
Abstract
To achieve minimum DNA input requirements for next-generation sequencing (NGS), pathologists visually estimate macrodissection and slide count decisions. Unfortunately, misestimation may cause tissue waste and increased laboratory costs. We developed an artificial intelligence (AI)-augmented smart pathology review system (SmartPath) to empower pathologists with quantitative metrics for accurately determining tissue extraction parameters. SmartPath uses two deep learning architectures, a U-Net based network for cell segmentation and a multi-field-of-view convolutional network for tumor area segmentation, to extract features from digitized H&E-stained formalin-fixed paraffin-embedded slides. From the segmented tumor area, SmartPath suggests a macrodissection area. To predict DNA yield per slide, the extracted features from within the macrodissection area are correlated with known DNA yields to fit a regularized linear model (R = 0.85). Then, a pathologist-defined target yield divided by the predicted DNA yield per slide gives the number of slides to scrape. Following model development, an internal validation trial was conducted within the Tempus Labs molecular sequencing laboratory. We evaluated our system on 501 clinical colorectal cancer slides, where half received SmartPath-augmented review and half traditional pathologist review. The SmartPath cohort had 25% more DNA yields within a desired target range of 100–2000 ng. The number of extraction attempts was statistically unchanged between cohorts. The SmartPath system recommended fewer slides to scrape for large tissue sections, saving tissue in these cases. Conversely, SmartPath recommended more slides to scrape for samples with scant tissue sections, especially those with degraded DNA, helping prevent costly re-extraction due to insufficient extraction yield. A statistical analysis was performed to measure the impact of covariates on the results, offering insights on how to improve future applications of SmartPath. With these improvements, AI-augmented histopathologic review has the potential to decrease tissue waste, sequencing time, and laboratory costs by optimizing DNA yields, especially for samples with scant tissue and/or degraded DNA.
Similar content being viewed by others


Introduction
Next-generation sequencing (NGS) has become an integral technique in the molecular diagnosis, prognosis, and treatment of cancer. To properly assess tumor tissue with NGS, solid samples must be dissected to meet minimum DNA input and tumor purity requirements1,2,3,4,5. In standard practice, pathologists visually inspect hematoxylin and eosin (H&E)-stained, formalin-fixed, paraffin-embedded (FFPE) slides to determine how much tissue should be dissected and whether macrodissection is necessary to enrich for tumor cells6,7. Besides meeting minimum input requirements, pathologists must also avoid recommending excessive dissection as tumor tissue is valuable and may be needed for further molecular tests. Tissue stewardship guidelines can help pathologists achieve this balance between sufficient and excessive dissection4. However, following these suggestions using manual dissection techniques is difficult, and thus, there is an increasing need to optimize tissue extraction procedures as NGS becomes more relevant in clinical practice.
NGS pipelines have undergone tremendous advancements in the past decade8,9,10, including the development of automated dissection systems for tissue extraction. Laser-capture microdissection was introduced about two decades ago11,12,13, but has not been widely adopted in clinical laboratories because precise dissection of single tumor cells from FFPE slides is rarely necessary for clinical testing14. Lower resolution mechanical macrodissection systems have also been developed as more clinically pragmatic alternatives15,16,17,18. These systems can be combined with digital slide marking (digitally guided macrodissection), enabling integration with computer vision models for tumor enrichment19,20. Several computer vision systems have been recently developed with the goal of estimating tumor-rich dissection areas from histopathology slides to meet tumor purity input requirements for molecular testing6,21,22,23,24. However, no recommendation systems exist for estimating tissue quantity for minimum DNA input requirements, and thus even automated dissection systems rely on a pathologist to determine how many slides should be scraped. Unfortunately, consequences of visual misestimation of tissue quantity include sequencing failure, tissue waste, and increased laboratory costs and turnaround times.
Here, we developed SmartPath: a computer vision-based method to empower pathologists with quantitative metrics, allowing them to accurately determine tissue input parameters for desired DNA yields. Echoing design principles of artificial intelligence (AI)-augmented pathology outlined by others25,26,27,28,29, SmartPath functions as a pathologist-in-the-loop system rather than a standalone predictor. Predictions are displayed in a browser-based user interface (UI) viewed during pathology review. The pathologist is free to recommend the predicted tissue input parameters as presented, or to modify them based on their expertise. We tested SmartPath in an internal trial to assess the impact of AI-augmented pathology review in a real-world clinical setting. We quantified immediate impacts of the AI-assistance on tissue usage and the extracted DNA content, as well as on two NGS workflow costs: the total number of extractions attempted and the DNA extraction-to-sequencing time (T-seq). A thorough statistical investigation of the impact of clinical covariates on these metrics was conducted, revealing factors that influence NGS success beyond tissue input parameters alone. Finally, recommendations are made how to improve the system for future applications.
Materials and methods
Model development
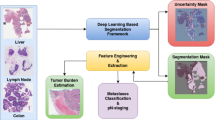
Before evaluating the SmartPath AI-augmented pathology review system in an internal trial (see Methods: Internal Model Evaluation Trial), we first developed the model through extensive validation experiments. The underlying models can be grouped into two categories: feature generation and DNA yield estimation. Feature generation aims to extract features from a single H&E-stained histopathology whole-slide image (WSI). These features are used to fit a DNA yield estimation model. At inference time, the full pipeline from feature generation to DNA yield estimation is run to produce predictions that augment pathologist tissue quantity selections to achieve a total extracted DNA mass within a user-defined target range. This modeling pipeline is summarized in Fig. 1A.

A SmartPath consists of several models which extract information from H&E-stained whole-slide images and make predictions used to augment pathologist decisions prior to DNA extraction. These models can be grouped into “Feature Generation” (blue boxes) and “DNA Yield Estimation” (green boxes). Arrows point in the direction of data flow between models. The feature generation pipeline receives inputs from pre-trained cell segmentation (U-Net) and tumor segmentation (multi-field-of view ResNet-18) models to generate features used for a DNA Yield Estimator. For samples that are macrodissected, these features are only computed from the macrodissection area by masking the slides, either by a U-Net-based ink detection (when run on archival slides for training) or by post-processing the tumor segmentation model output (when running on new slides during inference). The output of the feature generation pipeline is fed to a regularized linear model to predict the expected DNA yield per slide. During the trial, the predicted DNA yield per slide is used to estimate the total number of slides needed to achieve the following target yields: 100, 400, 1000 ng. These predictions, along with the predicted microdissection area, are output to a UI presented to the pathologist during review. B Samples either receives Traditional (“Trad”) or AI-augmented pathology review (“SmartPath”). In Trad review a pathologist reviewed a slide under a microscope and estimated the number of slides needed for DNA extraction. Slide scanning was not incorporated into the Trad workflow. In SmartPath review, slide scanning was incorporated into the workflow immediately after tissue was sliced. Slide scanning triggered an upload of the image to the cloud, where it was automatically processed by the SmartPath pipeline described in A. At the end of the Trad or Smart review process the pathologist made a review decision to recommend the number of slides scraped as well as the macrodissection area (if needed) for DNA mass extraction. Routine NGS proceeded after extraction, including library prep and DNA hybridization steps. If NGS failed at any step along the way the sample was re-extracted. Four metrics, marked by circled numbers in the diagram, were tracked to measure the impact of AI-assistance: 1) the number of slides scraped for extraction, 2) the extracted DNA mass, 3) the number of extraction attempts (referred to as “extraction count”), 4) the time elapsed from 1st extraction to 1st successful DNA sequencing (referred to as T-seq). All re-extracted samples were reassessed with traditional path review in order to minimize disruption of our existing clinical workflow.
Feature generation pipeline
Feature extraction relies on pretrained tissue and cell segmentation models. The tissue segmentation, based on a multi-field-of-view network with a fully convolutional ResNet-18 backbone30, produces segmentation maps of tumor-rich areas (AUC 0.947 for tumor classification, Section S1.1). The cell segmentation model, based on the U-Net architecture31, produces segmentations of cell nuclei throughout the whole image (Section S1.2). These models are combined to assign identities to tumor cells and lymphocytes (Section S1). Features are then generated from these model outputs, consisting of four feature groups: tumor shape, cell counts, cell nucleus shape, and cell nucleus texture, totaling 3,461 features from each slide (Section S2).
Tissue and cell segmentation model comparison to pathologist ground truth
Besides evaluating the classification accuracy of the tissue segmentation model on pathologist annotations (see Section S1.1), we also selected 334 slides (colorectal tumors metastasized to various tissues) which were previously reviewed by pathologists and assigned a visually estimated tumor percentage as part of our clinical NGS workflow. We computed the correlation coefficient between model predicted tumor percent and the visual estimate (Fig. 2A, R = 0.731). This modest correlation is close to the inter-pathologist correlation for such visual estimates of tumor percentage6. To evaluate the cell segmentation model, we asked three pathologists to annotate all cells as tumor cell, lymphocyte, or other within 300 fields of view sampled from 100 colorectal WSIs (see Section S1.2 for details). Correlation coefficients between predicted cell counts and the consensus of the annotated cell counts were R = 0.834 for all cells (Fig. 2B), R = 0.829 for tumor cells, and R = 0.728 for lymphocytes.

A The tumor percent predicted by tissue segmentation is plotted against the tumor percent visually estimated by pathologists from 334 WSIs. Four pathologists overall reviewed the samples, though each individual sample was reviewed by only one pathologist. Pearson R is computed, showing modest correlation. High correlation is not expected as visual estimates of tumor percent are not highly accurate, especially from a single rater. B Predicted total cell count is plotted against the pathologist-annotated cell count in 320 fields of view sampled from 108 WSIs. Each FOV was annotated by three pathologists and cell labels were assigned via a consensus voting scheme (Section S1.2). Pearson R is computed, showing relatively strong correlation. In this case the ground truth is highly accurate, as each cell was annotated by 3 pathologists, and thus strong correlation is expected. Predictions from the best performing DNA yield prediction linear model (log transform, L1, α = 0.01) strongly correlated to both the training set (C) and withheld validation set (D). Both axes are log-transformed. Each point represents an extraction attempt and is colored by macrodissection status (Light gray – macrodissected, dark gray – whole-slide dissected). Note that macrodissected slides tend to have higher mass per slide, because they tend to have larger surface area.
Macrodissection area masking during training and inference
In our normal clinical workflow, approximately 30% of CRC samples are macrodissected to ensure the extraction contains at least 20% tumor cells. For this subset of slides, feature extraction was restricted to tissue within the macrodissection area. Two strategies had to be developed for handling these samples because the retrospective slides used for training already had microdissection areas hand-drawn onto the slides by pathologists, while the prospective slides processed during inference had not yet been reviewed by a pathologist. To ensure that features from the training slides were only from the hand-drawn region, we developed an ink detection model which was post-processed to produce a macrodissection area mask (Section S3).
During inference the macrodissection area was estimated from tissue segmentation model predictions. The predicted tumor area was converted to a binary mask and post-processed to produce a contour mimicking hand-drawn macrodissection areas. Details on the implementation and validation of this method are in Section S4.
DNA Yield Prediction
Training and Validation Sets for DNA Yield Prediction
The core model underlying SmartPath is the prediction of DNA yield per slide using linear regression on extracted imaging features. To acquire a training set, the Tempus database was searched for slides scanned between January 2018 and January 2020 containing lung, breast, or colorectal cancer (CRC) primary tumor tissue. Three different cancer types were used for training because the average nuclear content per cell should be similar across cancers, and therefore extracted cell-count features should follow the same linear correlation with DNA yield regardless of cancer type. We confirmed this assumption by estimating the yield per cell for each cancer type (measured DNA yield per slide / predicted number of cells per slide) and finding no statistical difference between the means. Using the three cancer types also increased the training set size, which might help the model better generalize to unseen data in the future.
Aspirates and cytology specimens were excluded from the training set, as were slides with no recorded DNA mass or scraping, leaving a final training set of 1605 slides. Approximately 28% were previously macrodissected, reflecting the rate at which samples are macrodissected in our normal clinical workflow. Characteristics of the DNA yield prediction training set are shown in Table S1.
We also acquired a separate validation set of 332 retrospective samples from the same database, restricted to only CRC tissues, which was used for selecting a model with the best performing parameter combination (Table S2). The validation set was enriched for macrodissected cases (57% were macrodissected) to ensure thorough evaluation of our macrodissection estimation algorithm. Characteristics of the validation set are shown in Table S1.
Ground truth definition for DNA yield prediction model
The ground truth for training the DNA yield prediction model was taken as the extracted DNA yield from FFPE slides (see Section S5 for details on DNA extraction procedure). Each slide in the training and validation sets was an archival H&E slide representative of the unstained slides already extracted and sequenced by our NGS laboratory. Although most underwent only one DNA extraction attempt, some had multiple, in which case the imaged slide may have been closer to the tissues used for the 2nd extraction attempt. Therefore, the ground truth was defined as:
(DNA1 yield) / (Nslides), if only 1 extraction attempted
(DNA1 yield + DNA2 yield) / (Nslides for DNA1 + Nslides for DNA2), if > 1 extractions attempted
Section S6 contains more details on the ground truth definition.
Parameter and feature exploration for final model selection
For parameter exploration we used the full feature set (3641 features). Because the number of features was larger than the number of samples in the training set (1605), the linear model severely overfit and failed to generalize without regularization. Optimal regularization parameters were determined by parameter sweeping across L1 and L2 regularization strengths. Each regularization was tested with natural log and Box-Cox power transformations on features and ground truth. The parameter combination with the best validation set performance (R = 0.818) was a log transform and an L1 regularization with strength=0.01 (see Table S2 for full parameter exploration). Predictions of this optimal model are plotted against training and validation ground truths in Fig. 2C, D.
To confirm that including all 3641 features was advantageous, we performed a 200-fold cross validation using an 80/20 train/val split of the training set using the optimal parameter combination found from the exploration. We measured the mean coefficient magnitude across folds for each parameter. The top 10 features accounted for 93.4% of the model coefficient magnitude and were from a combination of feature groups (cell counts, tumor shape, cell shape, texture), with the total cell count having the highest importance (Fig. S2). Further confirmation of the usefulness of keeping all features was done in a 200-fold cross validation experiment where cumulatively more features were included. Starting with just 1 feature, then progressively adding features by group, performance on the cross-validated training set and the withheld validation set increased as more features were included (Fig. S3). This confirmed that the inclusion of all features gave the best performing model.
We also explored the inclusion of categorical features, such as procedure type, tissue site, and institution. However, the inclusion of categorical features did not offer any significant boost in the performance of cross-validated models, and their model coefficients were consistently pushed towards 0 by regularization. Because the final model was to be run in a real-world scenario, where image artifacts may cause some features to have infinite or non-numeric values, additional steps were also taken to ensure that such situations were handled smoothly in the inference pipeline (Section S7).
Target yields for number of slides prediction
The goal of SmartPath is to recommend the number of scraped slides needed to achieve a DNA yield between 100–2000 ng. To convert the predicted DNA yield per slide into a recommendation of how many slides to scrape, we divided a target yield by the predicted yield per slide and rounded down to the nearest integer. This target yield is a tunable operating point of the algorithm. During the trial, the SmartPath system presented the number of slides needed to achieve a target yield of at least 100, 400, or 1000ng. For details on how these target yields were selected, see Section S8.
We chose three target yields instead of one to give pathologists more flexibility. Because the relationship between number of slides scraped and DNA yield is linear, pathologists can also use the target yields to interpolate the recommended number of slides if they choose. This design choice emphasizes the principle of AI-augmented decision making, rather than AI automation.
Internal model evaluation trial
Trial design
To test the viability of our system in practice, we undertook an internal trial using clinical CRC samples to evaluate SmartPath compared to traditional (Trad) pathologist review. Trad and SmartPath workflows are summarized in Fig. 1B. Sample sizes of 250 SmartPath and 250 Trad samples were determined by power analysis at significance Level=0.01 and power = 0.8 (Section S9). The internal trial was designed to be run in tandem with standard clinical workflow, mirroring every step until pathologist review. Before pathologist review, each FFPE block was cut into 20 sections and affixed to glass slides. One slide midway through the levels was stained for H&E and designated for pathologist review, while the others were designated for scraping. If the tumor was CRC primary and met additional inclusion criteria (Table S3), the slides were flagged for trial enrollment and aggregated separately to avoid mixing with the rest of the clinical workflow. Enrolled samples were assigned in alternating order to Trad or SmartPath cohorts and assignments were recorded in a log, aiming to collect roughly equal numbers per day.
The Trad cohort H&E slides were a control group of samples which passed through our established pre-extraction workflow whereby a pathologist estimates tumor percentage by eye, marks a dissection area on the glass slide if needed, and recommends the number of slides to be scraped by an extraction technologist. All samples were still reviewed by a pathologist in a timely manner to not disturb the existing clinical sequencing workflow. Samples assigned to the Trad cohort were re-entered into the clinical workflow and path reviewed that same day.
The SmartPath cohort H&E slides were scanned on the Philips Ultra Fast Scanner (Philips, Eindhoven, The Netherlands) to produce a digitized WSI at ×40 base magnification level (0.25 µm/pixel). Slide scanning automatically triggered feature generation and DNA yield prediction on the WSI (Section S10). To minimize interference with existing clinical workflow, SmartPath-assisted review was conducted the morning after scanning, although in principle same day review is quite feasible because scanning and model deployment take only minutes to complete. After pathology review, the recommended number of slides were scraped, DNA was extracted (Section S5), and NGS was conducted using the Tempus xT platform8. This process was repeated daily over the span of several months until 249 SmartPath and 252 Trad cohort samples were accumulated.
During SmartPath-assisted review, the pathologist viewed a custom-built UI (Fig. S5) displaying recommendations for macrodissection area and number of slides needed to achieve at least three possible target DNA yields of 100, 400, or 1000 ng. The pathologist had the option to accept or reject model recommendations. If they chose to accept, the desired target yield was selected in the UI. If they disagreed, they recorded in the trial log if the rejection was driven by clinical reasoning or model performance. In cases where the pathologist disagreed with the predicted macrodissection area, the pathologist drew their own microdissection area. A summary of the pathologist decisions made during the trial is shown in Table S4.
Internal trial performance evaluation metrics
The impact of AI-assistance in the trial was evaluated by several metrics from two main categories: extraction metrics and NGS workflow costs.
Extraction metrics
-
Percent DNA yield within range: We defined the DNA yield target range as 100–2000 ng. The minimum of 100 ng was chosen to reflect the minimum input quantity of DNA requested by most NGS laboratories, which is between 50–200ng2,3. The maximum of 2000ng was chosen by collaborating pathologists as a reasonable cutoff indicating surplus of extracted DNA. The percent of samples below this range (<100 ng) is referred to as undershoot, and above this range (>2000ng) is referred to as overshoot.
-
Number of slides scraped (N slides): This measures the number of slides scraped for 1st DNA extraction attempt. We did not count slides scraped for later extraction attempts because additional attempts did not receive AI-assistance. This metric also excludes any slides scraped for RNA extractions or scraped after first successful DNA sequencing.
-
NGS workflow costs
-
Extraction count: This metric counts the number of extraction attempts made towards the first DNA sequencing attempt. It excludes extractions for RNA and extractions made after the first DNA sequencing attempt.
-
T-seq: DNA extraction-to-sequencing time, defined as the time elapsed from the first extraction attempt to the first successful DNA sequencing attempt (Fig. S6). This includes only the time period that can be influenced by AI-assistance, excluding RNA-sequencing and any subsequent DNA sequencing. This definition is restricted to the context of this study and is not reflective of Tempus’ operational turnaround time.
For each of these metrics, we also present the effect of two effect modifiers, the tissue area and extraction quality.
Effect Modifiers
-
Tissue area: Pathologists partially rely on tissue area to estimate how many slides should be scraped for extraction, where small tissues tend to have more slides scraped than large tissues. We split the tissue area at the 50th percentile of the distribution (85.46 mm2) for samples enrolled into the trial to obtain two groups: large (mean area 285.29 mm2) and small (mean area 18.53 mm2) (Fig. S7A).
-
Extraction quality: An in-house measure similar to other established methods32 for evaluating extracted DNA quality. Briefly, extracted nucleic acid is assessed with a Fragment Analyzer (Advanced Analytical Technologies, Ames, IA), which produces a distribution of nucleic acid fragment lengths, measured in base pairs. The fragment length distribution is split into custom-defined ranges corresponding to short, intermediate, and long fragments and the amount of the distribution in each of these ranges is quantified. To produce the extraction quality, the fragment data is combined with extracted DNA mass and binned into three quality levels: low, intermediate, or high. Low-quality samples generally have short fragments and low DNA yield (usually < 90 ng), while high-quality samples generally have long fragments and high DNA yield (usually > 400 ng).
Although small tissue samples can have high-quality DNA fragmentation, for most samples small tissue area correlates with low extraction quality Fig. S7B).
Internal trial data quality control
Of the 501 samples enrolled into the trial, 18 were rejected at pathology review due to insufficient tissue, 4 were erroneously enrolled either with incorrect cancer type or procedure type, 1 had an incorrect indication of number of slides scraped, and 2 were removed because their sequencing was delayed due to human error (Table S4). This left 476 samples for the overall analysis (233 Trad, 243 SmartPath). For analysis of T-seq, an additional 18 samples were dropped (7 Trad, 3.00% of population; 11 SmartPath, 4.53% of population) because they did not reach DNA sequencing due to failure downstream of extraction (either at library prep or hybridization steps, see Fig. 1B), and therefore did not have a defined sequencing time interval, leaving 458 samples (226 Trad, 232 SmartPath).
Statistical analysis of covariates
The FDA guidance for adjustment for clinical covariates in clinical trials (Docket number FDA-2019-D-0934) advises experimenters to identify the covariates expected to have an important influence on the primary outcome. The primary outcomes for the present work are the DNA yield and workflow costs. DNA yield may depend on sample age, as older samples may suffer from nucleotide degradation33. It may also depend on the individuals involved in the extraction (i.e., the pathologist and technicians). The day on which the sample is extracted could have an impact on workflow turnaround time due to weekly lab scheduling cycles. We included these sample-level measures as covariates (Table 1) and also recorded several patient-level characteristics commonly reported in cancer studies (Table S5). Covariate imbalance was measured by the chi-squared test computed from contingency tables. Contingency tables were computed by cross-tabulating counts for each characteristic and chi-squared tests were performed in Python 3.7 using scipy.chi2_contingency34.
Analysis of covariance using generalized linear models
An analysis of covariance (ANCOVA) allows researchers to dissociate contributions of additional covariates from the treatment to the total variance. For the present application the treatment variable is the trial cohort (Trad or SmartPath) and the dependent variables are the following: DNA mass undershoot boolean (1 if <100 ng, 0 otherwise), number of slides scraped (N slides), extraction count, and T-seq. Traditional ANCOVA is designed to run on normally distributed samples assuming linearity and homoscedasticity (constant variance across residuals). However, most of these metrics are not normally distributed, and thus appropriate distributions were chosen to model these dependent variables with generalized linear models (GLMs)35. All ANOVA analyses were performed in R version 0.4.436. For details on GLM selection see Section S11.
Scaling and encoding of covariates for GLMs
Covariates had to be appropriately encoded for the analysis. Trial cohort was dummy encoded as a binary indicator (0 - SmartPath, 1 - Trad). Extraction quality was numerically encoded as ordinal variables (0, 1, 2). Extraction day-of-week was encoded numerically from Monday to Sunday as 0–6. Sample age was log-transformed. Procedure type, pathologist, and extraction tech were dummy encoded, dropping one category from each to eliminate correlations. For more justification of these encoding choices see Section S12.
Univariate and multivariate GLMs
GLMs were fit only for the subset of the samples most in need of AI-assistance, namely small tissues with low extraction quality. Univariate GLMs were initially fit using the sample-level (Table 1) and patient-level characteristics (Table S5) as independent variables, but significant effects were not found for any of the patient-level characteristics. Multivariate models for each metric are built using only those variables with significant association in univariate tables (Tables S6–S9). For details on construction of univariate and multivariate models, see Section S13.
Results
Impact on extraction metrics
AI-assistance improved DNA yield within a target range of 100–2000 ng
The fraction of samples within the target range was significantly improved for the SmartPath cohort (Trad = 0.56+/− 0.064 vs SmartPath = 0.70+/− 0.058, P = 0.005, a 25% increase, Fig. 3A). This was primarily due to limiting over-extraction, as the fraction of samples with mass that overshot the desired range was also significantly improved (Trad = 0.32+/− 0.06 vs SmartPath = 0.18+/− 0.049, P = 0.001, a 14% decrease, Fig. 3A). The fraction of samples that undershot the desired range was not improved overall.

A The fraction of samples reaching the target (100–2000 ng), undershoot (<100 ng), or overshoot (>2000ng) DNA mass upon first extraction compared between SmartPath and Trad cohorts. Comparisons were then stratified by B tissue size and C extraction quality, where seven samples were excluded due to lack of extraction quality data. To compute statistics, each sample was assigned 1 or 0 whether it was inside or outside the indicated mass range. Error bars are 95% confidence intervals. Significance was determined by a chi-squared test for binary variables performed on the contingency table of counts split by Trad and SmartPath cohorts.
Tissue characteristics, such as tissue area and DNA fragment quality, are known to impact tissue extraction19,37. We confirm that these effects exist in our data as well. In Fig. 3B, we subset the data into large and small tissue area groups (defined in Methods), revealing that reduction in overshoot was restricted to large tissues. When further subset by extraction quality (defined in Methods), the reduced overshoot effect was primarily seen in large tissues with high extraction quality (Fig. 3C, top). Therefore, AI-assistance helped pathologists preserve tissue use for samples that were already likely to succeed NGS. Subsetting also revealed a trend in reduction of the undershoot fraction for small tissues with low extraction quality (Fig. 3C, bottom). Although the difference was not significant (chi-squared P = 0.088), there were only 50 samples in this subset and the sample size may be underpowered to measure the effect. As discussed in subsequent sections, however, subsetting by small tissue area and low extraction quality showed significant improvements in other metrics.
AI-assistance fosters more efficient use of tissue slides
Across all CRC samples from the trial (n = 476), DNA yields <100 ng almost always resulted in multiple extraction attempts (Fig. S10). These results demonstrate the importance of better metrics for scraping parameters, as more slides should be scraped initially when lower DNA yields are expected in order to avoid repeating extraction. While NGS laboratories typically scrape 5–10 FFPE slides per extraction38, our AI model recommended a broader distribution of slides for scraping compared with the Trad cohort (Fig. 4A). Large tissues in the SmartPath cohort usually had only one or two slides scraped, thus conserving tissue in this subset. On the other hand, small tissues in the SmartPath cohort usually had >10 slides scraped (Fig. 4A, B). Therefore, while the mean number of slides was not significantly different between SmartPath and Trad cohorts across all tissue sizes (Fig. 4B), slides in the SmartPath cohort were used more efficiently. However, because the distribution of N slides was not normal, the median should also be considered. While the overall mean number of slides scraped per sample in the SmartPath cohort was slightly higher than in the Trad cohort (7.7 ± 5.91 SmartPath vs 7.62 ± 3.0 Trad), the median in the SmartPath cohort was lower than in the Trad cohort (6 SmartPath vs 10 Trad).

A Distribution of N slides scraped plotted for all samples (left), only large tissues (middle), and only small tissues (right). Without AI-assistance, pathologists tended to recommend either 5 or 10 slides for scraping (Trad, orange), but with AI-assistance the distribution was much broader (SmartPath, blue). SmartPath distribution is shifted towards fewer slides for large tissues and more slides for small tissues. B Box plots comparing numbers of slides scraped for SmartPath and Trad cohorts grouped by large and small tissues. P-values were computed from t-tests on log-transformed data assuming unequal variance. White dots - mean. Horizontal black line - median C Truncated violin plots comparing number of sides scraped between SmartPath and Trad cohorts, grouped by large (top) and small (bottom) tissues and by extraction quality. Back boxes −25% and 75% percentiles. White dots – medians. Samples that did not have recorded numbers of slides scraped were dropped. P-values for Welch’s two-sided t-test assuming unequal variance are displayed above each group. Bimodal distributions for the Trad cohort (orange) correspond to 5 and 10 slides.
Further subsetting the data by extraction quality also shows that SmartPath recommended fewer slides for large tissues regardless of extraction quality (Fig. 4C, low quality P = 0.07, intermediate quality P«0.01, and high quality P«0.01). An opposite trend was observed for small tissues with low and intermediate extraction quality, where more slides were recommended in the SmartPath cohort, although there was no significant difference (Fig. 4C bottom). For the subset of small samples with high extraction quality, the SmartPath and Trad cohort means were very similar. This could be desirable as high-quality samples are already likely to succeed NGS.
Impact of AI-assistance on NGS workflow costs
Number of extraction attempts is similar between cohorts
The number of extraction attempts is an important metric for workflow improvement because re-extractions are financially and temporally costly. The distributions of N extractions for SmartPath and Trad cohorts are shown in Fig. 5A. As these distributions were not normal, they are represented with Poisson distributions for calculation of statistics (see Methods). The distributions are dominated by cases with only one extraction, which is already the optimum. Overall, no significant difference in mean number of extractions per sample was observed between the SmartPath and Trad cohorts Fig. 5B. Grouping by extraction quality reveals that all high-quality samples were already performing at optimum for this metric, with only one extraction per sample, and intermediate-quality samples were performing near optimum. On the other hand, the SmartPath cohort had a decreased mean extraction count for low-quality samples, albeit not significant (Poisson rate ratio P = 0.212).

A The numbers of extraction attempts needed to reach DNA sequencing are counted for SmartPath and Trad cohorts. Only 4 samples in the total dataset had extraction count=4, and only one had extraction count=5. B Mean number of extractions grouped by extraction quality. Error bars are 95% confidence intervals produced with 1000 bootstraps. High-quality samples have no error bars because they are already at optimum, with only one extraction per sample, regardless of cohort. To compute p-values, Poisson distributions were fit to the zeroed distributions (see Section S11) of SmartPath and Trad cohorts and a test was performed to assess if the ratio of the two Poisson rates is statistically different from 1. C Same as B but grouped by large and small tissues.
Further subsetting by tissue area reveals that the decrease in mean extraction count per sample approaches significance (Poisson rate ratio P = 0.052) for low-quality samples with small tissue area (Fig. 5C, bottom). This result suggests that AI-assistance may be useful in preventing re-extractions for low-quality samples with small tissue area, which are the samples most in need of improvement. However, there was a significant increase in mean extraction count for large intermediate-quality samples in the SmartPath cohort (Poisson rate ratio P = 0.017), caused by four samples which had >1 extraction count. The higher extraction count may be partly due to the age of these samples. SmartPath cohort samples were on average older than the Trad cohort (Fig. S12A), and for large samples in the SmartPath cohort, those with intermediate quality were also the oldest (Fig. S12B). Older samples correlate with higher extraction count (Fig. S12D), likely because they tend to be more degraded.
AI-assistance reduced DNA sequencing time for low quality samples with small tissue areas
Figure 6A shows the distribution of T-seq in the SmartPath and Trad cohorts. Similar to extraction counts, there was no significant difference in the mean T-seq between the two cohorts (Fig. 6B; Trad 3.74 ± 1.67 days, SmartPath 3.89 ± 1.67 days). We expect T-seq to follow a similar trend as extraction count because they are strongly correlated (Fig. S11). High-quality samples showed almost no difference in T-seq between cohorts, a reflection of the fact that extraction count is already optimal for high-quality samples. Intermediate quality samples showed a significant increase (P = 0.018) for the SmartPath cohort, likely due to the same samples that drove up extraction count for this group. However, when subset by tissue area the T-seq for small low-quality samples was almost 2 days shorter in the SmartPath cohort compared with Trad (Fig. 6C, bottom; Trad 6.90 ± 2.77 days, SmartPath 4.97 ± 2.06 days, P = 0.025).

A Distribution of time to first DNA sequencing (T-seq) for all samples in the SmartPath and Trad cohorts. B Time from first extraction to first DNA sequencing was measured for SmartPath and Trad cohorts. The plot shows the mean T-seq for SmartPath and Trad cohorts for all samples (left) as well grouped by extraction quality (low, intermediate, high). Error bars are the average of the 5–95% confidence intervals of Trad and SmartPath cohorts. C Same as B but grouped by large and small tissues. Significance between the cohort means was assessed by performing t-tests on the log-transformed T-seq.
Univariate analysis of covariates on full trial dataset
To determine if the effects observed were due entirely to the experimental condition (SmartPath vs. Trad) alone, we considered covariates of the study as detailed in Table 1. Despite an attempt to randomize samples by alternating assignment to SmartPath and Trad cohorts each day (see Methods), imbalances were detected. Moderate imbalance was detected for sample age (chi-squared P = 0.06), while strong imbalance was detected for pathologist (chi-squared P < 1e-60), extraction day-of-week (chi-squared P = 0.0008), and extraction tech (chi-squared P = 0.0001).
While dataset imbalance in covariates suggests other sources of variability besides the experimental condition, it alone does not prove that they impacted the dependent metrics. With univariate GLMs (see Methods), we quantified the correlation each covariate had with the following four trial metrics: DNA Mass undershoot boolean (True if <100 ng), number of slides scraped, extraction counts, and T-seq. Summary statistics of univariate GLMs for each covariate are presented in Tables S6–S9.
For three of the trial metrics, undershoot Boolean, number of slides scraped, and extraction count (Tables S6–S8), the extraction quality (P < 2e-16, P = 5.96e-05, and P < 2e-16, respectively) and tissue area (P = 4.01e-06, P < 2e-16, and P = 2.78e-06, respectively) were more predictive than any of the covariates. These same three metrics also were significantly correlated with procedure type. This is expected, as procedure type is strongly correlated with tissue area, where needle biopsies tend to have much smaller area than resections.
However, for T-seq (Table S9) the extraction day-of-week was the most predictive variable (P < 2e-16). This is a known effect, where sequencing times for samples extracted later in the week tend to be longer than samples extracted earlier in the week (Fig. S13B, see Discussion). Despite the randomized trial design, samples in the SmartPath cohort tended to be extracted later in the week than samples in the Trad cohort (Fig. S13A).
For all metrics except for N slides, the extraction tech group univariate GLMs showed significance (P < 0.05). Although this variable has high cardinality (25 categories), the Akaike Information Criterion (AIC) in these cases was lower than some of the other variables (Tables S6–S8), suggesting that these were not random correlations. Imbalance in the extraction tech group was also meant to be eliminated by trial design but persisted despite our efforts.
Only one of the metrics, N slides, was significantly predicted by sample age. None of the metrics had strong correlation with pathologist.
Multivariate analysis of covariates for subset of samples with small tissue area and low extraction quality
Here we investigate the impact these covariates had on the main effect of the experimental variable, the trial cohort. In Figs. 3–6 we identified that AI-assistance was most effective for samples with small tissue areas and low extraction quality. As this is the most interesting subset of samples, we restricted the following multivariate analysis to this subset (N = 50). For modeling each outcome metric, we chose only those covariates significantly associated (P < 0.05) with the outcome metrics in the univariate analysis (Tables S6–S9). We excluded procedure type, as it is already strongly correlated with tissue area and no surgical resections are present in this subset. We also excluded the extraction tech group as this variable has very high cardinality (25 categories) relative to the number of samples in this subset.
To measure the influence of these covariates on the main effect of AI-assistance, we compared a univariate GLM using trial cohort as the independent variable, to the multivariate GLMs for each outcome metric (see Methods). In the interest of brevity and focus, we limited the multivariate analysis to only 4 outcome metrics, dropping the target and overshoot fractions from the analysis. Summary statistics of these GLMs are shown in Table 2.
For the small tissue area and low extraction quality data subset, the trial cohort alone was significantly predictive for both extraction count and T-seq (P = 0.049 and 0.026, respectively). The other two metrics, undershoot and N slides, had univariate associations with trial cohort approaching significance (P = 0.055 and 0.075, respectively). In all outcome metrics, the inclusion of covariates in multivariate GLMs raised the trial cohort p-values, suggesting that the main effect can be partially explained by the covariates. However, the increase in trial cohort p-value was moderate.
Inclusion of covariates increased the AIC for multivariate GLMs of undershoot from 54.57 to 55.04 and N slides from 65.52 to 74.11, indicating that covariates carry little additional information about these metrics, adding complexity without proportionally improving the fit. For both NGS workflow metrics, though, the AIC was reduced (extraction count: from 120.21 to 118.91, T-seq: from 51.90 to 45.73), indicating that the covariates improved the model without adding unnecessary complexity.
The only significantly predictive covariate in the multivariate GLMs was extraction day-of-week, which strongly associated with T-seq (P = 0.006). The correlation of extraction day-of-week with T-seq is a known effect in our NGS laboratory (Fig. S13). Although the cohort imbalance in day-of-week was meant to be eliminated by alternating assignment of samples to SmartPath and Trad cohorts each day (see Methods), unfortunately the imbalance persisted (Table 1). Overall, the multivariate analysis shows that the covariates considered here have a measurable impact on the main effect of trial cohort. The adjusted effect of trial cohort is weaker upon inclusion of covariates; however it is difficult to ascertain if this fully explains the main effect as the main effect is itself underpowered (N = 50). Future trials of this tool should ensure that sufficiently high numbers of samples are available in the small tissue and low extraction quality regimes and should more rigorously control for covariate imbalances.
Discussion
Here, we developed SmartPath, a computer vision tool to assist pathologists in determining NGS tissue input parameters, and tested it in a real-world clinical setting. Compared to the group that received traditional pathology review, AI-assistance produced significantly more DNA yields falling within a target range of 100–2000 ng. The AI-assisted model also improved tissue stewardship by recommending scraping of more slides for samples with small tissue areas, likely preventing some re-extractions, but fewer slides for samples with large tissue area.
While scraping more slides for small tissues may seem counter to tissue stewardship, underpredicting could result in an insufficient yield, leading to re-extraction which is a further waste of tissue. There is a trade-off between initial tissue use and prevention of re-extraction. Our model design intention was to bias towards preventing re-extraction, but evidently it was not biased enough as overall undershoot fraction was not reduced in the SmartPath cohort. In retrospect, target yields of the model could have been tuned even higher to improve the undershoot fraction at the expense of the overshoot fraction. This was explored in a simulation (Fig. S9) which suggests that, had we scraped 1.4x more slides (scaling the target yields from 100, 400, 1000 to 140ng, 560, 1400 ng) the undershoot fraction for the SmartPath cohort could have dropped below that of the Trad cohort, while still maintaining a reduction in overshoot fraction. During the trial, the reviewing pathologists also noted that higher target yields would have been advantageous, and 82% of their selected target yields were for the highest available yield, 1000 ng (Table S4).
For the two NGS workflow costs, the extraction count and T-seq, no significant difference was found between the full populations of SmartPath and Trad cohorts, but improvements were seen in the subset of small samples with degraded DNA quality. It is known that NGS fails more often for smaller, poor-fragmentation-quality samples19,37, and pathologists will attempt to reject such samples for insufficient tissue. Notably, the SmartPath cohort had only 2 samples rejected for insufficient tissue, while the Trad cohort had 16, because pathologists were willing to observe SmartPath predictions before rejecting a sample (Table S4). Despite including samples in SmartPath review that likely would have been rejected in Trad review, the mean extraction count for small, low-quality samples was in fact reduced (albeit not statistically significant, Fig. 5C). Additionally, we observed a significant reduction in T-seq for small, low-quality samples (Fig. 6C). Furthermore, the subset of high-quality samples had only one extraction attempt regardless of SmartPath or Trad treatment (Fig. 5B), which is already optimal, so the extraction count could not be further improved for these samples. This subset also represented most of the samples in the entire trial (only ~13% were low quality). Therefore, lack of reduced NGS workflow costs in the full population is largely due to over-representation of high fragmentation quality samples in our cohort.
Colorectal cancer was chosen for the trial for internal workflow considerations, but we expect that the present algorithm can be generalized to other cancer types by replacing the underlying tissue and cell segmentation models with tissue-specific models already in existence. Other cancer types, such as non-small cell lung cancer and especially pancreatic cancer, have higher rates of low-fragmentation-quality samples (Fig. S15) and therefore may benefit even more from the AI-augmented pathology review system, although additional research must be conducted to determine if this is the case.
The present work does not evaluate the impact of SmartPath on genetic variant calling because its scope is restricted to evaluation of a workflow improvement tool. However, future applications could conceivably improve accuracy of variant calling by recommending tissue samples with optimized DNA yield and tumor enrichment. To evaluate such a system, a study could be designed where consecutive sections from the same samples are evenly split between SmartPath and control groups, and genetic variants between groups are compared.
Proper randomization is necessary to eliminate all biases in trials, but for many real-world trials like ours, this is not possible due to external constraints. Future tissue recommendation models could improve upon the current work by taking these sources of bias into account as variables in the model itself and/or explicitly eliminating these effects through trial design. One bias identified was the strong influence of extraction day-of-week on T-seq, likely due to weekly batch effects and staffing cycles which cause sequencing times to be longer for samples extracted later in the week. This resembles a well-documented weekly phenomenon in healthcare, termed the “weekend effect”39. The trial also took place in Aug-Dec 2020 during the lockdown period of the COVID-19 epidemic, and thus effects due to limited personnel were likely exaggerated. This bias was meant to be eliminated by the trial design by enrolling similar numbers of samples into SmartPath and Trad cohorts per day, but multivariate analysis showed that the AI-assistance effect was reduced after inclusion of extraction day-of-week as a predictor (Table 2).
Sample age has a known effect on FFPE sample extraction success. Evidence suggests that samples >7 years old are unsuitable for NGS33; however, much older samples have been successfully sequenced40. Exclusion of older samples can negatively impact patients’ lives, so our laboratory does not reject samples for sequencing due to old age. By chance, the SmartPath cohort samples were generally older than Trad samples (Fig. S12), and therefore a bias towards sequencing failure could have been introduced for the SmartPath cohort.
Imbalance was also detected when treating pathologist as a covariate. The SmartPath cohort was mostly reviewed by one pathologist who was trained to use the UI at the start of the trial. This was an operational constraint due to limited resources that were split between normal lab operation and conducting this trial. Our top priority was not to disturb our existing NGS clinical workflow. Despite this imbalance, the pathologist identity did not strongly associate with any of the outcome metrics (Table 2). In fact, the mean values of the outcome metrics were similar between the two pathologists that reviewed the largest number of SmartPath and Trad cohort samples (Fig. S14).
Several improvements to the existing DNA yield prediction strategy could also be made. Although feature extraction relied on neural networks, DNA yield prediction was accomplished with a relatively simple modeling approach using a regularized linear model on extracted features (primarily cell counts). This was done because the extracted features are easily interpretable to pathologists and because the relationship between number of slides and extracted DNA yield is inherently linear. However, future approaches may improve results by predicting DNA yield directly with a neural network.
While the present model relies mostly on extrinsic features such as cell counts and tumor area for correlation with tumor yields, more work could be done to investigate intrinsic features, such as slide preparation quality, and possibly even fragmentation quality with imaging means. An imaging-based predictor could potentially be trained to provide a prior on sample quality. Training data for an imaging-based quality predictor could come from a combination of established DNA quality metrics, including fragment analyzer data, qPCR assays to measure the amount of amplifiable DNA in a sample, the DNA Integrity Number, and Genomic Quality Number41.
Furthermore, the trial only incorporated AI into the initial screening of the sample but did not incorporate pathologist feedback for updating predictions, primarily to avoid disturbing the existing clinical workflow of our NGS laboratory. We envision a future pathologist-in-the-loop application, where pathologists may edit macrodissection areas and receive updated predictions in real time. Workflow improvements could also be made to maximize efficiency. For example, a model combining imaging-based measures with clinical data could flag samples up front that may need AI-assistance, while passing samples with high likelihood to succeed in NGS. Future models may also be trained to predict not only DNA yield, but RNA yield and library-prep success.
The SmartPath system was designed to support an existing NGS pipeline, which uses traditional scraping-based macrodissection to extract tissue within pathologist-defined tumor regions. A caveat of scraping-based macrodissection is that it may not always enrich the tumor percentage due to heterogeneity of cell types within tumor areas. Future applications of SmartPath could integrate with laser-capture microdissection systems to guide single-cell-based microdissection20.
By addressing these limitations, future applications of SmartPath may provide a viable alternative to manual estimation by accurately predicting tissue quantities needed for adequate DNA yield. SmartPath could be useful in circumstances where access to pathologists is scarce, or for laboratories processing large volumes of tissue. Coupled with a digital slide viewer, such a system can support fully remote pathology review of digitized WSIs, allowing NGS laboratories to widen their access to reviewing pathologists. Integration of SmartPath with automated microdissection systems15,16,19 could allow for tissue extraction workflows which are almost entirely automated, with a pathologist needed only to approve or modify input parameters, and potentially be economically and clinically beneficial for NGS laboratories.
Data availability
Most data generated or analyzed during this study are included in this article and its supplementary information files. Raw data are not made available due to their proprietary nature, but are available from the corresponding author on reasonable request.
References
McConnell, L, Houghton, O, Stewart, P, Gazdova, J, Srivastava, S, Kim, C, et al. A novel next generation sequencing approach to improve sarcoma diagnosis. Mod Pathol. 33, 1350–1359 (2020)
Austin, M C, Smith, C, Pritchard, C C, Tait, J F. DNA Yield From Tissue Samples in Surgical Pathology and Minimum Tissue Requirements for Molecular Testing. Arch Pathol Lab Med. 140, 130–133 (2015)
Arreaza, G, Qiu, P, Pang, L, Albright, A, Hong, L Z, Marton, M J, et al. Pre-Analytical Considerations for Successful Next-Generation Sequencing (NGS): Challenges and Opportunities for Formalin-Fixed and Paraffin-Embedded Tumor Tissue (FFPE) Samples. Int J Mol Sci 17, E1579 (2016)
Ascierto, P A, Bifulco, C, Palmieri, G, Peters, S, Sidiropoulos, N. Preanalytic Variables and Tissue Stewardship for Reliable Next-Generation Sequencing (NGS) Clinical Analysis. The J Mol Diagnostics. 21, 756–767 (2019)
Padmanabhan, V, Steinmetz, H B, Rizzo, E J, Erskine, A J, Fairbank, T L, de Abreu, F B, et al. Improving Adequacy of Small Biopsy and Fine-Needle Aspiration Specimens for Molecular Testing by Next-Generation Sequencing in Patients With Lung Cancer: A Quality Improvement Study at Dartmouth-Hitchcock Medical Center. Arch Pathol Lab Med. 141, 402–409 (2016)
Hamilton, P W, Wang, Y, Boyd, C, James, J A, Loughrey, M B, Hougton, J P, et al. Automated tumor analysis for molecular profiling in lung cancer. Oncotarget. 6, 27938–27952 (2015)
Roy-Chowdhuri, S, Chow, C, Kane, M K, Yao, H, Wistuba, I I, Krishnamurthy, S, et al. Optimizing the DNA yield for molecular analysis from cytologic preparations. Cancer Cytopathol. 124, 254–260 (2016)
Beaubier, N, Bontrager, M, Huether, R, Igartua, C, Lau, D, Tell, R, et al. Integrated genomic profiling expands clinical options for patients with cancer. Nat Biotechnol. 37, 1351–1360 (2019)
Wagle, P, Nikolić, M, Frommolt, P. QuickNGS elevates Next-Generation Sequencing data analysis to a new level of automation. BMC Genomics 16, 487 (2015)
Hess, J F, Koh, T A, Kotrová, M, Rönsch, K, Paprotka, T, Mohr, V, et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol Adv. 41, 107537 (2020)
Emmert-Buck, M R, Bonner, R F, Smith, P D, Chuaqui, R F, Zhuang, Z, Goldstein, S R, et al. Laser capture microdissection. Science. 274, 998–1001 (1996)
Hernández, S, Lloreta, J. Manual Versus Laser Micro-dissection in Molecular Biology. Ultrastructural Pathol. 30, 221–228 (2006)
de Bruin, E C, van de Pas, S, Lips, E H, van Eijk, R, van der Zee, M M C, Lombaerts, M, et al. Macrodissection versus microdissection of rectal carcinoma: minor influence of stroma cells to tumor cell gene expression profiles. BMC Genomics. 6, 142 (2005)
Krizman, D, Adey, N, Parry, R. Application of tissue mesodissection to molecular cancer diagnostics. J Clin Pathol. 68, 166–169 (2015)
Adey, N, Emery, D, Bosh, D, Callahan, S, Schreiner, J, Chen, Y, et al. A mill based instrument and software system for dissecting slide-mounted tissue that provides digital guidance and documentation. BMC Clin Pathol. 13, 29 (2013)
Going, J J. Histological microdissection in diagnostic and investigative pathology. Diagnostic Histopathol. 16, 43–48 (2010)
Qi, P, Bai, Q, Yao, Q, Yang, W, Zhou, X. Performance of Automated Dissection on Formalin-Fixed Paraffin-Embedded Tissue Sections for the 21-Gene Recurrence Score Assay. Technol Cancer Res Treat. 19, 1533033820960760 (2020)
Javey, M, Reinsch, C, Feldkamp, M, Siemann, S, Blüher, A, Woestmann, C, et al. Innovative Tumor Tissue Dissection Tool for Molecular Oncology Diagnostics. J Mol Diagnostics. 23, 399–406 (2021)
Geiersbach, K, Adey, N, Welker, N, Elsberry, D, Malmberg, E, Edwards, S, et al. Digitally guided microdissection aids somatic mutation detection in difficult to dissect tumors. Cancer Genet. 209, 42–49 (2016)
Hipp, J D, Johann, D J, Chen, Y, Madabhushi, A, Monaco, J, Cheng, J, et al. Computer-aided laser dissection: A microdissection workflow leveraging image analysis tools. J Pathol Inform. 9, 45 (2018)
Oner, M U, Chen, J, Revkov, E, James, A, Heng, S Y, Kaya, A N, et al. Obtaining spatially resolved tumor purity maps using deep multiple instance learning in a pan-cancer study. Patterns. 3, 100399 (2022)
Azimi, V, Chang, Y, Thibault, G, Smith, J, Tsujikawa, T, Kukull, B, et al. Breast cancer histopathology image analysis pipeline for tumor purity estimation. Proc IEEE Int Symp Biomed Imaging. 2017, 1137–1140 (2017)
Viray, H, Coulter, M, Li, K, Lane, K, Madan, A, Mitchell, K, et al. Automated objective determination of percentage of malignant nuclei for mutation testing. Appl Immunohistochem Mol Morphol. 22, 363–371 (2014)
Greene, C, O’Doherty, E, Sidi, F A, Bingham, V, Fisher, N C, Humphries, M P. The Potential of Digital Image Analysis to Determine Tumor Cell Content in Biobanked Formalin-Fixed, Paraffin-Embedded Tissue Samples. Biopreserv Biobank. 19, 324–331 (2021)
Cui, M, Zhang, D Y. Artificial intelligence and computational pathology. Lab Invest. 101, 412–422 (2021)
Bera, K, Schalper, K A, Rimm, D L, Velcheti, V, Madabhushi, A. Artificial intelligence in digital pathology — new tools for diagnosis and precision oncology. Nat Rev Clin Oncol. 16, 703–715 (2019)
Steiner, D F, MacDonald, R, Liu, Y, Truszkowski, P, Hipp, J D, Gammage, C, et al. Impact of Deep Learning Assistance on the Histopathologic Review of Lymph Nodes for Metastatic Breast Cancer. Am J Surg Pathol. 42, 1636–1646 (2018)
Chen, PHC, Gadepalli, K, MacDonald, R, Liu, Y, Kadowaki, S, Nagpal, K, et al. Microscope 2.0: An Augmented Reality Microscope with Real-time Artificial Intelligence Integration. Nat Med. 25, 1453–1457 (2019)
Fine, J L. 21st century workflow: A proposal. J Pathol Inf. 5, 44 (2014)
Sha, L, Osinski, B L, Ho, I Y, Tan, T L, Willis, C, Weiss, H, et al. Multi-field-of-view deep learning model predicts nonsmall cell lung cancer programmed death-ligand 1 status from whole-slide hematoxylin and eosin images. J Pathol Inform. 10, 24 (2019)
Ronneberger O, Fischer P, Brox T. U-Net: Convolutional Networks for Biomedical Image Segmentation. arXiv:1505.04597 [cs] (2015)
Wang, F, Wang, L, Briggs, C, Sicinska, E, Gaston, S M, Mamon, H, et al. DNA degradation test predicts success in whole-genome amplification from diverse clinical samples. J Mol Diagn. 9, 441–451 (2007)
Millán-Esteban, D, Reyes-García, D, García-Casado, Z, Bañuls, J, López-Guerrero, J A, Requena, C, et al. Suitability of melanoma FFPE samples for NGS libraries: time and quality thresholds for downstream molecular tests. Biotechniques. 65, 79–85 (2018)
Virtanen, P, Gommers, R, Oliphant, T E, Haberland, M, Reddy, T, Cournapeau, D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 17, 261–272 (2020)
Dobson A J. An Introduction to Generalized Linear Models, Fourth Edition. (Chapman and Hall/CRC, 2018). https://doi.org/10.1201/9781315182780
R Core Team. R: A language and environment for statistical computing. (R Foundation for Statistical Computing) (2013)
Kofanova, O, Bellora, C, Frasquilho, S G, Antunes, L, Hamot, G, Mathay, C, et al. Standardization of the preanalytical phase of DNA extraction from fixed tissue for next-generation sequencing analyses. N. Biotechnol. 54, 52–61 (2020)
Snow, A N, Stence, A A, Pruessner, J A, Bossler, A D, Ma, D. A simple and cost-effective method of DNA extraction from small formalin-fixed paraffin-embedded tissue for molecular oncologic testing. BMC Clin Pathol. 14, 30 (2014)
Clarke, M S, Wills, R-A, Bowman, R V, Zimmerman, P V, Fong, K M, Coory, M D, et al. Exploratory study of the ‘weekend effect’ for acute medical admissions to public hospitals in Queensland, Australia. Intern Med J. 40, 777–783 (2010)
Ludyga, N, Grünwald, B, Azimzadeh, O, Englert, S, Höfler, H, Tapio, S, et al. Nucleic acids from long-term preserved FFPE tissues are suitable for downstream analyses. Virchows Arch. 460, 131–140 (2012)
Brisco, M J, Latham, S, Bartley, P A, Morley, A A. Incorporation of measurement of DNA integrity into qPCR assays. Biotechniques. 49, 893–897 (2010)
Acknowledgements
We thank Timothy Baker for assisting with NGS workflow data acquisition, Dr. Lingdao Sha and Dr. Andrew Kruger for help generating handcrafted imaging features, Dr. Mark Carty for insightful discussions on the statistical analysis of covariates, Dr. Tim Taxter for participating in the validation trial, and Matthew Kase for rigorously reviewing the text and figures. Finally, we are grateful to Erin MCarthy, the Tempus histology lab, and all members of the Tempus NGS lab, who made the internal validation trial possible.
Funding
This work was supported by Tempus Labs.
Author information
Authors and Affiliations
Contributions
BLO, RDJ, AW, and BMM performed the study concepts and design. BLO, RJ, ABT, RPJ and MCS performed the development of methodology and writing, review, and revision of the paper. BLO and IH wrote and validated the algorithm code. MC, CW, and LS built the browser-based UI and coordinated deployment to production. RDJ and AW supervised the validation trial. RDJ and BMM reviewed samples in both SmartPath and Trad arms during the validation trial. BLO, ABT, and RPJ provided acquisition and analysis of the validation trial results. BLO, ABT, and RPJ provided interpretation of data and statistical analysis. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
All authors were employees and shareholders of Tempus Labs at the time this work was done.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Osinski, B.L., BenTaieb, A., Ho, I. et al. Artificial intelligence-augmented histopathologic review using image analysis to optimize DNA yield from formalin-fixed paraffin-embedded slides. Mod Pathol 35, 1791–1803 (2022). https://doi.org/10.1038/s41379-022-01161-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01161-0
