritic cell control of tolerogenic responses. Immunol. Rev. 241, 206–227 (2011).
Konkel, J. E. & Chen, W. Balancing acts: the role of TGF-β in the mucosal immune system. Trends Mol. Med. 17, 668–676 (2011).
Bergstrom, K. S. B. et al. Goblet cell derived RELM-β recruits CD4+ T cells during infectious colitis to promote protective intestinal epithelial cell proliferation. PLoS Pathog. 11, e1005108 (2015).
Taupin, D. & Podolsky, D. K. Trefoil factors: initiators of mucosal healing. Nat. Rev. Mol. Cell Biol. 4, 721–732 (2003).
Podolsky, D. K., Gerken, G., Eyking, A. & Cario, E. Colitis-associated variant of TLR2 causes impaired mucosal repair due to TFF3 deficiency. Gastroenterology 137, 209–220 (2009).
Haber, A. L. et al. A single-cell survey of the small intestinal epithelium. Nature 551, 333 (2017).
Zhao, X. et al. CCL9 is secreted by the follicle-associated epithelium and recruits dome region Peyer’s patch CD11b<sup> + </sup> dendritic cells. J. Immunol. 171, 2797 (2003).
Dartt, D. A. & Masli, S. Conjunctival epithelial and goblet cell function in chronic inflammation and ocular allergic inflammation. Curr. Opin. Allergy Clin. Immunol. 14, 464–470 (2014).
Kanno, H. et al. Cholinergic agonists transactivate EGFR and stimulate MAPK to induce goblet cell secretion. Am. J. Physiol.-Cell Physiol. 284, C988–C998 (2003).
Dartt, D. A. Interaction of EGF family growth factors and neurotransmitters in regulating lacrimal gland secretion. Exp. Eye Res. 78, 337–345 (2004).
Ota, I. et al. Alpha 1-adrenergic and cholinergic agonists activate MAPK by separate mechanisms to inhibit secretion in lacrimal gland. Am. J. Physiol. Cell. Physiol. 284, C168–C178 (2003).
Koff, J. L., Shao, M. X., Ueki, I. F. & Nadel, J. A. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L1068–L1075 (2008).
Contreras-Ruiz, L. & Masli, S. Immunomodulatory cross-talk between conjunctival goblet cells and dendritic cells. PLoS ONE 10, e0120284 (2015).
Tseng, S. C. G. et al. Possible mechanisms for the loss of goblet cells in mucin-deficient disorders. Ophthalmology 91, 545–552 (1984).
Dogru, M. et al. Ocular surface and MUC5AC alterations in atopic patients with corneal shield ulcers. Curr. Eye Res. 30, 897–908 (2005).
Dong, Q. et al. Diversity of bacteria at healthy human conjunctiva. Invest. Ophthalmol. Vis. Sci. 52, 5408–5413 (2011).
McGilligan, V. E. et al. Staphylococcus aureus activates the NLRP3 inflammasome in human and rat conjunctival goblet cells. PLoS ONE 8, e74010 (2013).
AAWT, Have-Opbroek, Otto-Verberne, C. J. M., Dubbeldam, J. A. & Dÿkman, J. H. The proximal border of the human respiratory unit, as shown by scanning and transmission electron microscopy and light microscopical cytochemistry. Anat. Rec. 229, 339–354 (1991).
Gong, J. L. et al. Intraepithelial airway dendritic cells: a distinct subset of pulmonary dendritic cells obtained by microdissection. J. Exp. Med. 175, 797–807 (1992).
Rajavelu, P. et al. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J. Clin. Invest. 125, 2021–2031 (2015).
Tanabe, T. & Rubin, B. K. Airway goblet cells secrete pro-inflammatory cytokines, chemokines, and growth factors. Chest 149, 714–720 (2016).
Tanabe, T., Shimokawaji, T., Kanoh, S. & Rubin, B. K. IL-33 stimulates CXCL8/IL-8 secretion in goblet cells but not normally differentiated airway cells. Clin. Exp. Allergy 44, 540–552 (2014).
Hilty, M. et al. Disordered microbial communities in asthmatic airways. PLoS ONE 5, e8578 (2010).
Dickson, R. P. & Huffnagle, G. B. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog. 11, e1004923 (2015).
Huffnagle, G. B., Dickson, R. P. & Lukacs, N. W. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 10, 299 (2016).
Kim, Y. S., & Ho, S. B.. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr. Gastroenterol. Rep. 12, 319–330 (2010).
Park, K.-S. et al. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J. Clin. Invest. 117, 978–988 (2007).
Artis, D. & Grencis, R. K. The intestinal epithelium: sensors to effectors in nematode infection. Mucosal Immunol. 1, 252 (2008).
Hauber, H.-P., Foley, S. C. & Hamid, Q. Mucin overproduction in chronic inflammatory lung disease. Can. Respir. J.: J. Can. Thorac. Soc. 13, 327–335 (2006).
Acknowledgements
The authors would like to thank Jenny K. Gustafsson, Devesha H. Kulkarni, Keely G. Mcdonald, and Phillip I. Tarr for insightful critiques and discussions. Supported by grants: DK109006-KAK, DK097317-RDN, AI112626-RDN, AI131342-RDN, DK05257 to R.D.N. and K.A.K., Children’s Discovery Institute Grant MD-II-2015-481 to R.D.N.
Author information
Authors and Affiliations
Contributions
K.A.K. and R.D.N. wrote the initial manuscript draft, reviewed, and discussed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Knoop, K.A., Newberry, R.D. Goblet cells: multifaceted players in immunity at mucosal surfaces. Mucosal Immunol 11, 1551–1557 (2018). https://doi.org/10.1038/s41385-018-0039-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-018-0039-y
This article is cited by
-
SLE serum induces altered goblet cell differentiation and leakiness in human intestinal organoids
EMBO Molecular Medicine (2024)
-
Rhinovirus induces airway remodeling: what are the physiological consequences?
Respiratory Research (2023)
-
Persea americana extract protects intestinal tissue from Eimeria papillata-induced murine Infection
BMC Veterinary Research (2023)
-
Analysis of cell-specific transcriptional responses in human colon tissue using CIBERSORTx
Scientific Reports (2023)
-
The protective role of conjunctival goblet cell mucin sialylation
Nature Communications (2023)
