
Abstract
Genetic investigations of people with impaired development of spoken language provide windows into key aspects of human biology. Over 15 years after FOXP2 was identified, most speech and language impairments remain unexplained at the molecular level. We sequenced whole genomes of nineteen unrelated individuals diagnosed with childhood apraxia of speech, a rare disorder enriched for causative mutations of large effect. Where DNA was available from unaffected parents, we discovered de novo mutations, implicating genes, including CHD3, SETD1A and WDR5. In other probands, we identified novel loss-of-function variants affecting KAT6A, SETBP1, ZFHX4, TNRC6B and MKL2, regulatory genes with links to neurodevelopment. Several of the new candidates interact with each other or with known speech-related genes. Moreover, they show significant clustering within a single co-expression module of genes highly expressed during early human brain development. This study highlights gene regulatory pathways in the developing brain that may contribute to acquisition of proficient speech.
Similar content being viewed by others
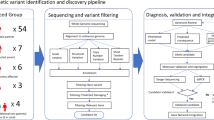


Introduction
The capacity to acquire complex spoken language appears to be unique to humans [1]. The majority of children, when exposed to linguistic input in their environment, develop skills to understand others, to convert thoughts into spoken utterances and to produce intelligible speech. Remarkably, this sophisticated suite of abilities emerges rapidly within the first few years of life without the need for formal teaching or special effort. Several complementary lines of biological research suggest that there are strong genetic underpinnings for such skills, ranging from evidence of significant heritability from twin and family studies to observations of gene associations in molecular studies of relevant traits [2]. Identification of the responsible genes not only sheds novel light on the pathways underlying the disorders, but can also greatly enhance our fundamental knowledge about the neurobiological mechanisms enabling humans to acquire language [3].
Most cases of developmental language impairments are likely to involve genetic complexity, resulting from the inheritance of multiple risk factors with small individual effect sizes [2]. Nonetheless, it has been established that disorders of speech and language sometimes occur in monogenic form. One relevant disorder that may be enriched for damaging gene variants of large effect size is childhood apraxia of speech (CAS), also known as developmental verbal dyspraxia. CAS is a rare severe developmental disorder characterized by difficulties with automatically and accurately sequencing speech sounds into syllables, syllables into words, and words into sentences with the correct prosody [4]. In CAS, it is thought that impairments in the neural planning and/or programming of spatiotemporal parameters of movement sequences result in errors in speech sound production and prosody. Diagnostic features of CAS include inconsistency in the types of speech errors that are made, and greater problems as the complexity and length of the utterance increase.
In 2001, studies of a large multigenerational pedigree named the KE family, along with an unrelated case with a chromosomal translocation, led to the discovery that disrupting one copy of the FOXP2 gene (on chromosome 7q31) is sufficient to cause CAS [5]. Multiple different cases of FOXP2 disruption have since been identified, including missense and nonsense mutations, insertion/deletions and chromosomal rearrangements of various kinds; CAS is the most consistent phenotypic consequence in the affected people [2, 6]. More than 15 years after the identification of FOXP2, progress in identifying additional genetic risk factors has been slow, and mainly driven by studies of chromosomal rearrangements. For example, deletion of a ~600-kb region in human chromosomal band 16p11.2, encompassing >25 genes, significantly increases risk of CAS, amongst other phenotypic consequences [7]. The BCL11A and ERC1 genes have been found to be disrupted by deletions at 2p16.1 and 12p13.33, respectively in children with CAS or with broader problems that also involve intellectual disability, motor difficulties, developmental problems (for BCL11A deletions) and psychiatric manifestations (for ERC1 deletions) [8, 9]. Recently, point mutations that disrupt BCL11A function have been implicated in a neurodevelopmental syndrome that includes language delays, although a diagnosis of speech apraxia was not specifically reported in these cases [10]. Still, the majority of speech apraxia cases do not seem to carry causal mutations in FOXP2, BCL11A or ERC1 [6, 11, 12].
The present study aimed to take advantage of cutting-edge genomic strategies to identify novel genes implicated in CAS, and thereby move the field significantly beyond FOXP2. In particular, we applied whole-genome sequencing (WGS) in 19 probands with a diagnosis of CAS, to identify single nucleotide variants (SNVs) and small insertions and deletions (indels) against a genome-wide background. Crucially, for half the WGS cohort of the present study (9 probands), we could also sequence the entire genomes of nuclear family members without CAS (both parents, and in one family a sibling as well), allowing us to directly pinpoint de novo variants, which are known to have an increased likelihood of being causal [13]. Moreover, given the established evidence that genes involved in speech and language disruptions cluster in related functional pathways [14, 15], we coupled our WGS findings to co-expression network analysis, identifying correlated expression patterns across developing human brain tissue samples. We further validated findings by comparison to WGS data from an independent set of healthy controls, analyzed using the same procedures as the CAS cohort. Our work uncovered a neural co-expression module of functionally-related genes, which brings together ERC1 and BCL11A with newly implicated candidate genes in CAS susceptibility, several of which have also been connected with neurodevelopment through studies of other disorders.
Patients and Methods
Ethics
The research in this study was approved by the appropriate review boards: the Social and Behavioral Sciences Institutional Review Board of the University of Wisconsin-Madison [Protocol 2013-0438], the Augustana College Institutional Review Board, and the Mayo Clinic Institutional Review Board [Protocol PR08-002372] (primary CAS cohort); the Melbourne Human Research Ethics Committee [project 27053] (Australian case); and the Basse-Normandie local ethics committee [reference CPP-2006-16] (control data set).
Participants
The primary data set comprised 19 probands who were ascertained based on a formal clinical diagnosis of CAS. Participants were recruited for a study of pediatric motor speech disorders at two collaborative sites, as described previously [11, 12]. All probands were evaluated using the Madison Speech Assessment Protocol and the Speech Disorders Classification System to identify and classify speakers’ speech status at assessment, including CAS (Supplementary Table 1) [16]. Medical genetic evaluations were not included in the assessment protocol. For 9 of the probands, 19 additional nuclear family members provided DNA samples for sequencing (i.e., unaffected parents for 9 children, plus one unaffected sibling). Blood samples were obtained from these 9 probands; blood and saliva samples (Oragene DNA OG-500 kit; DNA Genotek Inc., Kanata, Ontario, Canada) were obtained from the remaining 10 probands. All participants gave informed consent. The current study included 7 out of 10 probands from a previous WES-based study that primarily focused on known candidate genes, and 5 out of 12 probands previously studied using aCGH screening [11, 12]. However, neither of these prior studies included any analyses of parental/sibling DNA samples. Supplementary Table 2 summarizes the relationship with the two previously studied cohorts.
Whole-genome sequencing and variant calling
Novogene (Hong Kong) performed WGS of the CAS cohort using Illumina’s HiSeq Xten technology, involving paired-end sequencing, with reads of 150 base pairs long and a library insert size of 350 base pairs. Clean raw reads made up 97% of the total reads and were mapped onto the human reference genome (hg19) using Burrows–Wheeler Aligner (BWA) [17]. BAM files were sorted using SAMtools [18] and PCR duplicated reads were marked using Picard. Re-alignment around indels (insertion/deletions) and base quality control recalibration was performed using Genome Analysis Toolkit Software (GATK v3.4) [19]. Genetic variants in the sequence data were called as recommended by GATK best practices. We performed Variant Quality Score Recalibration (VQSR) on the WGS data, excluding variants with a VQSR score over 99%.
Anonymized primary data are deposited at The Language Archive (TLA: https://corpus1.mpi.nl/), a public data archive hosted by the Max Planck Institute for Psycholinguistics. Data are stored at the TLA under the node ID: MPI2535402#, and accessible with a persistent identifier: https://hdl.handle.net/1839/BAC29352-0AF4-4A09-B946-F4AC4865A67E@view. Access can be granted upon request.
Control data
WGS data of 22 unaffected unrelated individuals (12 female, 10 male), recruited as part of the BIL&GIN data set [20], were used as healthy controls to test for potential enrichment bias that might result from variant filtering procedures (see below). Informed consent was obtained from all participants. BGI (Hong Kong/Shenzhen) performed WGS on this data set using Illumina’s HiSeq Xten technology, involving paired-end sequencing with reads of 150 base pairs long (8 samples) or 90 base pairs long (14 samples). The same pipeline as that applied to the CAS cohort (updated to GATK v3.5, VEP v88 and Gemini v20.0) was used for WGS data alignment and variant calling, annotation and filtering.
Structural variant calling
We used a combination of two algorithms for the detection of structural variants on the autosomes: BreakDancer [21] (v1.1.2) and BIC-seq2 [22] (v0.7.2), using standard settings for both. BreakDancer bases its detection of structural variants on abnormal alignment of read pairs. BIC-seq2 bases its detection of structural variants on read depth. Structural variants detected by both BreakDancer and BIC-seq2 with a maximum two-fold size difference and maximal distance of 10 kb between predicted start or end sites were considered.
Variant annotation and filtering
Variant Effect Predictor [23] (v73) was used for annotation and Gemini [24] (v0.18.3) was used to select exonic variants in protein-coding genes from the data set. Variants located in known regions of genomic duplications, in regions with a sequencing depth below 10 reads and present in any of the unaffected nuclear family members were excluded. Variants were filtered further based on minor allele frequency (MAF), and expression of the gene in developing brain according to thresholds outlined below. In addition, in absence of information about de novo status, variants identified in the ten singleton probands for whom parental DNA was not available, were further filtered based on gene intolerance and predicted functional impact of the variant.
The threshold for MAF was 5 × 10−4 in 1000 genomes (1000G; phase 3) and the exome variant server (ESP; release ESP6500SI-V2) and a maximum tolerated allele count of 10 in the Exome Aggregation Consortium (ExAC; v0.3) database [25], based on a recent statistical framework that takes into account multiple factors, including disease prevalence, genetic heterogeneity, inheritance mode and penetrance [26].
Genes were considered intolerant based on a Residual Variation Intolerance Score (RVIS) <25 [27]. In addition, genes with loss-of-function (LoF) variants were considered if the probability of being LoF intolerant (pLi) was >0.9, and genes with missense variants were considered if the Z-score for missense constraint (MIS_Z) was >3 [25]. Missense variants with GERP++ >2, scaled CADD (v1.0) >15 and Polyphen and SIFT indicating a damaging effect were considered to have high impact.
As CAS is a neurodevelopmental disorder with early onset, only genes that are expressed in the developing human brain were included. Genes were considered expressed in the developing brain if transcripts were present in the developmental human RNA sequencing data set of Brainspan (http://www.brainspan.org/) in any brain tissue collected 8–24 weeks post conception, with >1 fragments per kilobase of exon per million reads mapped (FPKM). Established candidate genes for involvement in CAS (FOXP2, BCL11A and ERC1) and other speech/language-related developmental disorders (ATP2C2, AUTS2, CMIP, CNTNAP2, CTNND2, DCDC2, DOCK4, DYX1C1, FOXP1, GRIN2A, KIAA0319, NFXL1, ROBO1, SETBP1 and SRPX2) [2] all passed this threshold.
Variant interpretation
Phenotypes previously associated with similar variants occurring in the same gene were collected using searches in PubMed, the Online Mendelian Inheritance in Man (OMIM) database and the Human Gene Mutation Database (HGMD Professional, version 2016.3). Variants identified in the current WGS analysis were interpreted according to a five-tier system of classification for variants of Mendelian disorders into 1) pathogenic, 2) likely pathogenic, 3) uncertain significance, 4) likely benign and 5) benign variants [28].
All variants reported in the main manuscript were independently validated using Sanger sequencing.
Co-expression network
Brainspan RNA-sequencing data were used to construct a co-expression network. All cerebral brain samples from fetal, neonatal and infancy periods (8 weeks post conception up to 12 months of age) were included. A total of 224 samples collected from multiple regions of 23 human brains with high RNA quality (RIN > 9), and 14,442 genes with high expression (≥1 FPKM in at least 2 samples), and variable expression between samples (>0 FPKM in at least 50% of samples and coefficient of variance >0.25) were included. Motivated by the prior literature on neural correlates of CAS, as well as knowledge of brain regions impacted by known CAS-related genes [29,30,31,32,33,34], the samples used for calculating the co-expression network included not only cortical regions but also subcortical structures (discussed further in “Results” section). Co-expression analysis was carried out using the weighted correlation network analysis (WGCNA) R package [35]. A signed weighted adjacency matrix was calculated from the log-transformed gene expression data, using biweight midcorrelations to calculate co-expression similarity, and a soft thresholding power of 31, which was transformed into a topological overlap matrix. Modules were detected using the cutreeDynamic function of the WGCNA package, with hybrid tree cutting, and a minimal cluster size of 200 genes (settings method = hybrid, deepSplit = 2, minClusterSize = 200). To summarize the expression pattern of the genes in a module, module Eigengenes are calculated as the first principal component.
Gene set analysis
Enrichment of gene sets in co-expression modules was calculated using a two-sided Fisher exact test, followed by false discovery rate (FDR) correction for multiple-testing. For the enrichment analysis of the CAS candidate genes, we used a background set comprising the 2,143 genes of the genome that are intolerant to mutations and that are expressed in the developing brain, to compensate for the background enrichment pattern of intolerant genes in the network (Supplementary Fig. 1). In addition, we studied enrichment for lists of genes with de novo mutations in patients with intellectual disability (ID, n = 230), autism spectrum disorder (ASD, n = 2,760) and schizophrenia (n = 711) [36]. The enrichment of these gene lists has previously been studied in a co-expression network based only on cortical tissues [37].
To functionally interpret modules, gene ontology (GO) term enrichments were performed in DAVID 6.8 [38]. The Functional Classification Tool of DAVID was applied to group identified GO terms with low clustering stringency. Bonferroni-corrected geometrical mean p values for each group of GO terms were reported.
Results
Whole-genome sequencing was used to discover gene variants
Whole-genome sequencing (WGS) with on average 32.1 times sequencing depth was carried out for 38 DNA samples from 19 probands with CAS and 19 nuclear family members (Supplementary Tables 1 and 2). Before considering a genome-wide view, we used the data to assess potential contributions of three genes implicated in CAS in prior published work: FOXP2, BCL11A and ERC1 [5, 8, 9], as well as the FOXP2-paralogue FOXP1 that has been implicated in a broader speech-related neurodevelopmental phenotype [39]. Consistent with earlier limited studies of subsets of the present cohort, our analyses identified two rare missense variants: FOXP1 (ENST00000318789, c.322A>G) p.I107T in proband 01 and FOXP2 (ENST00000408937, c.1864A>C) p.N622H in proband 11. However, we found that the FOXP1 variant was in fact inherited from the proband’s unaffected mother, casting doubt on a causal role in CAS. Moreover, recent molecular and cellular assays of both variants indicate that they do not impact on the function of the encoded proteins [39, 40]. We, therefore, classified both as variants of uncertain significance. Thus, prominent CAS-related risk genes from the literature do not appear to account for the disorder in this WGS data set.
De novo variants disrupt CHD3, SETD1A and WDR5 in probands with CAS
We next took advantage of the trio/quartet design of part of our WGS sample to search for novel genes that might be implicated in CAS, the first systematic application of the de novo paradigm for assessing any speech disorder. Filtering on minor allele frequency of the variant in public databases (1000G, ESP and ExAC), and expression of the gene in the developing brain, yielded nine non-synonymous exonic de novo variants (Table 1; Supplementary Table 3). All nine variants were successfully validated and confirmed as de novo by Sanger sequencing. In addition, a 1.86 Mb deletion at 2q31.1 (chr2:172,788,173-174,646,059) in proband 06, detected previously in this proband using aCGH [11], was independently detected in our WGS analyses and now shown to represent a de novo structural variant. The deletion affects multiple contiguous genes (HAT1, METAP1D, DLX1, DLX2, ITGA6, PDK1, RAPGEF4, ZAK and CDCA7). Mutations of these deleted genes have not been implicated in a neurodevelopmental disorder in prior work, although common variation at DLX1 and DLX2 has been associated with increased risk of ASD [41]. Of note, all other structural variants and SNVs previously reported in cases 01–09 (Supplementary Table 2) [11, 12] were also found in unaffected nuclear family members of the probands, revealing that those variants are unlikely to be causal for CAS.
In each of probands 01, 04 and 07, a single exonic de novo variant was identified (Fig. 1). Proband 01 carries a de novo missense variant (p.R1228W in ENST00000380358; p.R1169W in ENST00000330494) in a key functional domain of CHD3, a chromatin remodeling factor. In proband 04, a de novo loss-of-function (LoF) variant (p.V553Wfs*110) was found disturbing SETD1A, a histone methyltransferase. In proband 07, a de novo missense variant (p.T208M) was found in WDR5, within the WD40-repeat domain of the encoded protein. In each case, the respective variant was predicted to be pathogenic and there was strong independent evidence supporting causality from prior studies of the gene in question. Following our discovery of the CHD3 variant in proband 01, de novo variants disrupting this gene have now been pinpointed in 34 other cases worldwide, with a variable neurodevelopmental disorder in which impaired speech and language is one of the phenotypic features (L. Snijders Blok, personal communication). In addition, LoF variants in SETD1A have been associated with neurodevelopmental disorders that include schizophrenia, ID and speech/language delays [42]. Lack of symptoms indicating developmental delay or a psychiatric disorder suggests that proband 04, who carries the SETD1A frameshift, has a mild form of SETD1A-associated disorder. Lastly, WDR5 and SETD1A have close functional connections, since the proteins they encode belong to the same protein complex that confers histone-3 lysine-4 methylation [43]. For probands 02 and 08, three de novo variants were identified in each case (Table 1), all classified as variants of unknown significance.

De novo pathogenic variants in CHD3, SETD1A and WDR5. a Sanger validation of de novo pathogenic variants in family 01, 04 and 07. b Conservation of the mutated amino acids across species. Blue boxes mark the mutated amino acid. The blue line indicates the site of the duplication. c Schematic representation of CHD3 (ENST00000380358) organization, with CHD C- and N-terminal domains in black, plant homeodomain (PHD) zinc finger domains in blue, chromatin organization modifier (CHROMO) domains in green, DEAD-like helicases superfamily (DEXDc) domain in yellow, a helicase superfamily c-terminal (HELICc) domain in red and two domains of unknown function (DUF) in gray. Blue line indicates site of the p.R1228W variant. d Schematic representation of SETD1A (ENST00000262519) protein, with an RNA recognition motif (RRM) in red, a complex proteins associated with Set1p (COMPASS) component N (N-SET) domain in yellow, a Su(var)3–9, Enhancer-of-zeste, Trithorax (SET) domain in green and a post-SET domain in blue. Blue line indicates site of the frameshift, and the blue shaded area indicates the deleted part of the protein. e Schematic representation of WDR5 (ENST00000358625), with WD40 repeats in green. Blue line indicates site of the p.T208M variant
Loss-of-function variants in CAS implicate known neurodevelopmental genes
We went on to study the genomes of the 10 singleton probands for whom parental DNA was not available. On searching for variants in any of the nine genes already shown to carry de novo variants (Table 1), proband 15 had a missense variant in HERC1 (c.2932-2933GA>TC, p.E978S). HERC1 is associated with a recessive type of ID [44], but it is unknown whether heterozygous variants could lead to CAS.
Broadening to a genome-wide view, strict filtering criteria based on minor allele frequency, gene intolerance and expression of the gene in developing brain were applied to obtain only the most likely causal variants. A total of seven LoF variants in five probands were identified (Table 2). Interestingly, four of these LoF variants disrupted genes previously implicated in neurodevelopmental disorders: KAT6A (c.1599-56_1621del in proband 10), SETBP1 (c.1781del, p.P594Lfs*36 in proband 13), TNRC6B (c.2040G>A, p.W680* in proband 15) and ZFHX4 (c.3646-1G>A in proband 14) [45,46,47,48]. These four mutations were therefore considered pathogenic. LoF variants in KAT6A are associated with severe speech delay, ID, hypotonia and facial dysmorphism [45]. To our knowledge, proband 10 of the present study represents the first case of a KAT6A LoF variant that only yields a speech phenotype, in absence of global developmental delay. Recurrent de novo LoF mutations were recently identified in TNRC6B in children with ASD in a large cohort of over 2,500 families [47], while deletions of ZFHX4 have been associated with ID and specific facial features [48]. The neurodevelopmental features of proband 13 (CAS, intellectual deficits and motor delay) overlap with the clinical picture associated with SETBP1 haploinsufficiency in previous reports [46]. In this proband, as well as the frameshift in SETBP1, another two LoF variants were present (in OPA1 and RAP1GAP), but considered of less significance for CAS, given prior literature on SETBP1 dysfunction and speech/language phenotypes. Lastly, proband 18 carries a splice acceptor variant in MKL2. A recent large-scale targeted sequencing study of ~300 brain-related genes in ASD identified rare variants of MKL2, but the relevance of these for the disorder was uncertain [49]. Based on the functional consequence of the MKL2 variant observed in proband 18 and the gene intolerance, we considered this variant as likely pathogenic.
A putative causal role for the multiple LoF mutations that we identified in the CAS cohort is further illustrated by comparison to WGS data from an independent control cohort of unaffected individuals, filtered in an identical manner (see “Patients and Methods” section). In these 22 unaffected controls, three putative LoF variants were identified in intolerant genes, none of which involved a gene previously identified as causal for a neurodevelopmental disorder (Supplementary Table 4).
Moreover, in independent exome sequencing of clinically-defined speech disorders, we identified an unrelated Australian case of CAS, together with oral apraxia, dysarthria, moderate ID, seizures and motor impairments, with a de novo LoF mutation disrupting SETBP1. This child carried a novel premature stop variant (c.C2665T, p.R889X) in SETBP1 that was not present in her unaffected parents.
We also assessed rare missense and in-frame indel variants in our singleton CAS probands, again strictly filtered based on minor allele frequency, gene intolerance, predicted impact of the variant, and expression of the gene in developing brain (see “Methods” section). A total of 21 (0–4 per sample) rare, predicted damaging missense variants in intolerant genes were found (Supplementary Table 5). Again, variants were found in genes previously implicated in neurodevelopmental disorders. In particular, proband 19 carried a p.D2155N (c.6463G>A) variant in TRIO, a gene which has been associated with ASD and ID [50], and proband 18 carried a deletion of five amino acids (c.1029_1043del, p.Ala345_Ala349del) in ARID1A, one of the causal genes for Coffin–Siris syndrome [51]. When we analyzed WGS data from the 22 unaffected individuals of the control cohort using the same filtering steps, 17 rare missense mutations in intolerant genes were identified, of which six lay in a gene previously identified as causal for a neurodevelopmental disorder (Supplementary Table 4). Therefore, in absence of segregation information and functional evidence, we considered all the missense variants of the singleton CAS probands as variants of uncertain significance.
541 structural variants were detected in the WGS data of the singleton CAS probands, of which only one disrupted exonic regions of an intolerant gene. This 127-kb deletion (chr7:16,693,649–16,821,881), found in proband 14, disrupts all but the first exon of BZW2 and the first five exons of TSPAN13. Neither gene has been associated with a neurodevelopmental disorder and no deletions of similar size at this location have been reported.
A CAS-related co-expression module in developing brain tissue
Prior independent findings of protein-protein interactions between FOXP2 and CHD3 [52] and between SETD1A and WDR5 [43] imply that genes mutated in monogenic forms of CAS may belong to molecular networks with shared functionality. We formally tested this hypothesis with respect to the set of ten genes with strongest evidence for carrying causal variants in our whole-genome sequencing CAS analyses: CHD3, SETD1A, WDR5, KAT6A, SETBP1, ZFHX4, TNRC6B, MKL2, ARID1A and TRIO. These include the eight genes with a mutation that was classified as pathogenic or likely pathogenic, as well as the two genes that had a missense mutation and that were previously implicated in neurodevelopmental disorders.
An unbiased co-expression approach was taken to assess shared functionality. For this, we analyzed correlated patterns in gene expression levels in RNA-sequencing data of 224 samples from multiple brain regions of 23 human brains collected from 8 weeks post conception up to 1 year. Subcortical samples were included a priori, contrasting with a previous developmental brain co-expression analysis that utilized only cortical samples [37]. The inclusion of subcortical regions was based on a growing appreciation in the literature of their importance for speech and language development, including the identification of altered subcortical structure and function in individuals with language-related disorders [29], especially in CAS [30, 31]. Indeed, established CAS-related genes such as FOXP2, BCL11A and ERC1 show high expression in subcortical tissues, and the effects of CAS mutations on basal ganglia function have been well documented [30,31,32,33,34].
A spatiotemporal co-expression network was calculated using weighted gene co-expression network analysis (WGCNA) [35] (Fig. 2a), yielding 16 co-expression modules, each comprising 265 to 1,365 genes. The genes that we had identified as potential risk factors in CAS were highly co-expressed in the developing human brain, with eight out of ten (all except ZFHX4 and MKL2) belonging to a single shared module (M3 module; Fisher’s exact test: p = 1.09 × 10−5; FDR-corrected p = 1.85 × 10−4). This significant enrichment signal remained even if we excluded ARID1A and TRIO as genes with missense variants of uncertain significance (six out of eight genes belong to M3 module; FDR-corrected p = 4.96 × 10−3). Remarkably, although FOXP2 could not be assigned to any module in this analysis, both BCL11A and ERC1 were found to be members of this same module shared by the new CAS candidate genes. Genes carrying de novo mutations in ID, ASD and schizophrenia showed evidence of enrichment in module M3 as well (Fig. 2b). Genes implicated in ASD showed similar evidence for enrichment in modules M9, M12 and M14, and genes implicated in ID were enriched in module M14. By contrast, no enrichment was found for genes with LoF and predicted damaging missense mutations in intolerant genes identified in our WGS control cohort.

Co-expression network analysis. a A co-expression network was calculated using gene expression data of brain samples collected between 8 weeks post conception up to 1 year of age from the cortex, hippocampus, amygdala, striatum and thalamus. A total of 16 modules were detected. Module 3 (indicated by black arrow) was highly enriched for the genes we implicated in CAS through whole-genome sequencing (CAS-WGS). b Enrichment of developmental disorder gene sets in the 16 modules. Gene sets included are the 10 genes implicated in CAS through WGS (CAS-WGS), and genes with de novo mutations in patients with the following (1) intellectual disability (ID, n = 230), (2) autism spectrum disorder (ASD, n = 2760), and (3) schizophrenia (SCZ, n = 711). Significant enrichments with False discovery rate (FDR)-corrected p value < 0.05 and odds ratio (OR) >1 are shown. Colors indicate FDR-corrected p values for enrichment. Numbers show OR
Genes belonging to module M3 have high expression during early and mid-embryonic development in most brain regions, except for the mediodorsal nucleus of the thalamus, for which expression decreases during late fetal development and after birth (Fig. 3a, b). Analysis of biological pathways, using gene ontology terms, indicated that this CAS-related module is highly enriched for a number of classes, including genes involved in nucleic acid binding (726 genes, Bonferroni-corrected p = 7.05 × 10−8), which encompasses multiple transcription factors and genes involved in histone modification (400 genes, Bonferroni-corrected p = 1.12 × 10−4) (Fig. 3c). This finding is consistent with prior studies that implicated molecular pathways related to neurodevelopment and plasticity in speech disorders [33, 53], as well as in other neurodevelopmental disorders [54, 55]. ASD/ID-related modules M9 and M12 are also enriched for genes involved in transcription regulation, while ID-related module M14 is enriched for genes encoding synaptic components (Supplementary Fig. 2).

Human brain expression pattern and functional enrichment of module M3. a Spatial expression pattern of the CAS-related module at 13–24 weeks post conception, as visualized by the M3 module’s Eigengene. Red shows high expression, blue shows low expression. Multiple samples per region were averaged. No expression data were available for gray regions. b Developmental brain expression pattern of the enriched module during development, as visualized by the module Eigengene. Each dot represents a brain sample, the black line is the loess curve fitted through the data points. The blue vertical line represents time of birth. Pcw: post conception week. c Gene functions enriched in the module identified through gene ontology (GO) term enrichment followed by clustering of GO terms using the functional annotation clustering tool in DAVID. The p values represent the geometric mean of Bonferroni-corrected p values of all GO terms underlying each function. The blue vertical line represents the threshold for significant enrichment (p = 0.05). Cx cortex, PF prefrontal
Discussion
Over a decade and a half since identifying a role for the FOXP2 gene in development of speech and language, we still have limited knowledge about other molecular factors that underlie these human capacities. Here we took advantage of advances in next-generation sequencing technologies as well as gene expression analyses of developing brain tissue, to reveal novel neurogenetic pathways implicated in speech development. Specifically, we applied whole-genome sequencing to a cohort of 19 probands ascertained on the basis of a rare severe developmental speech disorder with a distinct diagnosis, CAS. Based on prior studies of this rare phenotype, we hypothesized that CAS would be enriched for monogenic causes, involving disruptive mutations of large effect. In support of this hypothesis, we found that seven cases carried variants that were classified as pathogenic (in CHD3, SETD1A, WDR5, KAT6A, SETBP1, TNRC6B and ZFHX4) and another case carried a variant that was classified as likely pathogenic (in MKL2). These results strongly contrast with other more common language-related disorders that do not involve CAS. For example, a recent exome sequencing study on 43 unrelated probands with specific language impairment identified only a few possibly pathogenic mutations, and mainly supported a more complex pattern of inheritance, in which affected individuals carry multiple risk factors of modest effect size [56].
For half of the probands, whole-genome sequence data from both parents allowed for the isolation of de novo mutations, implicating the genes CHD3, SETD1A and WDR5 in the pathogenesis of CAS for the first time. Remarkably, a previous systematic yeast-two-hybrid screen for interaction partners of FOXP2, the most well-established CAS-related gene, identified the CHD3 protein as one of the top putative interactors of this transcription factor (see Table S1 in reference [52]). Our findings are consistent with the identification of multiple de novo mutations disrupting the same functional domain of CHD3 in other patients with neurodevelopmental disorders involving speech and language problems (L. Snijders Blok, personal communication). Beyond CHD3, another seven of the genes highlighted by our WGS analyses (SETD1A, KAT6A, SETBP1, TNRC6B, ZHFX4, ARID1A and TRIO) have been linked to related neurodevelopmental phenotypes in prior published work. Moreover, for a number of these genes, including SETD1A, KAT6A, SETBP1 and TRIO, the associated disorders, while being broader and/or more severe than CAS, have nevertheless been noted to include speech and language deficits. A particularly interesting example is SETBP1, haploinsufficiency of which has been previously associated with a phenotype including mild to severe ID, motor delay, facial dysmorphism and expressive language delay [46]. In the present study, we identified a SETBP1 frameshift mutation in our WGS cohort. A de novo LoF variant disrupting SETBP1 in an additional case with CAS, which we identified through independent exome sequencing, confirmed the relevance of this gene for developmental speech deficits. In addition, in a recent genome-wide screen of a geographically isolated Russian population with high prevalence of developmental language disorder, common variants of SETBP1 were significantly associated with complexity of linguistic output [57].
It is well established that diverse neurodevelopmental consequences can result from mutations in the same gene [58], hence it is perhaps not surprising that genes involved in other neurodevelopmental phenotypes may be implicated in primary speech disorders [8,9,10]. This holds even for FOXP2, the most-well studied gene in the language sciences, mutations of which were recently shown to cause a range of phenotypic profiles in different cases: various types of speech and language impairment with or without mild cognitive impairment or mild delays in motor development [59]. Our findings are consistent with this picture—while CAS was the major feature used to diagnose probands in the present study, some of the affected children also showed signs of reduced cognitive function and/or deficits in motor development. CAS may therefore be considered as a part of a range of neurodevelopmental brain disorders with a shared genetic foundation, instead of a pure isolated phenotype involving genes that are exclusively related to speech disorders.
Using expression data from multiple regions of the developing human brain, we discovered that the genes identified in our CAS whole-genome screening have highly correlated gene expression patterns; eight out of ten (all but ZHFX4 and MKL2) are members of a single co-expression module, including mostly transcription factors and chromatin remodelers with high expression during early and mid-fetal brain development. This finding is in line with the known molecular functions of the genes that we identified through WGS, since most are directly involved in regulating gene expression, and are functionally connected with each other. CHD3 encodes a chromatin remodeler that is part of a protein complex (the NuRD complex) regulating gene repression [60], and both SETBP1 and ZHFX4 encode transcription factors that can interact with the NuRD complex [61, 62]. As noted above, FOXP2 interacts with CHD3 [52], providing a direct link between the novel genes and earlier established pathways in CAS. SETD1A and WDR5 encode different parts of a protein histone methyltransferase complex named SET1/MLL [43]. KAT6A codes for the histone-acetyl-transferase of the MOZ complex that establishes gene activation, also through interaction with SET1/MLL [63]. The protein encoded by ARID1A is part of a large chromatin remodeling complex (called SNF/SWI) [64], and MKL2 encodes a subunit of the stimulus-dependent transcription factor SRF [65]. Finally, TNRC6B is involved in micro-RNA-directed RNA processing [66], playing a more indirect role in gene expression regulation.
The enrichment of CAS-associated genes in the co-expression module suggests that regulation of gene expression during early human brain development plays a role in susceptibility to this disorder. Genes implicated in ID, ASD and schizophrenia are also enriched in module M3, in line with previous reports highlighting the involvement of chromatin remodelers and transcription factors in these neurodevelopmental disorders [37, 54, 55]. However, the degree of enrichment is much lower than that observed for the CAS candidate genes. In addition, genes related to ID and ASD show enrichment in the synaptic gene module M14, which is consistent with previous reports linking synaptic gene function to autistic phenotypes [37, 54]. Thus, we propose that a subset of genes involved in neurodevelopmental disorders—transcription factors and chromatin remodelers mostly expressed during early brain development—are particularly relevant for speech development. Dynamic chromatin-level modifications of the genome are crucial for coordination of multiple stages of neural development [67]. For example, several of the CAS-related proteins in module M3 belong to chromatin remodeling and/or transcription factor complexes that have been shown to play key roles in neuronal differentiation and cortical layer specification in mouse models [68, 69].
Given the well-established involvement of cortical and subcortical brain structures in pathology of CAS [30, 31], we calculated the co-expression network for this study based on both cortical and subcortical tissues. As a post hoc analysis, we compared our network findings to a network from a prior ASD investigation that used the same primary source of RNA data, but limited to only cortical samples [37] (Supplementary Table 6). While the inclusion or exclusion of subcortical structures obviously yields differences between the derived networks of the two studies, the clustering of our CAS candidates is robust. Five genes (CHD3, SETD1A, SETBP1, ARID1A and TRIO) are part of module M2 from the cortical-only study and the enrichment is significant (FDR-corrected p value 2.0 × 10−4). Module M2 (containing 1,036 genes) is the closest match in the cortical-only network to module M3 (containing 901 genes) of our cortical-subcortical network, with 295 overlapping genes. These modules show similar developmental expression trajectories and GO term enrichments.
Similarity in function and developmental brain expression pattern provided support for a causal relation between the variants we identified through WGS and speech deficits. However, interpretation of WGS results is challenging in cases when recurrent mutations cannot provide final proof for a causal relation between the identified genes and disorder [70]. Our report is the first to associate heterozygous variants of WDR5 with a neurodevelopmental disorder, therefore further evidence of causative variants in children with CAS or a related phenotype will be important to confidently implicate this gene. ARID1A and TRIO were already associated with neurodevelopmental disorder, and the missense mutations in these genes were selected as potential causal variants in proband 18 and 19, respectively. Disease-causing missense variants in TRIO have so far been described mostly in the first Dbl homology-Pleckstrin homology (DH-PH) domain [50], while the p.D2155N variant identified in case 19 is located adjacent to the second DH-PH domain. In addition, mostly LoF mutations have been described in ARID1A, and the impact of missense mutations is difficult to predict without functional studies. Analyses of our control cohort suggested that the strict filtering criteria might have increased the chance of finding missense mutations in genes associated with neurodevelopmental disorders. Therefore, we treat these two genes with some degree of caution at this point. Our WGS analysis identified another 19 missense variants that were predicted as damaging and located in intolerant genes. Despite very strict filtering criteria, we cannot infer causality for such variants without additional cases. For example, in HERC1, recurrent missense mutations were identified: a de novo p.S3485N variant in proband 08, and a p.E978S variant in proband 15. To date, only recessive mutations in HERC1 have been found as causal for intellectual disability [44], and no phenotype has been described for the parents harboring a single HERC1 variant, so the relevance of HERC1 missense variants for CAS cannot currently be determined. Lastly, we cannot exclude a more complex pattern of inheritance in the probands who carry more than one variant that was predicted to be damaging.
In summary, by analyzing whole-genome sequences from 19 probands ascertained through a CAS diagnosis, we identified de novo mutations in CHD3, SETD1A and WDR5 and LoF mutations in SETBP1, KAT6A, TNRC6B and ZFHX4 that were all classified as pathogenic. Moreover, we implicated a network of functionally connected genes, part of a coordinated expression module in the embryonic human brain, in the development of proficient speech skills. A significant number of these genes (CHD3, SETD1A, KAT6A, SETBP1, TNRC6B, ZFHX4, ARID1A and TRIO) have been associated with neurodevelopmental disorders with or without speech problems [42, 45,46,47,48,49,50,51]. These results indicate that the genetic underpinnings of CAS are—at least in part—shared with those of other brain-related syndromes, but that genes relevant for speech are clustered within particular functional networks. Our work identifies molecular pathways involved in regulation of gene expression during early brain development that may be critical for the acquisition of fluent spoken language.
References
Fisher SE, Marcus GF. The eloquent ape: genes, brains and the evolution of language. Nat Rev Genet. 2006;7:9–20.
Graham SA, Fisher SE. Understanding language from a genomic perspective. Annu Rev Genet. 2015;49:131–60.
Fisher SE, Scharff C. FOXP2 as a molecular window into speech and language. Trends Genet: TIG. 2009;25:166–77.
American Speech-Language-Hearing Association. Childhood apraxia of speech. 2007. http://www.asha.org Accessed in April 2017.
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–23.
Morgan A, Fisher SE, Scheffer I, Hildebrand M. FOXP2-related speech and language disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al, editors. Gene reviews(R). Seattle (WA):University of Washington; 2017.
Fedorenko E, Morgan A, Murray E, Cardinaux A, Mei C, Tager-Flusberg H, et al. A highly penetrant form of childhood apraxia of speech due to deletion of 16p11.2. Eur J Human Genet. 2016;24:302–6.
Thevenon J, Callier P, Andrieux J, Delobel B, David A, Sukno S, et al. 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. Eur J Human Genet. 2013;21:82–8.
Peter B, Matsushita M, Oda K, Raskind W. De novo microdeletion of BCL11A is associated with severe speech sound disorder. Am J Med Genet A. 2014;164A:2091–6.
Dias C, Estruch SB, Graham SA, McRae J, Sawiak SJ, Hurst JA, et al. BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am J Hum Genet. 2016;99:253–74.
Laffin JJ, Raca G, Jackson CA, Strand EA, Jakielski KJ, Shriberg LD. Novel candidate genes and regions for childhood apraxia of speech identified by array comparative genomic hybridization. Genet Med: Off J Am Coll Med Genet. 2012;14:928–36.
Worthey EA, Raca G, Laffin JJ, Wilk BM, Harris JM, Jakielski KJ, et al. Whole-exome sequencing supports genetic heterogeneity in childhood apraxia of speech. J Neurodev Disord. 2013;5:29.
Deriziotis P, O’Roak BJ, Graham SA, Estruch SB, Dimitropoulou D, Bernier RA, et al. De novo TBR1 mutations in sporadic autism disrupt protein functions. Nat Commun. 2014;5:4954.
Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359:2337–45.
Kato M, Okanoya K, Koike T, Sasaki E, Okano H, Watanabe S, et al. Human speech- and reading-related genes display partially overlapping expression patterns in the marmoset brain. Brain Lang. 2014;133:26–38.
Shriberg LD, Fourakis M, Hall SD, Karlsson HB, Lohmeier HL, McSweeny JL, et al. Extensions to the Speech Disorders Classification System (SDCS). Clin Linguist Phon. 2010;24:795–824.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Mazoyer B, Zago L, Jobard G, Crivello F, Joliot M, Perchey G, et al. Gaussian mixture modeling of hemispheric lateralization for language in a large sample of healthy individuals balanced for handedness. PLoS ONE. 2014;9:e101165.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81.
Xi R, Lee S, Xia Y, Kim TM, Park PJ. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res. 2016;44:6274–86.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122.
Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9:e1003153.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med: Off J Am Coll Med Genet. 2017;19:1151–8.
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med: Off J Am Coll Med Genet. 2015;17:405–24.
Krishnan S, Watkins KE, Bishop DV. Neurobiological basis of language learning difficulties. Trends Cogn Sci. 2016;20:701–14.
Belton E, Salmond CH, Watkins KE, Vargha-Khadem F, Gadian DG. Bilateral brain abnormalities associated with dominantly inherited verbal and orofacial dyspraxia. Hum Brain Mapp. 2003;18:194–200.
Liégeois F, Baldeweg T, Connelly A, Gadian DG, Mishkin M, Vargha-Khadem F. Language fMRI abnormalities associated with FOXP2 gene mutation. Nat Neurosci. 2003;6:1230–7.
Groszer M, Keays DA, Deacon RM, de Bono JP, Prasad-Mulcare S, Gaub S, et al. Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol. 2008;18:354–62.
Vernes SC, Oliver PL, Spiteri E, Lockstone HE, Puliyadi R, Taylor JM, et al. Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLoS Genet. 2011;7:e1002145.
French CA, Jin X, Campbell TG, Gerfen E, Groszer M, Fisher SE, et al. An aetiological Foxp2 mutation causes aberrant striatal activity and alters plasticity during skill learning. Mol Psychiatry. 2012;17:1077–85.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:559.
Shohat S, Ben-David E, Shifman S. Varying intolerance of gene pathways to mutational classes explain genetic convergence across neuropsychiatric disorders. Cell Rep. 2017;18:2217–27.
Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–21.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Sollis E, Graham SA, Vino A, Froehlich H, Vreeburg M, Dimitropoulou D, et al. Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Hum Mol Genet. 2016;25:546–57.
Estruch SB, Graham SA, Chinnappa SM, Deriziotis P, Fisher SE. Functional characterization of rare FOXP2 variants in neurodevelopmental disorder. J Neurodev Disord. 2016;8:44.
Liu X, Novosedlik N, Wang A, Hudson ML, Cohen IL, Chudley AE, et al. The DLX1 and DLX2 genes and susceptibility to autism spectrum disorders. Eur J Human Genet. 2009;17:228–35.
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–7.
Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–9.
Ortega-Recalde O, Beltran OI, Galvez JM, Palma-Montero A, Restrepo CM, Mateus HE, et al. Biallelic HERC1 mutations in a syndromic form of overgrowth and intellectual disability. Clin Genet. 2015;88:e1–3.
Tham E, Lindstrand A, Santani A, Malmgren H, Nesbitt A, Dubbs HA, et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am J Hum Genet. 2015;96:507–13.
Filges I, Shimojima K, Okamoto N, Rothlisberger B, Weber P, Huber AR, et al. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J Med Genet. 2011;48:117–22.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
Palomares M, Delicado A, Mansilla E, de Torres ML, Vallespin E, Fernandez L, et al. Characterization of a 8q21.11 microdeletion syndrome associated with intellectual disability and a recognizable phenotype. Am J Hum Genet. 2011;89:295–301.
Li J, Wang L, Guo H, Shi L, Zhang K, Tang M, et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol Psychiatry. 2017;22:1282–90.
Sadybekov A, Tian C, Arnesano C, Katritch V, Herring BE. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat Commun. 2017;8:601.
Kosho T, Okamoto N, Coffin-Siris Syndrome International C. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet. 2014;166C:262–75.
Estruch SB, Graham SA, Deriziotis P, Fisher SE. The language-related transcription factor FOXP2 is post-translationally modified with small ubiquitin-like modifiers. Sci Rep. 2016;6:20911.
Vernes SC, Spiteri E, Nicod J, Groszer M, Taylor JM, Davies KE, et al. High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. Am J Hum Genet. 2007;81:1232–50.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15.
McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19:652–8.
Chen XS, Reader RH, Hoischen A, Veltman JA, Simpson NH, Francks C, et al. Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci Rep. 2017;7:46105.
Kornilov SA, Rakhlin N, Koposov R, Lee M, Yrigollen C, Caglayan AO et al. Genome-wide association and exome sequencing study of language disorder in an isolated population. Pediatrics 2016;137:e20152469
Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013;12:406–14.
Reuter MS, Riess A, Moog U, Briggs TA, Chandler KE, Rauch A, et al. FOXP2 variants in 14 individuals with developmental speech and language disorders broaden the mutational and clinical spectrum. J Med Genet. 2016;54:64–72.
Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–61.
Vishwakarma BA, Nguyen N, Makishima H, Hosono N, Gudmundsson KO, Negi V, et al. Runx1 repression by histone deacetylation is critical for Setbp1-induced mouse myeloid leukemia development. Leukemia. 2016;30:200–8.
Chudnovsky Y, Kim D, Zheng S, Whyte WA, Bansal M, Bray MA, et al. ZFHX4 interacts with the NuRD core member CHD4 and regulates the glioblastoma tumor-initiating cell state. Cell Rep. 2014;6:313–24.
Paggetti J, Largeot A, Aucagne R, Jacquel A, Lagrange B, Yang XJ, et al. Crosstalk between leukemia-associated proteins MOZ and MLL regulates HOX gene expression in human cord blood CD34 + cells. Oncogene. 2010;29:5019–31.
Dallas PB, Cheney IW, Liao DW, Bowrin V, Byam W, Pacchione S, et al. p300/CREB binding protein-related protein p270 is a component of mammalian SWI/SNF complexes. Mol Cell Biol. 1998;18:3596–603.
Selvaraj A, Prywes R. Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J Biol Chem. 2003;278:41977–87.
Baillat D, Shiekhattar R. Functional dissection of the human TNRC6 (GW182-related) family of proteins. Mol Cell Biol. 2009;29:4144–55.
Tyssowski K, Kishi Y, Gotoh Y. Chromatin regulators of neural development. Neuroscience. 2014;264:4–16.
Nitarska J, Smith JG, Sherlock WT, Hillege MM, Nott A, Barshop WD, et al. A functional switch of NuRD chromatin remodeling complex subunits regulates mouse cortical development. Cell Rep. 2016;17:1683–98.
Wiegreffe C, Simon R, Peschkes K, Kling C, Strehle M, Cheng J, et al. Bcl11a (Ctip1) controls migration of cortical projection neurons through regulation of Sema3c. Neuron. 2015;87:311–25.
MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–76.
Acknowledgements
We are grateful to Sarah Graham for assistance with handling of DNA samples and to Heather Mabie for assistance with the phenotype materials. EE, AC-C, AV and SEF were supported by the Max Planck Society. LDS was supported by a National Institute on Deafness and Other Communication Disorders Grant (DC000496) and a core grant to the Waisman Center from the National Institute of Child Health and Development (Grant HD03352). ATM, MSH, IES, MB and SEF are supported by a National Health and Medical Research Council (NHMRC) Centre of Research Excellence in Speech and Language Neurobiology (ID: 1116976). ATM, MSH and SEF are supported by an NHMRC Project grant (1127144). ATM, MSH, IES, and MB are supported by the March of Dimes, United States. ATM was supported by an NHMRC Practitioner Fellowship (1105008). MB was supported by an NHMRC Program Grant (1054618) and NHMRC Senior Research Fellowship (1102971). IES is supported by an NHMRC Program Grant (1091593) and NHMRC Practitioner Fellowship (1006110). This work was supported by the Victorian Government’s Operational Infrastructure Support Program and Australian Government NHMRC Independent Research Institute Infrastructure Support Scheme (IRIISS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eising, E., Carrion-Castillo, A., Vino, A. et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatry 24, 1065–1078 (2019). https://doi.org/10.1038/s41380-018-0020-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-018-0020-x
This article is cited by
-
Structural rearrangements as a recurrent pathogenic mechanism for SETBP1 haploinsufficiency
Human Genomics (2024)
-
Genetic architecture of childhood speech disorder: a review
Molecular Psychiatry (2024)
-
The value of genomic testing in severe childhood speech disorders
European Journal of Human Genetics (2024)
-
Whole exome sequencing and polygenic assessment of a Swedish cohort with severe developmental language disorder
Human Genetics (2024)
-
Screening autism-associated environmental factors in differentiating human neural progenitors with fractional factorial design-based transcriptomics
Scientific Reports (2023)
