
Abstract
Autoantibodies of the IgG class against N-methyl-D-aspartate-receptor subunit-NR1 (NMDAR1-AB) were considered pathognomonic for anti-NMDAR encephalitis. This view has been challenged by the age-dependent seroprevalence (up to >20%) of functional NMDAR1-AB of all immunoglobulin classes found in >5000 individuals, healthy or affected by different diseases. These findings question a merely encephalitogenic role of NMDAR1-AB. Here, we show that NMDAR1-AB belong to the normal autoimmune repertoire of dogs, cats, rats, mice, baboons, and rhesus macaques, and are functional in the NMDAR1 internalization assay based on human IPSC-derived cortical neurons. The age dependence of seroprevalence is lost in nonhuman primates in captivity and in human migrants, raising the intriguing possibility that chronic life stress may be related to NMDAR1-AB formation, predominantly of the IgA class. Active immunization of ApoE−/− and ApoE+/+ mice against four peptides of the extracellular NMDAR1 domain or ovalbumin (control) leads to high circulating levels of specific AB. After 4 weeks, the endogenously formed NMDAR1-AB (IgG) induce psychosis-like symptoms upon MK-801 challenge in ApoE−/− mice, characterized by an open blood–brain barrier, but not in their ApoE+/+ littermates, which are indistinguishable from ovalbumin controls. Importantly, NMDAR1-AB do not induce any sign of inflammation in the brain. Immunohistochemical staining for microglial activation markers and T lymphocytes in the hippocampus yields comparable results in ApoE−/− and ApoE+/+ mice, irrespective of immunization against NMDAR1 or ovalbumin. These data suggest that NMDAR1-AB of the IgG class shape behavioral phenotypes upon access to the brain but do not cause brain inflammation on their own.
Similar content being viewed by others


Introduction
Autoantibodies (AB) of the immunoglobulin G (IgG) class against the N-methyl-D-aspartate-receptor subunit-NR1 (NMDAR1) were originally interpreted as pathognomonic for a condition called “anti-NMDAR encephalitis”, characterized by high serum and cerebrospinal fluid (CSF) titers of these AB, as well as a variably favorable response to immunosuppressive therapy. The reported syndrome, reflecting typical NMDAR1 antagonistic actions, consisted of psychosis, epileptic seizures, dyskinesia, cognitive decline, reduced consciousness, and autonomic dysregulation [1,2,3,4]. However, work on >5000 individuals, healthy or affected by different diseases, consistently revealed overall comparable age-dependent seroprevalence of functional NMDAR1-AB of all Ig classes, nurturing serious doubts regarding a purely pathological role of NMDAR1-AB of any Ig class [5,6,7,8,9,10].
NMDAR1-AB apparently belong to a pre-existing autoimmune repertoire [11,12,13,14,15,16,17], where Ig isotypes are determined by extracellular vs. intracellular antigen location [6]. This may explain the rarity of the IgG class among AB directed against extracellular epitopes, e.g., NMDAR1, MOG, and CASPR2. In contrast, AB that recognize intracellular antigens, e.g., amphiphysin, ARHGAP26, or GAD65, show predominance of IgG [6]. Despite this apparent “physiological autoimmunity”, no report exists that systematically screened mammals other than humans for the presence of NMDAR1-AB. In recent work, we found that all naturally occurring NMDAR1-AB are functional and thus have pathogenic potential irrespective of epitope and Ig class [10]. Pathophysiological significance may emerge in conditions of compromised blood–brain barrier (BBB), for instance, upon injury, infection, inflammation, or genetic predisposition (APOE4 haplotype), which then allows substantial access of circulating NMDAR1-AB to the brain where they act as NMDAR antagonists [5, 9, 18,19,20]. Alternatively, AB-specific plasma cells may reside or settle in the brain and produce large amounts of AB intrathecally [14, 21]. The question whether abundant endogenously produced NMDAR1-AB of the IgG class can—upon access to the brain—induce inflammation and thus “anti-NMDAR1 encephalitis” has never been experimentally addressed.
The present paper has therefore been designed to (i) systematically screen mammals other than humans for seroprevalence of functional NMDAR1-AB and (ii) study mice with open BBB behavioral and morphological consequences of high circulating levels of endogenous NMDAR1-AB of the IgG class formed in response to immunization.
Materials and methods
Ethical approvals
Ethics committees of Georg-August University, Göttingen, and collaborating centers approved the Göttingen Research Association for Schizophrenia (GRAS) data collection and other studies “extended GRAS” acquiring human data, serum samples, and IPSC [5, 6, 8, 9, 22, 23]. Hannover Medical School Ethics Committee approved the neurosurgical specimen collection. Studies comply with Helsinki Declaration. Patients gave written informed consent. Mouse studies were approved by Animal Ethics (LAVES, Oldenburg) following German Animal Protection Law.
Notes: All experiments were performed by researchers unaware of group assignment. The new nomenclature GluN1 for NMDAR1 is mostly disregarded here for consistency with the respective literature.
Human samples
GRAS and “extended GRAS”
The GRAS [22, 23] subsample used here consists of deep-phenotyped patients (N = 970; age 39.29 ± 0.40 years; 66.3% men), diagnosed with schizophrenia or schizoaffective disorder according to DSM-IV-TR [24]. Subjects of “extended GRAS” (N = 4933; age 43.29 ± 0.24 years; 56.9% men) comprise healthy individuals and patients with different neuropsychiatric diagnoses, including schizophrenia, affective disorders, multiple sclerosis, Parkinson, ALS, stroke, and personality disorders (detailed description in [5, 6, 8, 9]). For this study, subjects are dichotomously classified as nonmigrants or migrants comprising first (patient migrated) and second generation (parents migrated). Identified migrants (N = 301/N = 4933) are from Europe (49.8%), Asia (36.9%), Africa (9%), North America (2%), South America (0.7%), or mixed (1.6%).
Neurosurgical patients
A total of N = 72 paired samples of serum and ventricular CSF were available from patients (N = 45 women; age 55.9 ± 2.2 years; N = 27 men; age 60.2 ± 2.7 years) undergoing neurosurgery for various reasons: meningiomas, metastases, and other brain tumors (N = 25); intracerebral/subarachnoid hemorrhages (N = 20); hydrocephalus (N = 12); arterial aneurysms (N = 7); trigeminal neuralgia (N = 4); and others (N = 4). Most pairs were taken simultaneously at the time point of surgery, i.e., <5 min (N = 64) or <30 min (N = 8) apart.
Other mammals
Dogs and cats
Serum samples from dogs and cats of different breeds were prospectively collected during routine (health check/vaccination) or diagnostic (spectrum of different disorders) workup of outpatients in the Small Animal Clinic, University of Veterinary Medicine, Hannover.
Monkeys
Serum samples from healthy baboons and rhesus macaques were obtained through routine checkups at the Leibniz Institute for Primate Research, Göttingen.
Rodents
Serum samples from healthy rats and mice were obtained at the Max Planck Institute of Experimental Medicine and the Institute for Multiple Sclerosis Research, Göttingen.
Serological analyses
NMDAR1-AB determination by clinical standard procedures
Human serum and ventricular CSF were tested for NMDAR1-AB positivity using commercially available kits, based on HEK293T cells transfected with NMDAR1 and secondary AB against human IgG, IgM, or IgA (Euroimmun, Lübeck, Germany) [2, 25]. Mouse serum was analyzed using the same assay with secondary AB against mouse IgG, IgM, or IgA (M31001, A-31570, A-21042; Thermo Fisher, Rockford, USA).
NMDAR1-AB IgM screening in monkey samples
HEK293T cells (50,000) cultured at 37 °C/8% CO2 in DMEM (high glucose, Life Technologies, Carlsbad, USA) were seeded on a 35 -mm dish, grown for 3 days, and transfected with 3 µg of myc-His-tagged GluN1-1b cloned into pcDNA4/TO/myc-His A (Invitrogen, Carlsbad, USA) using Metafectene-Pro (Biontex, Munich, Germany) [10]. One day post transfection, cells were split onto five poly-D-lysine-coated coverslips in a 35 -mm dish and 1 day later, they were fixed with 5% paraformaldehyde (PFA) for 20 min, washed 5× (PBS), permeabilized with 0.1% Triton X-100 for 5 min, again washed 5× (PBS), and blocked with 5% normal goat serum (NGS; Sigma-Aldrich, Munich, Germany) for 1 h. After five PBS washes, cells were incubated with serum and monoclonal mouse anti-myc IgG (clone 9E10, Hollmann-Lab, Bochum) for 1 h, washed with 10× (PBS), incubated for 1 h with fluorescein-labeled goat anti-monkey IgM (072-11-031; KPL, Gaithersburg, USA) and AlexaFluor®594-labeled goat anti-mouse IgG (A11005; Thermo Fisher) secondary AB, and PBS washed 5×. Cells were mounted in Fluoromount-G (Southern Biotech, Birmingham, USA) and analyzed via TCS-SP2-AOBS confocal microscope (63× oil immersion objective; Leica-Microsystems, Wetzlar, Germany). The results were independently assessed by three investigators.
Protein-A assay
Human serum (for cross-validating clinical standard procedure and protein-A method), as well as dog, cat, rat, and monkey serum were labeled with protein-A from Staphylococcus aureus, binding the Fc portion of immunoglobulins of different species [26]. Plasma (50 μl) and 25 μg of FITC-conjugated protein-A (Sigma-Aldrich) were incubated for 2 h in the dark at room temperature (RT). The mixture was then diluted to 250 μl (PBS) and unbound FITC–Protein-A was removed using 100- kDa Amicon filter units (Sartorius, Göttingen, Germany), reconcentrating to ~50 μl [27]. NMDAR1-AB seropositivity was determined using Euroimmun assay combined with commercial monoclonal mouse NMDAR1-AB (114011; M68, SYSY, Göttingen, Germany). Samples showing distinct double labeling were rated “positive” (>98% consensus of three investigators).
Endocytosis assay
Functional studies were conducted with sera following ammonium-sulfate precipitation of immunoglobulins [28] and dialysis (Slide-A-Lyzer® Mini Dialysis Units, 10,000 MWCO Plus Float, Thermo Fisher). To assess AB functionality, human IPSC-derived neurons were exposed to dialyzed serum [10]. For each species, arbitrarily selected seronegative (N = 1) and seropositive samples (N = 2–3) were analyzed. Briefly, cells were precooled on ice and washed prior to incubation in cold HBSS with 1:50 diluted dialyzed sera, control NMDAR1-AB (M68-SYSY), or HBSS alone (negative control) for 30 min/4 oC. After washing to remove unbound AB, neurons were returned to their media and incubated for 20 min at 37 oC (three coverslips/sample, endocytosis) or 4 °C (one coverslip/sample, endocytosis control). The remaining surface NMDAR1 was exposed to mouse anti-human NMDAR1-AB (N-terminal; ab134308; Abcam, Cambridge, UK, 1:100), followed by labeling with secondary donkey anti-mouse IgG (A10036; Life Technologies, AlexaFluor®546, 1:100). Neurons were fixed with ice-cold 4% PFA and double stained with chicken anti-NeuN-AB (266006; SYSY, 1:500) and secondary donkey anti-chicken AB (703-546-155; Life Technologies, AlexaFluor®488, 1:250). Nuclei were visualized using DAPI (Sigma-Aldrich, 0.01 µg/ml). After PBS wash, coverslips were mounted on SuperFrost®-Plus slides with Mowiol mounting media (Sigma-Aldrich). Confocal laser-scanning microscopy was used to quantify NMDAR1 density at the membrane (63× glycerol objective; TCS-SP5 Leica-Microsystems, Mannheim, Germany). From each coverslip, Z series of optical sections (0.5 μm apart) covering the three-dimensional extension of neurons were acquired (sequential scanning mode, identical acquisition parameters). FIJI-ImageJ software [29] was used to randomly select NeuN+ cells and determine soma profile. Fluorescence intensity/cell surface area (AlexaFluor546) was automatically measured as readout of NMDAR1 surface expression. After background subtraction, the mean intensity for each coverslip was determined and fluorescence intensity ratio (37/4 °C) was calculated.
BBB-integrity testing
BBB integrity of 12-month-old ApoE−/− (N = 5) and ApoE+/+ (N = 5) mice was determined using two different fluorescent tracers, Evans blue (50 mg/g body weight) [30] and sodium fluorescein (200 mg/g body weight). A detailed description of this method will be published elsewhere [31]. Briefly, for tracer quantification in the brain at 4 h after intravenous injection in the tail vein, animals were PBS perfused to remove the circulating tracer. Brains were dissected, immediately frozen on dry ice, weighed, and stored at −80 °C. Tissue was lyophilized at −56 °C for 24 h under vacuum of 0.2 mBar (Christ LMC-1-BETA-1-16, Osterode, Germany). For tracer extraction, hemispheres were incubated with shaking in 10 ml formamide/mg brain at 57 °C for 24 h. Integrated density of tracer fluorescence was determined in triplicates on a fluorescent microscope (Observer Z2, Zeiss, Germany), equipped with AxioCamMRc3, 1×Camera-Adapter, and ZEN2012 blue-edition software, recorded at 10× magnification (Plan-Apochromat 10×/0.45M27). Tracer concentration was calculated using a standard curve and normalized to controls (set to 1).
Mouse immunization
Mice (12-month-old C57BL/6 littermates: ApoE−/−N = 20 and ApoE+/+N = 23; genders balanced) were immunized with a mixture of GluN1 extracellular peptides and/or chicken ovalbumin (Sigma-Aldrich), and emulsified in equal volume of complete Freund’s Adjuvant (Mycobacterium tuberculosis H37RA plus incomplete Freund’s Adjuvant; Becton-Dickinson, Sparks, USA) at a final concentration of 1 mg/ml [32]. At the tail base, 50 μg of GluN1 peptides and/or 20 μg of ovalbumin were injected subcutaneously (each side one).
Enzyme-linked immunosorbent assay (ELISA)
Orbital sinus blood of immunized mice was stored as EDTA plasma at −80 °C. ELISA plates (96 well) were coated with 0.5 μg of GluN1 peptide mixture or 0.2 μg of chicken ovalbumin in 50 μl PBS/well overnight at 4 °C and blocked with 5% BSA/PBS (Carl Roth, Karlsruhe, Germany). Mouse plasma (1:1000 or 1:50,000 5% BSA/PBS 50 μl/well) was added for 2 h at RT. The signal was amplified with horseradish peroxidase-linked anti-IgG (Sigma-Aldrich), and 3,3′,5,5′-Tetramethylbenzidine as colorimetric substrate (BD Biosciences, San Jose, USA). Absorbance at 450 nm was measured by microplate reader (Tecan-Trading AG, Männedorf, Switzerland).
Basic behavioral screening
The behavioral test battery was performed as described previously [33,34,35,36]. Starting at age 5 months, experimentally naïve ApoE−/− and ApoE+/+ littermates underwent (during light phase) tests of anxiety, activity and exploratory behavior (elevated plus-maze, open field, hole-board), motor (rotarod, grip strength) and sensory function (visual cliff, olfaction, hearing, hot plate), sensorimotor gating (prepulse inhibition), pheromone-based social preference, and cognitive performance (IntelliCage place/reversal learning). Males and females were tested separately.
Baseline and post MK-801 locomotion in the open field
The open-field apparatus consisted of a gray circular Perspex-arena (120 cm diameter; wall height 25 cm). Indirect white light illumination ensured constant light intensity of 120 lux in the center. Locomotion was measured using automated tracking software (Viewer2-Biobserve, Bonn, Germany). ApoE−/− and ApoE+/+ littermates received four baseline measurements preimmunization and post immunization (15 min each), the last followed by intraperitoneal MK-801 (Dizocilpine-[5S,10R]-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate; 0.3 μg/10 μl PBS/g Sigma-Aldrich). MK-801 is a noncompetitive NMDAR antagonist, acting as a use-dependent ion-channel blocker, and known to induce psychosis-like hyperactivity in the open field (loss of inhibition) [37]. Directly post injection, locomotor activity in open field was analyzed (4 min intervals), with the first 4 min defined as reference locomotion to express changes over 120 min as % reference.
Immunohistochemistry
Mice were anesthetized with Avertin (2,2,2-Tribromoethanol, Sigma-Aldrich), and transcardially perfused with 4% PFA/Ringer solution (Braun-Melsungen, Germany). Brains were removed, postfixed in 4% PFA overnight at 4 °C, and incubated in 30% sucrose/PBS for 2 days at 4 °C. Brains were cryosectioned coronally into 30 µm slices and stored in 25% ethylene glycol and 25% glycerol/PBS at −20 °C. Frozen sections (three/mouse; rostral hippocampus), mounted on SuperFrost®-Plus slides (Thermo Fisher, Waltham, USA), were used for cell quantification. For CD3 staining, sections were microwaved 3× for 4 min in citrate buffer (1 mM, pH 6) and blocked with 5% normal horse serum (NHS), and 0.5% Triton X-100/PBS for 1 h at RT. Incubation with rat anti-mouse CD3 (MCA1477; BioRad, Hercules, USA; 1:100) diluted in 5% NHS, and 0.5% Triton X-100/PBS was performed for two nights/4 °C, followed by incubation with goat anti-rat AlexaFluor®647 (A-21247; Thermo Fisher, Schwerte, Germany; 1:1000) diluted in 5% NHS, and 0.5% Triton X-100/PBS for 2 h at RT. For Iba1, GFAP, CD68, and MHC-II staining, sections were blocked with 5% NGS and/or 5% NHS in 0.5% Triton X-100/PBS for 1 h at RT. Incubation with rabbit anti-mouse Iba1 (019-19741; Wako-Chemicals GmbH, Neuss, Germany; 1:1000), or mouse anti-mouse GFAP (NCL-GFAP-GA5; Novocastra-Leica, Newcastle upon Tyne, UK; 1:500), diluted in 3% NGS or 3% NHS, and 0.5% Triton X-100/PBS, was performed overnight, and incubation with rat anti-mouse CD68 (MCA1957GA; BioRad GmbH, München, Germany, 1:400) and rat anti-mouse MHC-II (14-5321; eBioscience, San Diego, USA, 1:100) diluted in 3% NGS and 3% NHS, and 0.5% Triton X-100/PBS, was performed over two nights, all at 4 °C. Incubation with secondary antibodies was performed with goat anti-rabbit AlexaFluor®555 (A-21428; Thermo Fisher; 1:500) diluted in 3% NGS, 0.5% Triton X-100/PBS, or donkey anti-rabbit AlexaFluor®488 (A-21206; Thermo Fisher, 1:500) or donkey anti-mouse AlexaFluor488 (A21202; Thermo Fisher, 1:500) or goat anti-rat AlexaFluor®647 (A-21247; Thermo Fisher, 1:500), diluted in 3% NGS or 3% NHS, and 0.5% Triton X-100/PBS for 1.5 h at RT. Nuclei were counterstained with DAPI (Sigma-Aldrich, 0.01 µg/ml) and sections were mounted using Aqua-Poly/Mount (Polysciences, Warrington, USA). Tile scans of hippocampus were acquired using Leica-DMI6000 epifluorescence microscope (20× objective; Leica) and Iba1+ and CD3+ cells were counted using cell counter plug-in of FIJI-ImageJ software [29]. GFAP+ cells in the hippocampus were quantified densitometrically upon uniform thresholding (expressed as % respective area).
Statistical analyses
Statistical analyses were performed using SPSSv.17 (IBM-Deutschland-GmbH, Munich, Germany) or Prism4 (GraphPad Software, San Diego, California, USA). Group differences in categorical and continuous variables were assessed using χ2, Mann–Whitney U, or Student's t-tests depending on data distribution/variance homogeneity. ANOVA was employed as indicated in display item legends. All p-values are two tailed; significance is set to p < 0.05; data are presented as mean ± S.E.M.
Results
Cross-validation of NMDAR1-AB detection methods
To determine NMDAR1-AB seropositivity in mammals other than humans, we had to validate the protein-A detection method [27]. For that, N = 72 paired human serum and ventricular CSF samples, prospectively collected from random neurosurgical patients, were analyzed by the usual cell-based assay, employing specific secondary AB for all Ig classes. A total of N = 5 sera turned out NMDAR1-AB positive (titers ≤ 1:100; 3× IgM; 2× IgA; 0× IgG). Ventricular CSF samples were all negative. For cross-validation of NMDAR1-AB of the IgG class, we used serum of a seropositive stroke patient [8]. Application of protein-A method combined with double labeling for NMDAR1-AB M68 confirmed positive and negative results (Fig. 1a).

NMDAR1-AB seropositivity and functionality across mammalian species. a Cross-validation of assays: paired serum and intraventricular CSF samples from neurosurgical patients were tested using a HEK293T cell-based clinical standard assay for NMDAR1-AB seropositivity (Euroimmun biochip). For step 1, fluorescently labeled IgA-specific, IgM-specific, and IgG-specific secondary AB were used; for method cross-validation (step 2), NMDAR1-AB seropositive and seronegative samples of each Ig class from step 1 were labeled with protein-A–FITC conjugate and tested for colocalization (yellow) of protein-A–FITC+ (green) and M68+ (monoclonal mouse NMDAR1-AB followed by Alexa555 donkey anti-mouse IgG red). Representative pictures of both methods using the same seropositive samples (IgA, IgM, and IgG) are displayed on the right: upper row step 1/lower row step 2. b NMDAR-AB seropositivity (%) of young and old mammals for all Ig classes combined (#protein-A–FITC/Euroimmun) or for individual classes (+Euroimmun; *protein-A–FITC/Euroimmun and cross-validation with Euroimmun/monkey IgM) presented in the bars; color codes used for consistency and kept also in c and d; age given in months (m) or years (y); χ2 or Fisher’s exact test. c NMDAR-AB seropositivity of subjects with migration (first and second generation) vs. nonmigration history (GRAS data collection); all Ig classes presented; age split at 35 years; χ2 test. d NMDAR1-AB course by Ig classes in serum over age groups in migrants vs. nonmigrants of the extended GRAS data collection. Note the different course particularly for IgA. eFunctionality testing of NMDAR1-AB in human IPSC-derived cortical neurons: degree of internalization expressed as a ratio of fluorescence intensity measured at 37 and 4 °C; number of neurons and sera (N) given; Mann–Whitney U test
High seroprevalence of NMDAR1-AB across mammalian species
We next analyzed by protein-A method serum samples of dogs, cats, rats, baboons, and rhesus macaques. Strikingly, all mammalian species, independent of their respective life expectancy, show high NMDAR1-AB seropositivity (Fig. 1b). Mouse samples were analyzed using specific AB against murine IgA, IgM, and IgG. As known for humans [6], NMDAR1-AB of the IgG class were the rarest. For another cross-validation, all monkey samples (N = 100) were analyzed in blinded fashion by an independent lab (Bochum; using specific anti-monkey IgM). IgM-positive results coincided with the protein-A positivity by >97% (76 of 78). The fraction of protein-A positive but IgM-negative monkey samples (total 22%) likely presents NMDAR1-AB of IgA class and IgG class where specific AB were not available.
Age-dependent NMDAR1-AB seroprevalence except for nonhuman primates and human migrants
All species revealed age dependence of NMDAR1-AB seroprevalence (χ2 test; dogs: χ2(1) = 11.5, p = 0.01; cats: χ2(1) = 4.8, p = 0.03; rats: χ2(1) = 9.5, p = 0.002; and mice: Fisher’s exact test p = 0.032) as for humans [5, 8] with the exception of baboons (χ2(1) = 1.0, p = 0.3), where already >50% of young animals were seropositive. This surprising result made us investigate another monkey species, rhesus macaques, showing again high seroprevalence in old and young animals (χ2(1) = 0.2, p = 0.6) (Fig. 1b). We wondered what the difference between humans, dogs, cats, mice, and rats, on one hand, and monkeys, on the other hand, could be, leading to loss of the usual age pattern regarding seroprevalence. Postulating that captivity/nondomestication of young monkeys might induce chronic life stress due to maladaptation to the environment, we investigated in a hypothesis-driven way whether young human migrants would display a similar increase in NMDAR1-AB seropositivity. Of the GRAS data collection, detailed information on migration was available in a subsample of N = 970 individuals. While nonmigrants show the typical age association of NMDAR1-AB seroprevalence (χ2(1) = 10.7, p = 0.001), migrants do not (χ2(1) = 0.6, p = 0.4) (Fig. 1c). Seroprevalence in young migrants is significantly higher as compared to young nonmigrants (χ2(1) = 5.381, p = 0.020). In both monkey species and migrants, the IgM fraction still follows the expected age trend, while IgA seems to account for the early increase in NMDAR1-AB seroprevalence (Fig. 1c). Presentation of NMDAR1-AB by Ig class in the extended GRAS sample (N = 4933), with N = 4632 of likely nonmigrants (available information less detailed) and N = 301 known migrants, illustrates the abnormal course of IgA vs. IgM/IgG seroprevalence over age in migrants (Fig. 1d).
Functionality of NMDAR1-AB from different mammalian species
To assess whether NMDAR1-AB of the tested species are functional, our endocytosis assay using IPSC-derived human cortical neurons [10] was employed. All positive sera provoked NMDAR1 internalization, verifying functionality (Mann–Whitney U; all p < 0.001) (Fig. 1e).
BBB dysfunction but normal behavior of ApoE −/− mice
We next induced endogenous NMDAR1-AB formation in a mouse model of BBB dysfunction, ApoE−/− mice vs. WT littermates, ApoE+/+. Before that, we confirmed in 12-month-old mice (age of immunization) BBB leakiness using two fluorescent tracers. While brain water content was similar in both genotypes, pointing against inflammation, ApoE−/− mice showed increased tracer extravasation, confirming BBB dysfunction (Student’s t-test: EB: t(8) = −10.66, p < 0.001; NaFl: t(8) = −8.97, p < 0.001) (Fig. 2a). We wondered whether this compromised BBB would by itself lead to behavioral abnormalities in ApoE−/− mice. A comprehensive behavioral battery, including tests for anxiety, activity, exploratory behavior, motor and sensory function, sensorimotor gating, pheromone-based social preference, and cognitive performance did not reveal any differences between genotypes (Table 1).

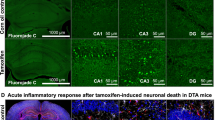
Behavioral and morphological effects of endogenous NMDAR1-AB of the IgG class in a mouse model with open BBB. a Demonstration of BBB leakiness in ApoE−/− mice using an intravenously injected mixture of Evans blue (EB) and sodium fluorescein (NaFl): After brain cryopreservation/lyophilization, tracers were extracted with formamide and quantified; Student’s t-test; b Experimental outline; c Immunization: Left: GluN1 peptides (P1–P4) located in the extracellular part of the receptor were used for immunization (compare Fig. 3); middle and right: Time course of anti-ovalbumin and anti-GluN1-AB (IgG) upon immunization in ApoE−/− and ApoE+/+ mice; optical density at dilution 1:1000 shown; titers after day 10 reach up to 1:50,000; d Effect of MK-801 injection on activity in the open field; results presented as % change from baseline (first 4 min post MK-801 set to 100%); no difference in MK-801-induced hyperactivity between genotypes after ovalbumin immunization (one-way repeated measures ANOVA: treatment × group interaction: F(1,17) = 0.2; p = 0.7); increase in hyperactivity (during rise, plateau, decline, and after-effect phases) upon MK-801 in ApoE−/− but not ApoE+/+ mice immunized against GluN1 (one-way repeated measures ANOVA: treatment × group interaction: F(1,22) = 5.6; p = 0.028). e Quantification of Iba1+ and CD3+ cells in the hippocampus to assess inflammation in the brain; one-way ANOVA; representative pictures of Iba1 (left) and CD3 (right) stainings in the middle
Immunization of ApoE −/− and ApoE +/+ mice against NMDAR1-peptides
To explore whether endogenously formed NMDAR1-AB would lead to measurable behavioral and morphological effects, we immunized 12-month-old ApoE−/− and ApoE+/+ littermates against four peptides of the extracellular NMDAR1/GluN1-domain (including NTD-G7; N368/G369) and ovalbumin or against ovalbumin alone as immunization control (Fig. 2b–c). GluN1 shows >99% sequence homology among all here-tested mammalian species, with immunizing peptides being 100% homologous (Fig. 3). Immunization led to high circulating levels of specific IgG (titers up to 1:50,000). Efficacy of immunization and time course of IgG appearance as determined by ELISA were comparable for NMDAR1-peptides and ovalbumin across genotypes, making a simple boosting effect of NMDAR1-peptides on pre-existing NMDAR1-specific B cell clones rather improbable (Fig. 2c).

Alignment of GluN1-1b receptor amino acid sequence across all mammalian species tested. Regions containing the four peptide sequences (peptides 1–4: P1: AA35–53, P2: AA361–376, P3: AA385–399, and P4: AA660–811) used in the immunization experiment are highlighted in yellow and light brown (compare three-dimensional presentation in Fig. 2c) and nonhomologous amino acids in pink. SP signal peptide, S1, S2 segments of the ligand-binding domain, TMD A transmembrane domain A, TMD B transmembrane domain B, TMD C transmembrane domain C
Psychosis-related behavior of ApoE −/− mice upon MK-801 challenge
Open-field tests measuring baseline preimmunization and postimmunization locomotion did not reveal any differences between genotypes and/or immunization groups (Fig. 2b; not shown). After 4 weeks, the endogenously formed NMDAR1-AB of the IgG class induced strong hyperactivity (psychosis-like symptoms [37]) upon MK-801 challenge in ApoE−/− mice only. In contrast, ApoE+/+ mice behaved comparably to ovalbumin-only immunized mice of both genotypes (Fig. 2d; all p > 0.5). Thus, an open BBB together with sufficiently high titers of AB (to reach a threshold loss of NMDAR1 surface expression) is a prerequisite for the observed behavioral perturbation upon MK-801.
No inflammation in hippocampus of immunized ApoE −/− and ApoE +/+ mice
Immunohistochemistry did not show any evidence of inflammation in either genotype and/or immunization group. Numbers of Iba1+ and CD3+ cells as markers of microglia and T cells, respectively, were comparable for total hippocampus (one-way ANOVA: Iba1: F(3,18) = 0.3; p = 0.8; CD3: F(3,18) = 0.4; p = 0.8) (Fig. 2e) and for all hippocampal subfields separately (all p-values > 0.2; not shown). Also, staining for microglial activity markers, CD68 and MHCII, was essentially negative and identical across groups. Moreover, staining for GFAP did not reveal any appreciable density increase in the hippocampus, and thus no sign of astrogliosis (data not shown).
Discussion
The present work demonstrates high seroprevalence of functional NMDAR1-AB of all Ig classes across mammals, indicating that these AB are part of a pre-existing autoimmune repertoire [11,12,13,14,15,16,17]. As in humans, NMDAR1-AB of the IgG class are the least frequent [6, 20]. The age related up to >50% NMDAR1-AB seropositivity is independent of the respective species’ life expectancy, indicating that the aging process itself rather than years of exposure to a certain environment triggers NMDAR1-AB formation. However, our knowledge on predisposing factors and inducing mechanisms is limited. Specific autoimmune-reactive B cells may get repeatedly boosted by, e.g., infections, neoplasms, or the microbiome, and less efficiently suppressed over a lifespan likely owing to a gradual loss of immune tolerance upon aging [14].
Unexpectedly, we find the age-dependence lost in nonhuman primates and in human migrants that all display an early-life rise in NMDAR1-AB seropositivity, mainly of IgA. The intriguing possibility that chronic life stress, known to be present in human migrants [38] and animals in captivity [39], acts as a trigger of early NMDAR1-AB formation is worth pursuing experimentally in the future. A large proportion of migrants in our human samples are suffering from neuropsychiatric illness. This may additionally support our chronic stress hypothesis since migration is recognized as an environmental stressor predisposing to mental disease [40]. Further studies should screen wild-life monkeys and species in zoos for NMDAR1-AB. Experimental confirmation of our findings provided, NMDAR1-AB (IgA) may even serve as stress markers. In fact, earlier reports show that total serum-Ig of all classes, most prominently IgA, respond to psychological stress [41]. NMDAR1-AB might thus belong to a set of stress-boosted AB. Interestingly, we also find accumulated seroprevalence of 23 other brain-directed AB [6] in young migrants vs. nonmigrants increased (data not shown), suggesting a global inducer role of chronic stress in humoral autoimmunity.
Earlier work has shown that AB against brain antigens in general are common among mammals [42], but no study has so far systematically screened nonhuman mammals for NMDAR1-AB. As an exception, a recent report described “anti-NMDAR1 encephalitis” in the young polar bear Knut [27]. Based on the present findings, Knut may have belonged to those nondomesticated species in captivity—comparable to monkey species investigated here—that are affected by chronic early-life stress, inducing NMDAR1-AB seropositivity. Pre-existing NMDAR1-AB of this bear may have ultimately shaped the clinical picture of an encephalitis of unexplained origin (likely infectious according to the zoo’s pathology reports) where an epileptic seizure led to drowning [27].
This interpretation is supported by our novel autoimmune model, namely, mice immunized against NMDAR1-peptides. Even high titers of endogenously formed NMDAR1-AB (IgG; up to 1:50,000) that induce psychosis-like behavior upon MK-801 challenge in ApoE−/− mice, with here-confirmed open BBB, do not lead to any appreciable signs of encephalitis. This dissociation of behavioral/symptomatic consequences and inflammation in the brain is of major importance for clinicians [14]. For instance, earlier studies reported an influence of NMDAR1-AB infusions into the hippocampus on learning and memory in mice [43], and others found increased NMDAR1-AB seroprevalence in patients with mild cognitive impairment and Alzheimer's disease [44, 45]. However, while all naturally occurring NMDAR1-AB that have pathogenic potential irrespective of epitope and Ig class [10], and upon entry to the brain (or via intrathecal production) can shape brain functions in the sense of NMDAR antagonism, only a fraction of individuals happens to have underlying encephalitis of various etiologies, which is then called anti-NMDAR encephalitis. The highly variable neuropathology and response to immunosuppression of this condition [2, 3, 46] may point to a broad range of possible encephalitogenic mechanisms (from infection to oncology or genetics) which need to be diagnosed and specifically treated [14].
Even though it is unclear how NMDAR1-AB are generated by chronic stress, it should be considered that NMDAR1 are not only expressed in the brain but also by peripheral organs and tissues, including adrenal glands and gut [47] which may be involved in triggering NMDAR1-AB formation but may also be functionally modulated by them. Since NMDAR antagonists are increasingly recognized as antidepressant, anxiolytic, and anti-inflammatory agents [48,49,50,51,52], we speculate that stress-induced NMDAR1-AB could serve as endogenous stress protectants. Remarkably, also in stroke, NMDAR1-AB can be protective [8].
In conclusion, the widespread occurrence of NMDAR1-AB across mammals, as well as the failure of even high titers of endogenously formed NMDAR1-AB of the IgG class to induce any signs of brain inflammation should lead to rethinking current concepts that link NMDAR1-AB to neuropsychiatric disease including encephalitis.
References
Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36.
Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–8.
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10:63–74.
Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–65.
Hammer C, Stepniak B, Schneider A, Papiol S, Tantra M, Begemann M, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry. 2014;19:1143–9.
Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, Saschenbrecker S, et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. 2014;76:82–94.
Steiner J, Teegen B, Schiltz K, Bernstein HG, Stoecker W, Bogerts B. Prevalence of N-methyl-D-aspartate receptor autoantibodies in the peripheral blood: healthy control samples revisited. JAMA Psychiatry. 2014;71:838–9.
Zerche M, Weissenborn K, Ott C, Dere E, Asif AR, Worthmann H, et al. Preexisting serum autoantibodies against the NMDAR subunit NR1 modulate evolution of lesion size in acute ischemic stroke. Stroke. 2015;46:1180–6.
Castillo-Gomez E, Kastner A, Steiner J, Schneider A, Hettling B, Poggi G, et al. The brain as immunoprecipitator of serum autoantibodies against N-Methyl-D-aspartate receptor subunit NR1. Ann Neurol. 2016;79:144–51.
Castillo-Gomez E, Oliveira B, Tapken D, Bertrand S, Klein-Schmidt C, Pan H et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol Psychiatry. 2016;22:1776–178.
Abramson J, Husebye ES. Autoimmune regulator and self-tolerance—molecular and clinical aspects. Immunol Rev. 2016;271:127–40.
Cohen I, Young D. Autoimmunity, microbial immunity and the immunological homunculus. Immunol Today. 1991;12:105–10.
Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812–8.
Ehrenreich H. Autoantibodies against the N-methyl-d-aspartate receptor subunit NR1: untangling apparent inconsistencies for clinical practice. Front Immunol. 2017;8:181.
Lobo PI. Role of natural autoantibodies and natural IgM anti-leucocyte autoantibodies in health and disease. Front Immunol. 2016;7:198.
Mader S, Brimberg L, Diamond B. The role of brain-reactive autoantibodies in brain pathology and cognitive impairment. Front Immunol. 2017;8:1101.
Nguyen TT, Baumgarth N. Natural IgM and the development of b cell-mediated autoimmune diseases. Crit Rev Immunol. 2016;36:163–77.
Coutinho E, Harrison P, Vincent A. Do neuronal autoantibodies cause psychosis? A neuroimmunological perspective. Biol Psychiatry. 2014;75:269–75.
Diamond B, Huerta P, Mina-Osorio P, Kowal C, Volpe B. Losing your nerves? Maybe it’s the antibodies. Nat Rev Immunol. 2009;9:449–56.
Hammer C, Zerche M, Schneider A, Begemann M, Nave KA, Ehrenreich H. Apolipoprotein E4 carrier status plus circulating anti-NMDAR1 autoantibodies: association with schizoaffective disorder. Mol Psychiatry. 2014;19:1054–6.
Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain. 2016;139:2641–52.
Begemann MGS, Papiol S, Malzahn D, Krampe H, Ribbe K, et al. Modification of cognitive performance in schizophrenia by complexin 2 gene polymorphisms. Arch Gen Psychiatry. 2010;67:879–88.
Ribbe. K, Friedrichs H, Begemann M, Grube S, Papiol S, Kästner. A, et al. The cross-sectional GRAS sample: a comprehensive phenotypical data collection of schizophrenic patients. BMC Psychiatry. 2010;10:0–20.
American Psychiatric Association. (2000) Diagnostic and statistical manual of mental disorders. 4th edn American Psychiatric Press, Washington, DC. .
Wandinger KP, Saschenbrecker S, Stoecker W, Dalmau J. Anti-NMDA-receptor encephalitis: a severe, multistage, treafa disorder presenting with psychosis. J Neuroimmunol. 2011;231:86–91.
Boyle MDP, Reis KJ. Bacterial Fc receptors. Nat Biotechnol. 1987;5:697–703.
Pruss H, Leubner J, Wenke NK, Czirjak GA, Szentiks CA, Greenwood AD. Anti-NMDA receptor encephalitis in the polar bear (Ursus maritimus) Knut. Sci Rep. 2015;5:1–7.
Toyka KBD, Pestronk A, Kao I. Myasthenia gravis: passive transfer from man to mouse. Science. 1975;190:397–9.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Berghoff SA, Gerndt N, Winchenbach J, Stumpf SK, Hosang L, Odoardi F, et al. Dietary cholesterol promotes repair of demyelinated lesions in the adult brain. Nat Commun. 2017;8:1–15.
Berghoff SA, Düking T, Spieth L, Winchenbach J, Stumpf SK, Gerndt N, et al. Blood-brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol Commun. 2017;5:94.
Wust S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol. 2008;180:8434–43.
Dere E, Dahm L, Lu D, Hammerschmidt K, Ju A, Tantra M, et al. Heterozygous ambra1 deficiency in mice: a genetic trait with autism-like behavior restricted to the female gender. Front Behav Neurosci. 2014;8:181.
Dere E, Winkler D, Ritter C, Ronnenberg A, Poggi G, Patzig J, et al. Gpm6b deficiency impairs sensorimotor gating and modulates the behavioral response to a 5-HT2A/C receptor agonist. Behav Brain Res. 2015;277:254–63.
Netrakanti PR, Cooper BH, Dere E, Poggi G, Winkler D, Brose N, et al. Fast cerebellar reflex circuitry requires synaptic vesicle priming by munc13-3. Cerebellum. 2015;14:264–83.
Winkler D, Daher F, Wustefeld L, Hammerschmidt K, Poggi G, Seelbach A et al. Hypersocial behavior and biological redundancy in mice with reduced expression of PSD95 or PSD93. Behav Brain Res. 2017. https://doi.org/10.1016/j.bbr.2017.02.011 (e-pub ahead of print).
Radyushkin K, El-Kordi A, Boretius S, Castaneda S, Ronnenberg A, Reim K, et al. Complexin2 null mutation requires a ‘second hit’ for induction of phenotypic changes relevant to schizophrenia. Genes Brain Behav. 2010;9:592–602.
Akdeniz C, Tost H, Streit F, Haddad L, Wust S, Schafer A, et al. Neuroimaging evidence for a role of neural social stress processing in ethnic minority-associated environmental risk. JAMA Psychiatry. 2014;71:672–80.
Jacobson SL, Ross SR, Bloomsmith MA. Characterizing abnormal behavior in a large population of zoo-housed chimpanzees: prevalence and potential influencing factors. PeerJ. 2016;4:e2225.
Stepniak B, Papiol S, Hammer C, Ramin A, Everts S, Hennig L, et al. Accumulated environmental risk determining age at schizophrenia onset: a deep phenotyping-based study. Lancet Psychiatry. 2014;1:444–53.
Maes M, Hendriks D, Gastel AV, Demedts P, Wauters A, Neels H, et al. Effects of psychological stress on serum immunoglobulin, complement and acute phase protein concentrationsin normal volunteers. Psychoneuroendocrinology. 1997;2:397–409.
Nagele EP, Han M, Acharya NK, DeMarshall C, Kosciuk MC, Nagele RG. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS ONE. 2013;8:4.
Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain. 2015;138:94–109.
Busse S, Brix B, Kunschmann R, Bogerts B, Stoecker W, Busse M. N-methyl-d-aspartate glutamate receptor (NMDA-R) antibodies in mild cognitive impairment and dementias. Neurosci Res. 2014;85:58–64.
Doss S, Wandinger KP, Hyman BT, Panzer JA, Synofzik M, Dickerson B, et al. High prevalence of NMDA receptor IgA/IgM antibodies in different dementia types. Ann Clin Transl Neurol. 2014;1:822–32.
Tuzun E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol. 2009;118:737–43.
Du J, Li XH, Li YJ. Glutamate in peripheral organs: biology and pharmacology. Eur J Pharmacol. 2016;784:42–8.
Gerhard DM, Wohleb ES, Duman RS. Emerging treatment mechanisms for depression: focus on glutamate and synaptic plasticity. Drug Discov Today. 2016;21:454–64.
Abdallah CG, Adams TG, Kelmendi B, Esterlis I, Sanacora G, Krystal JH. Ketamine’s mechanism of action: a path to rapid-acting antidepressants. Depress Anxiety. 2016;33:689–97.
Alexander JK, DeVries AC, Kigerl KA, Dahlman JM, Popovich PG. Stress exacerbates neuropathic pain via glucocorticoid and NMDA receptor activation. Brain Behav Immun. 2009;23:851–60.
Rubio-Casillas A, Fernandez-Guasti A. The dose makes the poison: from glutamate-mediated neurogenesis to neuronal atrophy and depression. Rev Neurosci. 2016;27:599–622.
Nair A, Hunzeker J, Bonneau RH. Modulation of microglia and CD8(+) T cell activation during the development of stress-induced herpes simplex virus type-1 encephalitis. Brain Behav Immun. 2007;21:791–806.
Acknowledgements
This work was supported by the Max Planck Society, the Max Planck Förderstiftung, the DFG (CNMPB), EXTRABRAIN EU-FP7, and the Niedersachsen-Research Network on Neuroinfectiology (N-RENNT). The authors thank all subjects for participating in the study, and the many colleagues who have contributed over the past decade to the extended GRAS data collection.
Author contributions
Concept, design, and supervision of the study: HE; Data acquisition/analysis/interpretation: HP, BO, ED, DT, MM, JS, JW, DW, CKS, AR, KS, RT, KMR, StB, YAK, HM, MB, WS, GS, FJK, RM, SB, KAN, JKK, MH, FL, and HE; Drafting manuscript: HE, with the help of BO and HP; Drafting display items: HE and BO, with the help of HP, MM, DT, and JS. All authors read and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
WS is a member of the board and holds stocks in Euroimmun AG. HM is a full-time employee of Synaptic Systems GmbH. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hong Pan, Bárbara Oliveira, and Gesine Saher contributed equally to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Pan, H., Oliveira, B., Saher, G. et al. Uncoupling the widespread occurrence of anti-NMDAR1 autoantibodies from neuropsychiatric disease in a novel autoimmune model. Mol Psychiatry 24, 1489–1501 (2019). https://doi.org/10.1038/s41380-017-0011-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-017-0011-3
This article is cited by
-
CPLX2 is a novel tumor suppressor and improves the prognosis in glioma
Journal of Neuro-Oncology (2024)
-
Movement disorders in autoimmune encephalitis: an update
Journal of Neurology (2023)
-
NMDAR1 autoantibodies amplify behavioral phenotypes of genetic white matter inflammation: a mild encephalitis model with neuropsychiatric relevance
Molecular Psychiatry (2022)
-
Anti-NMDAR encephalitis induced in mice by active immunization with a peptide from the amino-terminal domain of the GluN1 subunit
Journal of Neuroinflammation (2021)
-
Inducing sterile pyramidal neuronal death in mice to model distinct aspects of gray matter encephalitis
Acta Neuropathologica Communications (2021)
