
Abstract
Pancreatic intraductal tubulopapillary neoplasm (ITPN) is a recently recognized intraductal neoplasm. This study aimed to clarify the clinicopathologic and molecular features of this entity, based on a multi-institutional cohort of 16 pancreatic ITPNs and associated adenocarcinomas. The genomic profiles were analyzed using histology-driven multi-regional sequencing to provide insight on tumor heterogeneity and evolution. Furthermore, an exploratory transcriptomic characterization was performed on eight invasive adenocarcinomas. The clinicopathologic parameters and molecular alterations were further analyzed based on survival indices. The main findings were as follows: 1) the concomitant adenocarcinomas, present in 75% of cases, were always molecularly associated with the intraductal components. These data definitively establish ITPN as origin of invasive pancreatic adenocarcinoma; 2) alterations restricted to infiltrative components included mutations in chromatin remodeling genes ARID2, ASXL1, and PBRM1, and ERBB2-P3H4 fusion; 3) pancreatic ITPN can arise in the context of genetic syndromes, such as BRCA-germline and Peutz–Jeghers syndrome; 4) mutational profile: mutations in the classical PDAC drivers are present, but less frequently, in pancreatic ITPN; 5) novel genomic alterations were observed, including amplification of the Cyclin and NOTCH family genes and ERBB2, fusions involving RET and ERBB2, and RB1 disruptive variation; 6) chromosomal alterations: the most common was 1q gain (75% of cases); 7) by transcriptome analysis, ITPN-associated adenocarcinomas clustered into three subtypes that correlate with the activation of signaling mechanism pathways and tumor microenvironment, displaying squamous features in their majority; and 8) TP53 mutational status is a marker for adverse prognosis. ITPNs are precursor lesions of pancreatic cancer with a high malignant transformation risk. A personalized approach for patients with ITPN should recognize that such neoplasms could arise in the context of genetic syndromes. BRCA alterations, ERBB2 and RET fusions, and ERBB2 amplification are novel targets in precision oncology. The TP53 mutation status can be used as a prognostic biomarker.
Similar content being viewed by others

Introduction
Pancreatic intraductal tubulopapillary neoplasm (ITPN) is recognized as a subtype of pancreatic neoplasms that form a heterogeneous group of intraductal lesions, which also includes intraductal papillary mucinous neoplasm (IPMN) and intraductal oncocytic papillary neoplasm (IOPN)1. ITPN accounts for up to 3–5% of all intraductal pancreatic neoplasms1,2,3,4.
Similar to IPMN, ITPN shows various intraductal growth degrees. However, compared to IPMN, ITPN is less frequently cystic, forming instead fleshy and solid masses in the involved ducts4,5. Histologically, ITPNs are hypercellular tumors comprising nodules of back-to-back tubular glands with absent or very scant mucin formation1,3,6,7,8. The tubular areas are predominant, whereas papillary components are limited. In addition to architectural complexity, ITPN displays uniform high-grade cytological atypia with numerous mitotic figures and frequent foci of necrosis. Intra-cytoplasmic and extra-cellular mucins are consistently absent4,6,7,8.
Pancreatic ITPN is a presumed precursor of invasive ductal adenocarcinoma, although definitive evidence is still lacking. Concomitant adenocarcinomas have been reported in up to 70% of cases at diagnosis8. Despite the high-grade cytological and architectural features and the frequent association with concomitant invasive cancer, ITPN usually has a more favorable prognosis than conventional pancreatic ductal adenocarcinoma (PDAC), even when associated infiltrative lesions are present. However, a small subset of patients presents with locally advanced or metastatic disease at diagnosis or will develop local recurrence or distant metastases after surgical resection; thus, better comprehension of this lesion type is warranted.
ITPN has a distinct mucin immunohistochemical profile, rendering immunohistochemistry (IHC) an important supportive tool in the ITPN diagnosis. ITPNs are usually characterized by the expression of MUC1 and MUC6 and generally lack expression of the MUC5AC and MUC2 proteins1,7. Moreover, pancreatic ITPN is molecularly distinct from IPMN and conventional ductal adenocarcinoma, showing rare (but not absent) mutations in the KRAS and TP53 genes and more common PI3KCA mutations and FGFR2 fusions9,10,11,12,13.
In the present study, we performed a multi-institutional analysis of the molecular profile of different ITPN components (tubular and papillary areas) and concomitant invasive cancers through histology-driven multi-regional sequencing. This study aimed to clarify the genomic features of pancreatic ITPN, including tumor heterogeneity and the molecular progression to invasive cancers. Based on the results of our analyses, we provide specific insights into molecular markers with clinical impact and suggest possible novel targets for precision oncology.
Materials and methods
Case selection and clinicopathologic analysis
The following electronic databases were searched for pancreatic ITPN cases: Verona University and Hospital Trust (Verona, Italy), National Cancer Center Research Institute (Tokyo, Japan), Asan Medical Center (Seoul, South Korea), University Medical Center (Utrecht, The Netherlands), and Indiana University (Indianapolis, IN, USA). Cases with material available for molecular analysis were selected. Our cohort comprised 16 cases, which were subsequently confirmed by histology performed by two pancreatic pathologists. All cases were negative for BCL10, chromogranin A, and synaptophysin. Medical records and electronic databases were used to obtain supplementary clinicopathologic data, including prognostic outcomes. Cases were staged using the American Joint Committee on Cancer staging, 8th edition14.
Multi-regional massive parallel DNA sequencing
To understand better tumor heterogeneity and evolution, a multi-regional sequencing approach for genomic analysis was adopted. The most representative inclusion from each case were selected for analysis. The tubular area and the papillary region for the 16 ITPNs were then selected. Co-occurring adenocarcinomas, when present, were also analyzed. Genomic DNA was obtained from formalin-fixed, paraffin-embedded tissues after enrichment for neoplastic cellularity, using manual microdissection. DNA was extracted and quantified as previously described15, using the GeneRead DNA FFPE kit (Qiagen - Hilden, Germany) according to the manufacturer’s instructions.
DNA sequencing was performed for both tubular and papillary tumor components, following the previously described SureSelectXT HS CD Glasgow Cancer Core assay (www.agilent.com), hereafter referred to as CORE16,17. The CORE panel spans 1.8 Mb of the genome and searches 174 genes for somatic mutations, copy number alterations, and structural rearrangements. The details of the targeted genes are reported in Supplementary Table 1. Sequencing was performed on a NextSeq 500 (Illumina, San Diego, CA, USA) loaded with two captured library pools using a high-output flow cell and 2 × 75 bp paired-end sequencing.
CORE panel analysis started with demultiplexing performed with FASTQ Generation v1.0.0 on the BaseSpace Sequence Hub (https://basespace.illumina.com, last access 11/16/2021). Forward and reverse reads from each demultiplexed sample were aligned to the human reference genome (version hg38/GRCh38) using Burrows-Wheeler Aligner version 0.7.17-r118818. Mapped reads were subjected to PCR duplication removal and indexed, using biobambam2 v2.0.146 (https://gitlab.com/german.tischler/biobambam2.git; last access 11/16/2021)19. Coverage statistics were calculated using the same software20. Single nucleotide variants were identified using shearwater21. Small (<200 bp) insertions and deletions were identified using Pindel version 0.2.5b822. All candidate mutations were manually reviewed using the Integrative Genomics Viewer version 2.4 to exclude sequencing artifacts23.
Microsatellite instability was calculated using the method described by Papke et al.24. Copy number alterations of targeted genes were detected using the GeneCN software (https://github.com/wwcrc/geneCN; last access 06/30/2021). Structural rearrangements were detected using the BRASS software25, and visually reviewed using the Integrative Genomics Viewer, version 2.423.
Tumor variants were classified as benign (class 1), likely benign (class 2), variant of uncertain significance (class 3), likely pathogenic (class 4), or pathogenic (class 5), according to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology26.
Transcriptome analysis
Gene-expression analysis of 20,815 human genes was performed on the co-occurring adenocarcinomas to obtain their transcriptomic profile, according to previously described methods27. Briefly, libraries were prepared using the Ampliseq Transcriptome Human Gene Expression Kit (Thermo Fisher Scientific, Waltham, MA, USA) with 1 µg of retrotranscribed RNA for each multiplex PCR amplification. The AmpliSeqRNA plugin generated each sample’s expression data (counts per transcript). Counts were normalized and transformed using the DESeq2 package for R28. Visualization and clustering were performed using the ComplexHeatmap package for R29. The NbClust package was adopted to estimate the best number of clusters. Then, a hybrid hierarchical k-means approach was used to perform principal component analysis and to design a dendrogram showing the relationships between samples. To verify the resulting associations between samples, unsupervised consensus clustering was performed using ConsensusClusterPlus. For tumor classification, pancreatic cancer signatures were retrieved from studies performed by Bailey et al.30, Collisson et al.31, and Moffitt et al.32, and cluster-specific enriched gene sets were determined using the normalized count matrix. We applied gene set enrichment analysis (GSEA) using the GAGE-R package between clusters to obtain significant pairwise up- and down-regulated pathways33. We performed z-score normalization of pathway scores in each cluster.
Chromogenic multiplex IHC and additional IHC
Adenocarcinoma gene expression profiling related to immune microenvironment composition was cross-validated using chromogenic multiplex IHC analysis as previously described27. Based on the results of the transcriptome analysis, two T-lymphocyte markers, CD4 (labeled in red) and CD8 (DAB), and the class 2 macrophage marker CD163 (green) were selected for this study. Cells were considered “positive” when the cell membrane was stained. The expression of these markers was evaluated as previously reported, using a semi-quantitative (0–5) scoring system: 0 = negative (no stained cells), 1 = rare (1–10 positive cells per high-power field, HPF; 400× magnification), 2 = low (11–20 positive cells per HPF), 3 = moderate (21–30 positive cells per HPF), 4 = high (31–50 positive cells per HPF), and 5 = very high (>50 positive cells per HPF)27.
In the case of ERBB2 amplification, a specific IHC analysis for Her2 (Hercep test, Dako, Germany) was performed. Finally, all cases were tested for p53 with IHC (clone: DO-7, 1:50 dilution, Novocastra, UK).
Survival analysis
Univariate and multivariate Cox regression analyses were performed to investigate any association between clinicopathologic and molecular data, and survival outcomes. The outcomes considered were overall survival, cancer-specific survival, disease-free survival, and composite outcome. Multivariable analysis was planned using the factors significantly associated with the survival outcomes of interest with a p-value < 0.10 in the univariate analyses. Data from the Cox regression analyses were graphically reported using Kaplan–Meier curves. The results were presented as hazard ratios with a 95% confidence interval. Statistical analyses were performed using SPSS version 20.0 (Chicago, IL, USA).
Results
Clinicopathologic analysis
The crucial clinicopathological features of the 16 cases are summarized in Table 1. Five patients were men (31.2%) and 11 were women (68.8%), with an average age at diagnosis of 63.2 years (range 47–76). Three cases (18.8%) were incidentally diagnosed in asymptomatic individuals; of these, two were diagnosed during routine follow-up for genetic syndromes, such as hereditary breast and ovarian cancer syndrome (HBOC) and Peutz–Jeghers syndrome.
At diagnosis, co-occurring invasive adenocarcinoma was present in 12 cases (75%), represented by glandular/tubular adenocarcinoma. Regarding tumor stage, four cases (25%) were resected at stage 0 (i.e., non-invasive), four (25%) at stage I, four (25%) at stage II, three (18.8%) at stage III, and one case (6.2%) at stage IV due to the presence of a single liver metastasis.
Follow-up data were available for 15/16 patients. The majority (10, 62.5%) were alive and disease-free at the last follow-up (average follow-up time: 27.9 months). Two pancreatic lesions were analyzed in one patient; an initial lesion during surgical resection for a non-invasive ITPN (case #7a), and a later lesion during local adenocarcinoma tumor relapse (case #7b), observed 38 months after the surgical resection. After surgical re-intervention for relapse, the patient remains alive and disease-free at the most recent follow-up (8 months after re-intervention).
Molecular analysis
Multiregional massive parallel sequencing
All cases were investigated using multi-regional sequencing to assess their genomic profiles. In the single metastatic case, we investigated the invasive pancreatic adenocarcinoma and the liver metastasis in addition to the papillary and tubular intraductal components. Thus, we provided the molecular characterization of tubular and of papillary intraductal components of six cases, whereas in the remaining 10 cases, the co-occurring adenocarcinoma was also investigated (Fig. 1). For two cases (#1 and #3), the invasive component was not suitable for molecular analysis. The mutational profiles and copy number variations are summarized in Table 2 and structural alterations are shown in Table 3.

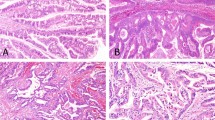
Histological images of the tumor areas selected for multi-regional sequencing: (A) tubular component; (B) papillary component; (C) adenocarcinoma. Hematoxylin-eosin staining at 10× magnification for observation of structures. The graphs show the results of the genomic analysis of all patients at diagnosis, represented per tumor component. Each case is identified with a number followed by an acronym, indicating the specific tumor region (TUB tubular, PAP papillary, AC adenocarcinoma).
Sequencing revealed recurrent mutations in the classical PDAC drivers: KRAS mutations in four cases (25%), in both the ITPN and the concomitant infiltrating adenocarcinoma; TP53 mutations in four cases (25%), three of which had a co-occurring adenocarcinoma; SMAD4 mutations in two cases (12.5%), restricted to the tubular area and not altered in either the papillary or the adenocarcinoma, the latter present in only one case; BRAF was mutated in two cases (12.5%), both displaying the same V600E mutation; RNF43 was mutated only in the papillary component of a noninvasive case; and no mutations were detected in CDKN2A or GNAS in any of the cases. None of the ITPN samples showed microsatellite instability. The four cases harboring TP53 mutations showed aberrant staining pattern in p53 IHC, with strong and diffuse nuclear positivity of >90% tumor cells (Supplementary Fig. 1). Other cases were interpreted as wild type.
Two ITPNs associated with concomitant adenocarcinoma (case #9 and #14) were detected as part of the spectrum of familial cancer syndromes. One case was diagnosed in a patient with Peutz–Jeghers syndrome; the germline variation was associated with LOH of STK11. The other case was detected in a patient with HBOC syndrome carrying a BRCA2 germline variation coupled with LOH on chromosome 13.
Copy number variations of the Cyclin family genes were noted in three cases (18.75% of cases), in particular, CCNE1 was amplified in two cases (12.5%) and CCND3 in one case (6.25%). Gene gain/amplification is frequently observed in the NOTCH and FGFR families in ITPN. Here, alterations in FGFR involved two cases with gene gain (12.5%) and one with amplification (6.25%), and in NOTCH, two cases with gain (12.5%) and two with amplification (12.5%). We also observed NTRK1 amplification in all the components in two cases (12.5%); one was the relapsing case and the alteration was maintained in the recurrent neoplasm. Finally, ERBB2 was amplified in all the components of one ITPN sample with concomitant adenocarcinoma. In IHC staining, Her2 expression showed a heterogeneous pattern, from a weak to a focally strong positivity (although there are no specific guidelines for assessing Her2 in pancreatic tumors; Supplementary Fig. 2).
Six cases harbored structural genomic alterations (Table 3). Among these, five showed gene fusions and one showed translocation. RET was fused with C14orf93 or TRIM24, FGFR2 with STCP1 or HSD17B4, and ERBB2 with P3H4. Translocation by asymmetric breakdown and repair of chromosome 1 in LMNA (1q22) and chromosome 13 in RB1 (13q14.2) resulted in dicentric and acentric chromosomes, respectively, containing the distal parts of the q arms welded together. This generated truncated proteins with 3′-3′ and 5′-5′ junctions, resulting in loss of function.
Altogether, the genomic profiling data, including mutations, variants of unknown significance (Supplementary Table 2), copy number variants, and gene fusion, clarified that the co-occurring adenocarcinomas were derived from the intraductal precursors and shared with them the majority of somatic alterations. Three cases harbored additional alterations restricted to the invasive components, such as mutations affecting the chromatin remodeling genes ARID2, ASXL1, and PBRM1, observed in three cases, and the ERBB2-P3H4 fusion in the case with ASXL1 mutation.
Chromosomal alterations were observed in all samples (Fig. 2). Chromosomal gains were detected in 15 cases (93.7%), whereas chromosomal loss was observed in all cases. The most common alterations were 1q gain, detected in 12 cases (75%), and 1p, 6q, or 18q losses found in 8 (50%), 9 (56.2%), and 9 (56.2%) cases, respectively. Loss of heterozygosity (LOH) followed by reduplication, leading to copy-neutral LOH or LOH with additional gain, was observed in 9 (56.2%) cases.

In this figure, chromosomes are presented in increasing order. Note: for chromosomal alterations, the adenocarcinoma of case #14 was not analyzed due to the low cellularity of the sample.
Transcriptome analysis
Overall, eight ITPN-concomitant adenocarcinomas were investigated using transcriptome analyses (Fig. 3). A hybrid hierarchical k-means approach (k = 3) was used to perform principal component analysis and design a dendrogram showing the relationships between samples. The resulting consensus matrix obtained from the unsupervised consensus clustering confirmed the associations obtained by the principal component analysis and the dendrogram.

Upper panel: gene expression heatmap stratified by the three consensus clusters (A, B, and C) derived from the transcriptome analysis of the cohort adenocarcinomas. Annotations for clinicopathologic variables are also provided. Lower panel: genomic alterations for essential tumor-related genes found in all three clusters.
The three identified clusters, A, B, and C (Fig. 2), included four and three samples, and one sample, respectively. Pairwise differential expression analysis was performed for all identified clusters. Cluster A showed 21 differentially expressed (DE) genes (Supplementary Table 3), in which no genes with cluster-based statistical significance were identified in clusters B and C. Comparison with the mutational analysis results showed that cluster A was enriched with 3 cases (75%) containing KRAS mutations and ARID2/PBMR1 chromatin remodeling. Cluster B included one case (33%) with a BRAF mutation. The single case in cluster C showed a BRCA alteration (germline BRCA2 mutation coupled with LOH).
The comparison between the expression profiles of each cluster and current PDAC classifiers highlighted that cluster A showed squamous-like signatures, similar to Moffitt’s basal-like and active stroma profiles and Collisson’s quasi-mesenchymal profile. In contrast, cluster C showed features of the classical pancreatic subtype similar to the exocrine profile. No statistically significant associations were identified for cluster B; nonetheless, we noted a trend for similarity with Moffitt’s basal-like subgroup (squamous-like profile) (Fig. 4A). Furthermore, using the GSEA-based approach, we identified differential biological processes among the three clusters. Based on the z-score, cluster A showed enrichment for induction of the epithelial-to-mesenchymal (EMT) pathway and KRAS signaling. In contrast, cluster B presented enrichment for the phosphatase and tensin homolog (PTEN) regulation pathways, whereas NOTCH signaling was enriched in cluster C (Fig. 4B).

A Heatmap showing similar statistically significant transcriptomic profiles among the clusters identified in the current study with the existing molecular subgroups of pancreatic ductal adenocarcinoma (Moffit’s, Collisson’s, and Bailey’s subgroups); B Activation of different biological mechanisms in the three clusters. Statistically significant values are shown. C Heatmap of the immune subpopulations inferred by gene expression of immune-related metagenes significantly enriched in any of the three clusters.
Using deconvolution analysis of the different clusters, statistically significant differences in immune cell populations were identified. Cluster A showed enrichment in CD8+ T-cells, M1-class macrophages, and cancer-associated fibroblasts (CAFs). Cluster B was enriched in CD4+ T-cells and M2-class macrophages, while cluster C was enriched in inflammatory cells implicated in the innate immune response (Fig. 4C).
Chromogenic multiplex IHC for CD4, CD8, and CD163 confirmed these findings, showing predominant CD8+ T-lymphocyte infiltration in cluster A (mean scores: CD8 = 3.8; CD4 = 1.6; CD163 = 1.2), predominance of class 2 macrophages and CD4+ T-lymphocytes in cluster B (mean scores: CD8 = 1.2; CD4 = 3.4; CD163 = 4.2), and low presence of cells positive for these markers in cluster C (mean scores: CD8 = 1.2; CD4 = 1.2; macrophages = 0.8).
Integrative multi-regional genomic and transcriptome analysis of ITPN, adenocarcinoma, and liver metastasis
Integrative genomic and transcriptome analyses were performed in the case of metastatic ITPN; in particular, on the tubular and papillary components of ITPN, concomitant pancreatic adenocarcinoma, and liver metastasis. The genomic analysis showed the presence of a truncating mutation in PBRM1 in the pancreatic adenocarcinoma. In the transcriptome analysis, statistical analysis showed no DE genes between the tubular and papillary components. By comparison, up-regulation of 15 genes and down-regulation of 8 genes were detected in the adenocarcinoma (Supplementary Table 3). The case with the liver metastasis had an even more variable profile: 135 DE genes (113 up-regulated and 22 down-regulated) between the metastatic and intraductal areas and 156 DE genes (103 up-regulated and 53 down-regulated) between the primary and metastatic adenocarcinoma were detected (Supplementary Table 3). On the basis of the highest and statistically significant values of correlation to the current PDAC signatures (Supplementary Fig. 3), the tubular and papillary components were very similar and showed a classical pancreatic profile (Collisson’s classical)31. In contrast, the adenocarcinoma showed features of the squamous profile, with positive enrichment for Collisson’s quasi-mesenchymal subtype31. By comparison, the transcriptomic profile of liver metastasis showed classical pancreatic features, with positive correlation with Bailey’s immunogenic profile30.
Survival analysis
In the survival analysis, the only parameter that showed statistically significant association with prognostic outcomes was the TP53 mutation, associated with an increased risk of death or recurrence (hazard ratio = 10.359, 95% confidence interval 1.911–117.776, p = 0.039; Fig. 5). The statistical significance of the TP53 mutation was maintained in the cases with concomitant adenocarcinoma (hazard ratio = 9.569, 95% confidence interval 1.861–106.371, p = 0.046).

Kaplan–Meier curves show patients’ survival in relation to the presence of TP53 mutation (blue: TP53 wild type; green: mutated TP53).
Discussion
In this study, we performed a comprehensive characterization of pancreatic ITPN and concomitant invasive adenocarcinoma in 16 cases. Below, we summarize our major findings. 1) Clinicopathologic features: concomitant adenocarcinoma was present in 75% of cases, represented by glandular/tubular adenocarcinomas; 2) ITPN as a precursor of pancreatic cancer: at the molecular level, the co-occurring adenocarcinoma was always associated with pancreatic intraductal components, establishing ITPN as a definitive precursor of pancreatic cancer; 3) tumor progression: mutations of chromatin remodeling genes represented a late event during ITPN oncogenesis. Indeed, mutations affecting such genes have been detected only in the invasive component of three different cases; 4) clinical genetics: ITPN can arise in the context of genetic syndromes, such as HBOC and Peutz–Jeghers, with direct implications for screening, therapy and genetic counseling; 5) mutational profile: mutations in the classical PDAC drivers are less frequent in pancreatic ITPN; 6) copy number variation: recurrent amplifications were observed for the Cyclin (3/16 cases, 18.75%) and NOTCH family genes (2/16 cases, 12.5%), whereas ERBB2, a potential target for molecular-based therapies, was amplified in one case; 7) chromosomal alterations: the most commonly observed were 1q gain (75% of cases) and 1p, 6q or 18q loss (approximately 50% of cases); 8) structural variations: common fusions involved the recently identified RET and FGFR2; 9) transcriptome analysis of ITPN-associated adenocarcinoma: three different clusters were identified, with the majority of cases displaying squamous-like features, differential activation of EMT, KRAS-signaling, and PTEN pathways, and variable immune microenvironment composition; and 10) survival analysis: the TP53 mutational status emerged as a hallmark of adverse prognosis.
At the molecular level, a critical finding emerged from the comparative analysis between intraductal components and concomitant adenocarcinoma: invasive cancers were present in 75% of cases and were always molecularly associated with intraductal components. Indeed, in our case series, all ITPN and co-occurring adenocarcinomas shared most of the genomic alterations. These data provide definitive evidence of ITPN as origin of invasive pancreatic adenocarcinoma. By contrast, a previous study found that co-occurring IPMN and adenocarcinomas were independent (i.e., not molecularly associated) in approximately 20% of cases34,35. Interestingly, we found that acquisition of the invasive phenotype in ITPN was always accompanied by alterations in the infiltrative lesion.
Mutations in the chromatin remodeling ARID2, ASXL1 or PBRM1 were observed only in the invasive component of three different cases. Alterations in the same class of genes have also been reported in the biliary counterpart of these neoplasms36; overall, present and previous findings suggest a potential role of this gene class in tumor progression and invasion. Alterations in chromatin remodeling genes have also been reported in the most comprehensive report published to-date on the molecular landscape of pancreatic ITPN13. Chromosome 1p loss and 1q gain in the majority of cases are additional common findings. However, some differences between the two studies are evident. First, Basturk et al.13 found chromatin remodeling genes with mutations or amplifications in a substantial subset of their cases (30–40% of cases); alterations were relatively rare in our study (approximately 20%). Second, in the earlier study, alterations were commonly detected in MLL; no such alterations were observed in our material. Nonetheless, the picture that emerges from these studies confirms that chromosomal alterations and mutations in chromatin remodeling genes are important components in the ITPN molecular landscape, with a potential role in acquiring invasiveness.
This study is the first to report that pancreatic ITPN can arise as part of the spectrum of genetic syndromes, a finding confirmed by molecular analysis. In our cases, neoplasms arose in the context of HBOC syndrome due to BRCA2 alteration and in the context of Peutz–Jeghers syndrome. Both neoplasms had infiltrative components. These findings have immediate implications for tumor screening and genetic counseling for patients with pancreatic ITPN and may influence clinical management (e.g., platinum-based chemotherapy and PARP-inhibitors for BRCA-tumors)37. These results emphasize the importance of a thorough anamnesis, including family history of cancer, of all patients presenting with pancreatic ITPN.
The present study confirmed the genomic distinctiveness of ITPN by showing that typical PDAC drivers, including KRAS, TP53, SMAD4, and CDKN2A, are less frequently altered in this lesion in comparison with conventional PDAC. Alterations in PDAC drivers, at similar or lower frequency, have already been reported in previous studies of pancreatic ITPNs38. The relative paucity of PDAC alterations in this case series highlights the molecular differences with conventional PDAC, but it should be acknowledged that KRAS alterations are still present in a not-negligible subset of cases (4/16 cases in this series, 25%). This indicates that pancreatic ITPN cannot be considered as a KRAS-independent entity, also taking into account that MAPK-pathway can be activated in this tumor type also through BRAF alterations (case #7).
Although the genomic landscape of pancreatic ITPN appears largely heterogeneous, notable common events are represented by gene amplification and fusion. Recurrent amplifications were found in genes of the Cyclin and NOTCH families. Amplification in ERBB2 in these neoplasms represents a novel finding and a potential target for precision oncology. It should be noted that ERBB2 is considered one of the most important targets for tailored treatments in breast and gastric cancer39, and our findings suggest new promising perspectives in treatment strategies for pancreatic cancer. Gene fusions commonly involved RET and FGFR2. Fusions involving FGFR2 have already been reported in two pancreatic ITPNs13 but with different partners from those reported here. Conversely, fusions involving RET represent a novel finding in pancreatic ITPN, detected here in two cases. Furthermore, we reported a novel ERBB2-P3H4 fusion and a newly established translocation involving LMNA and RB1, resulting in gene loss. All these detected rearrangements should be considered in molecular-based therapies, already approved for other cancer types40,41,42. The new molecular targets merit particular consideration as potential therapy targets in patients with ITPN-associated pancreatic cancer, especially in the metastatic setting.
Unsupervised clustering of DE genes in ITPN-associated adenocarcinomas identified three different clusters; however, the analysis at this stage should be considered exploratory due to the small sample size. Cluster A showed activation of KRAS signaling and EMT, and displayed squamous features, and enrichment in CD8+ T-cells, M1-class macrophages, and CAFs. Cluster B showed positive correlation with PTEN regulation, similar features to the PDAC squamous-like subgroup, and was enriched with CD4+ T-cells and M2-class macrophages. Cluster C showed activation of NOTCH signaling and a transcriptomic profile toward classical-pancreatic features. Although most cases displayed squamous features, the tumor microenvironment and biological processes activated in the tumors showed substantial differences. These aspects highlight the heterogeneity of tumor microenvironment in pancreatic ITPN and should be considered in future studies to indicate personalized therapeutic approaches43. Along these lines, a recent study demonstrated that concurrent loss of Arid1a and Pten in adult pancreatic ductal cells induced ITPN and ITPN-derived PDAC in mice44. In our cohort, the majority of cases studied by transcriptome analysis did show enrichment in the activation of EMT and PTEN-regulation pathway. Moreover, an ARID-gene mutation was detected in the invasive component in one case. Overall, our study extend results from animal studies to human disease and confirms the role of PTEN and ARID in pancreatic ITPN and associated cancers.
Interestingly, the analysis of a primary ITPN coupled with invasive and metastatic sites highlighted that the pancreatic transcriptional program can be plastic across different tumor stages. Despite genomic relatedness, the intraductal components featured the classical pancreatic subtype, whereas squamous-like characteristics were presented in the invasive adenocarcinoma and classical-pancreatic features in distant metastasis. This finding can be best appreciated in view of recent pioneering studies that found evidence of subtype switching during tumor progression45,46,47. Although the mechanism in PDAC is still not fully understood, our initial analysis of a pancreatic ITPN case and the associated primary and metastatic adenocarcinoma suggests that subtype switching may be necessary for intraductal lesions to acquire infiltrating and further metastatic capability48.
Finally, a finding that merits attention is the role of TP53 mutational status in adverse prognosis; importantly, the TP53 mutational status was maintained in the multivariable analysis that comprised cases with invasive adenocarcinoma. Association of TP53 mutations with an adverse prognosis is commonly encountered in different cancer types, including colorectal and ampullary adenocarcinomas in the gastrointestinal tract49,50. This finding may help stratify patients with ITPN at diagnosis. The TP53 mutational status could, thus, be adopted as a potential prognostic biomarker to identify high-risk lesions requiring aggressive therapeutic and surgical strategies. As demonstrated here, IHC is a valuable supportive tool for detecting TP53-mutated cases; potential applications of IHC in detecting this biomarker during routine diagnostic activity could be adopted.
It is important to acknowledge that this study has some limitations. First, the genomic analysis did not investigate the whole genome of the lesions; thus, potentially significant molecular events could have been missed. Nonetheless, the CORE panel we adopted was based on previously reported whole-genome sequencing focused on clinically relevant alterations. Furthermore, although the results of the transcriptomic analysis represent a novelty in the ITPN-context, they are based on eight cases and should be considered as exploratory rather than conclusive. We must also acknowledge that, despite the relatively small sample size, the multicenter design of the current study is a concrete answer to the difficulties of collecting large case series of rare neoplasms.
In conclusion, in this study we provided an integrative clinicopathologic and molecular characterization of a series of pancreatic ITPNs and associated adenocarcinomas. Our findings highlight that these lesions represent a distinct entity among pancreatic neoplasms. In the context of pancreatic intraductal/cystic lesions, correct identification of ITPNs is crucial given their distinctive clinicopathologic features, genomic and transcriptomic profiles, and potential for target-enrichment strategies for precision oncology.
Data availability
All data/information are available in the manuscript and in the Supplementary Material.
References
WHO Classification of Tumours Editorial Board. Digestive system tumours. International Agency for Research on Cancer; 2019.
Suda, K, Hirai, S, Matsumoto, Y, Mogaki, M, Oyama, T, Mitsui, T, et al. Variant of intraductal carcinoma (with scant mucin production) is of main pancreatic duct origin: a clinicopathological study of four patients. Am J Gastroenterol 91, 798–800 (1996).
Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of tumours of the digestive system. Lyon: IARC Press; 2010.
Yamaguchi, H, Shimizu, M, Ban, S, Koyama, I, Hatori, T, Fujita, I, et al. Intraductal tubulopapillary neoplasms of the pancreas distinct from pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol 33, 1164–1172 (2009).
Date, K, Okabayashi, T, Shima, Y, Iwata, J, Sumiyoshi, T, Kozuki, A, et al. Clinicopathological features and surgical outcomes of intraductal tubulopapillary neoplasm of the pancreas: a systematic review. Langenbecks Arch Surg 401, 439–447 (2016).
Tajiri, T, Tate, G, Kunimura, T, Inoue, K, Mitsuya, T, Yoshiba, M, et al. Histologic and immunohistochemical comparison of intraductal tubular carcinoma, intraductal papillary-mucinous carcinoma, and ductal adenocarcinoma of the pancreas. Pancreas 29, 116–122 (2004).
Kim, JY, Hong, SM. Precursor lesions of pancreatic Cancer. Oncol Res Treat 41, 603–610 (2018).
Basturk, O, Adsay, V, Askan, G, Dhall, D, Zamboni, G, Shimizu, M, et al. Intraductal tubulopapillary neoplasm of the pancreas: a clinicopathologic and immunohistochemical analysis of 33 cases. Am J Surg Pathol 41, 313–325 (2017).
Amato, E, Molin, MD, Mafficini, A, Yu, J, Malleo, G, Rusev, BC, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol 233, 217–227 (2014).
Hosoda, W, Sasaki, E, Murakami, Y, Yamao, K, Shimizu, Y, Yatabe, Y. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows Arch 466, 665–674 (2015).
Yamaguchi, H, Kuboki, Y, Hatori, T, Yamamoto, M, Shiratori, K, Kawamura, S, et al. Somatic mutations in PIK3CA and activation of AKT in intraductal tubulopapillary neoplasms of the pancreas. Am J Surg Pathol 35, 1812–1817 (2011).
Fischer, CG, Wood, LD. From somatic mutation to early detection: insights from molecular characterization of pancreatic cancer precursor lesions. J Pathol 246, 395–404 (2018).
Basturk, O, Berger, MF, Yamaguchi, H, Adsay, V, Askan, G, Bhanot, U, et al. Pancreatic intraductal tubulopapillary neoplasm is genetically distinct from intraductal papillary mucinous neoplasm and ductal adenocarcinoma. Mod Pathol 30, 1760–1772 (2017).
Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al. AJCC cancer staging manual. Eighth ed. New York: Springer; 2017.
Simbolo, M, Gottardi, M, Corbo, V, Fassan, M, Mafficini, A, Malpeli, G, et al. DNA qualification workflow for next generation sequencing of histopathological samples. PLOS ONE 8, e62692 (2013).
Beer, PA, Cooke, SL, Chang, DK, Biankin, AV. Defining the clinical genomic landscape for real-world precision oncology. Genomics 112, 5324–5330 (2020).
Lawlor, RT, Mafficini, A, Sciammarella, C, Cantù, C, Rusev, BC, Piredda, ML, et al. Genomic characterization of hepatoid tumors: context matters. Hum Pathol 118, 30–41 (2021).
Li, H, Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Tischler, G, Leonard, S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol Med 9, 13 (2014).
Li, H, Handsaker, B, Wysoker, A, Fennell, T, Ruan, J, Homer, N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Gerstung, M, Papaemmanuil, E, Campbell, PJ. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 30, 1198–1204 (2014).
Ye, K, Schulz, MH, Long, Q, Apweiler, R, Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Robinson, JT, Thorvaldsdóttir, H, Winckler, W, Guttman, M, Lander, ES, Getz, G, et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011).
Papke, DJ, Nowak, JA, Yurgelun, MB, Frieden, A, Srivastava, A, Lindeman, NI, et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod Pathol 31, 1882–1890 (2018).
Ahdesmäki, MJ, Chapman, BA, Cingolani, P, Hofmann, O, Sidoruk, A, Lai, Z, et al. Prioritisation of structural variant calls in cancer genomes. PeerJ 5, e3166 (2017).
Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Simbolo, M, Bersani, S, Vicentini, C, Taormina, SV, Ciaparrone, C, Bagante, F, et al. Molecular characterization of extrahepatic cholangiocarcinoma: perihilar and distal tumors display divergent genomic and transcriptomic profiles. Expert Opin Ther Targets 25, 1095–1105 (2021).
Love, MI, Huber, W, Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014).
Gu, Z, Eils, R, Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Bailey, P, Chang, DK, Nones, K, Johns, AL, Patch, A-M, Gingras, M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016).
Collisson, EA, Bailey, P, Chang, DK, Biankin, AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 16, 207–220 (2019).
Moffitt, RA, Marayati, R, Flate, EL, Volmar, KE, Herrera Loeza, G, Hoadley, KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47, 1168–1178 (2015).
Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinform 10, 161 (2009).
Felsenstein, M, Noë, M, Masica, DL, Hosoda, W, Chiachiano, P, Fischer, CG, et al. IPMNs with co-occurring invasive cancers: neighbours but not always relatives. Gut 67, 1652–1662 (2018).
Scarpa, A, Real, FX, Luchini, C. Genetic unrelatedness of co-occurring pancreatic adenocarcinomas and IPMNs challenges current views of clinical management. Gut 67, 1561–1563 (2018).
Gross, C, Engleitner, T, Lange, S, Weber, J, Jesinghaus, M, Konukiewitz, B, et al. Whole exome sequencing of biliary tubulopapillary neoplasms reveals common mutations in chromatin remodeling genes. Cancers 13, 2742 (2021).
Golan, T, Hammel, P. Management of BRCA mutation carriers with pancreatic adenocarcinoma. J Natl Compr Canc Netw 19, 469–473 (2021).
Paolino G, Esposito I, Hong SM, Basturk O, Mattiolo P, Kaneko T, et al. Intraductal tubulopapillary neoplasm (ITPN) of the pancreas: a distinct entity among pancreatic tumors. Histopathology (2022). https://doi.org/10.1111/his.14698.
Cen, S, Liu, Z, Pan, H, Han, W. Clinicopathologic features and treatment advances in cancers with HER2 alterations. Biochim Biophys Acta Rev Cancer 1876, 188605 (2021).
Weaver, A, Bossaer, JB. Fibroblast growth factor receptor (FGFR) inhibitors: a review of a novel therapeutic class. J Oncol Pharm Pr 27, 702–710 (2021).
Belli, C, Penault-Llorca, F, Ladanyi, M, Normanno, N, Scoazec, J-Y, Lacroix, L, et al. ESMO recommendations on the standard methods to detect RET fusions and mutations in daily practice and clinical research. Ann Oncol 32, 337–350 (2021).
Kim, J, Bradford, D, Larkins, E, Pai-Scherf, LH, Chatterjee, S, Mishra-Kalyani, PS, et al. FDA approval summary: pralsetinib for the treatment of lung and thyroid cancers with RET gene mutations or fusions. Clin Cancer Res 27, 5452–5456 (2021).
Raghavan, S, Winter, PS, Navia, AW, Williams, HL, DenAdel, A, Lowder, KE, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119–6137.e26 (2021).
Fukunaga, Y, Fukuda, A, Omatsu, M, Namikawa, M, Sono, M, Masuda, T, et al. Loss of Arid1a and Pten in pancreatic ductal cells induces intraductal tubulopapillary neoplasm via the YAP/TAZ pathway. Gastroenterology S0016-5085, 00432-2 (2022).
Miyabayashi, K, Baker, LA, Deschênes, A, Traub, B, Caligiuri, G, Plenker, D, et al. Intraductal transplantation models of human pancreatic ductal adenocarcinoma reveal progressive transition of molecular subtypes. Cancer Disco 10, 1566–1589 (2020).
Pickering, KA, Morton, JP. Environment influences tumor progression and transcriptional subtype in a new model of pancreatic cancer. Cancer Disco 10, 1448–1450 (2020).
Chan-Seng-Yue, M, Kim, JC, Wilson, GW, Ng, K, Flores Figueroa, E, O’Kane, GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 52, 231–240 (2020).
Malinova, A, Veghini, L, Real, FX, Corbo, V. Cell lineage infidelity in PDAC progression and therapy resistance. Front Cell Dev Biol 9, 795251 (2021).
Mafficini, A, Amato, E, Cataldo, I, Rusev, BC, Bertoncello, L, Corbo, V, et al. Ampulla of Vater carcinoma: sequencing analysis identifies TP53 status as a novel independent prognostic factor and potentially actionable ERBB, PI3K, and WNT pathways gene mutations. Ann Surg 267, 149–156 (2018).
Pan, M, Jiang, C, Tse, P, Achacoso, N, Alexeeff, S, Solorzano, AV, et al. TP53 gain-of-function and non-gain-of-function mutations are differentially associated with sidedness-dependent prognosis in metastatic colorectal cancer. J Clin Oncol 40, 171–179 (2022).
Funding
This study is supported by Associazione Italiana Ricerca sul Cancro (AIRC IG n. 26343); Fondazione Cariverona: Oncology Biobank Project “Antonio Schiavi” (prot. 203885/2017); Fondazione Italiana Malattie Pancreas (FIMP-Ministero Salute J38D19000690001); Italian Ministry of Health (RF CO-2019-12369662: CUP: B39C21000370001).
Author information
Authors and Affiliations
Contributions
CL: study conception and design; TS, S-MH, LAB, LC, GMar, GMal, AP, RS, NH, CJ, AS, CL: provided original material for the study; TS, S-MH, LAB, LC, GMar, GMal, AP, RS, NH, PM, CJ, MM, AS, CL: clinical analysis; TS, S-MH, LAB, LC, NH, VA, AS, CL: histological analysis; AM, MS, DA, CS, CC, RTL, AS, CL: molecular analysis; all authors: data elaboration, discussion and interpretation; AM, MS, CL: paper writing; all authors: final editing and approval of the present version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
This study has been approved by the Verona Ethics Committee, date of approval: 04–08-2020, project 2610-CESC, code: MN-2019.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mafficini, A., Simbolo, M., Shibata, T. et al. Integrative characterization of intraductal tubulopapillary neoplasm (ITPN) of the pancreas and associated invasive adenocarcinoma. Mod Pathol 35, 1929–1943 (2022). https://doi.org/10.1038/s41379-022-01143-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01143-2
