
Abstract
Neuroendocrine carcinomas (NEC) of the breast are exceedingly rare tumors, which are classified in the WHO system as small cell (SCNEC) and large cell (LCNEC) carcinoma based on indistinguishable features from their lung counterparts. In contrast to lung and enteropancreatic NEC, the genomics of breast NEC have not been well-characterized. In this study, we examined the clinicopathologic, immunohistochemical, and genetic features of 13 breast NEC (7 SCNEC, 4 LCNEC, 2 NEC with ambiguous small versus large cell morphology [ANEC]). Co-alterations of TP53 and RB1 were identified in 86% (6/7) SCNEC, 100% (2/2) ANEC, and 50% (2/4) LCNEC. The one SCNEC without TP53/RB1 alteration had other p53 pathway aberrations (MDM2 and MDM4 amplification) and was immunohistochemically RB negative. PIK3CA/PTEN pathway alterations and ZNF703 amplifications were each identified in 46% (6/13) NEC. Two tumors (1 SCNEC, 1 LCNEC) were CDH1 mutated. By immunohistochemistry, 100% SCNEC (6/6) and ANEC (2/2) and 50% (2/4) LCNEC (83% NEC) showed RB loss, compared to 0% (0/8) grade 3 neuroendocrine tumors (NET) (p < 0.001) and 38% (36/95) grade 3 invasive ductal carcinomas of no special type (IDC-NST) (p = 0.004). NEC were also more often p53 aberrant (60% vs 0%, p = 0.013), ER negative (69% vs 0%, p = 0.005), and GATA3 negative (67% vs 0%, p = 0.013) than grade 3 NET. Two mixed NEC had IDC-NST components, and 69% (9/13) of tumors were associated with carcinoma in situ (6 neuroendocrine DCIS, 2 non-neuroendocrine DCIS, 1 non-neuroendocrine LCIS). NEC and IDC-NST components of mixed tumors were clonally related and immunophenotypically distinct, lacking ER and GATA3 expression in NEC relative to IDC-NST, with RB loss only in NEC of one ANEC. The findings provide insight into the pathogenesis of breast NEC, underscore their classification as a distinct tumor type, and highlight genetic similarities to extramammary NEC, including highly prevalent p53/RB pathway aberrations in SCNEC.
Similar content being viewed by others
Introduction
Neuroendocrine carcinomas (NEC) of the breast are rare high-grade malignancies that are poorly understood biologically and clinically. Although neuroendocrine differentiation of breast tumors has long been recognized, classification has been problematic and has continued to shift over the years, with the most recent World Health Organization (WHO) classification (5th edition) based on a consensus that terminology should be more uniform across anatomic sites1,2,3,4. Accordingly, the WHO has defined NEC of the breast as small cell neuroendocrine carcinoma (SCNEC) and large cell neuroendocrine carcinoma (LCNEC), emphasizing that these tumors are histologically and immunohistochemically indistinguishable from their respective lung counterparts2,5. SCNEC has been recognized as a distinct and clinically aggressive breast cancer subtype for many years, although most published literature is based on case reports and small series6,7,8,9. The classification of LCNEC as a separate entity in the breast is again acknowledged in the most recent WHO edition, yet debated10, and clinical and pathologic features of these rare tumors remain largely uncharacterized.
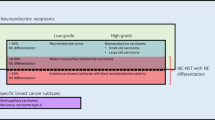
NEC of the breast should be distinguished from neuroendocrine tumors (NET), which are morphologically distinct from NEC and are usually Nottingham grade 1 or 25,11. However, it is recognized that some Nottingham grade 3 neuroendocrine neoplasms (NEN) of the breast do not resemble SCNEC or LCNEC morphologically1. These tumors lack high-grade nuclei, necrosis, and other nucleocytologic features of SCNEC or LCNEC despite areas of high mitotic activity and diffuse neuroendocrine marker expression. It is unclear if such tumors should be classified as NEC or grade 3 NET in the current WHO classification. In the enteropancreatic system, it has been well-established that high-grade (G3) NET can be challenging to differentiate from NEC histopathologically, yet these neoplasms are biologically and clinically distinct from one another12,13,14,15. Whether a similar paradigm exists for NEC and these other grade 3 NEN in the breast is unknown.
In this study, we comprehensively characterized a cohort of breast NEC (SCNEC, LCNEC, and NEC with features ambiguous between small and large cell morphology) by capture-based next-generation sequencing (NGS) and immunohistochemistry in order to identify molecular drivers and to determine if these rare aggressive tumors share pathogenetic features with NEC arising in other sites. NEC and invasive ductal carcinoma of no special type (IDC-NST) components of mixed tumors were separately analyzed to assess shared clonality and immunophenotypic divergence between the components. The immunoprofiles of NEC were additionally compared to other grade 3 NEN that morphologically were not considered to meet NEC criteria (including high-grade NET), and to grade 3 IDC-NST. Our results provide novel insights into the pathogenesis of breast SCNEC and LCNEC and highlight genetic similarities to extramammary NEC, including highly prevalent p53/RB pathway aberrations in SCNEC. Overall the findings support the separate classification of breast NEC, especially SCNEC, and suggest that LCNEC may be a more heterogeneous group.
Materials and methods
Study population and tumor classification
With institutional review board approval, the pathology archives of University of California San Francisco (UCSF), Stanford University, and Kaiser Permanente (San Francisco, CA) were searched for cases of Nottingham grade 3 NEN of the breast. This was supplemented by the consultation service of one of the authors (S.J.S.); three SCNEC were described in part in a prior report6. NEC (n = 11) and NEC components of mixed NEC/IDC-NST tumors (n = 2) comprising the study population were classified as SCNEC or LCNEC based on independent review by two pulmonary pathologists experienced in the diagnosis of neuroendocrine carcinoma (A.U. and K.D.J.), who classified them based on morphologic criteria used in the lung. Two tumors with discordant classification as SCNEC or LCNEC were classified as NEC with features ambiguous for small cell versus large cell morphology (ANEC1 and ANEC2). All NEC expressed at least one neuroendocrine marker, and diffuse (>90%) staining with synaptophysin and/or chromogranin was required for LCNEC. Of these 13 tumors, eleven (6 SCNEC, 2 ANEC, and 3 LCNEC) were analyzed by DNA sequencing using the UCSF500 assay, and two (1 SCNEC, 1 LCNEC) were submitted for FoundationOne tumor-only sequencing for clinical purposes (Foundation Medicine, Cambridge, MA).
Eight Nottingham grade 3 NEN with diffuse neuroendocrine morphology and extensive (>90%) synaptophysin and/or chromogranin expression that did not show characteristic cytomorphologic features of either SCNEC or LCNEC of the lung were identified. We opted to classify these tumors as grade 3 NET for purposes of comparison to NEC (see Results for additional histologic description of these tumors). Nottingham grade 3 invasive carcinomas with less than diffuse neuroendocrine differentiation, including <90% synaptophysin and/or chromogranin expression, were classified as invasive ductal carcinomas with neuroendocrine differentiation (IDC-NED) (n = 2) or invasive lobular carcinoma with neuroendocrine differentiation, solid pattern (ILC-NED) (n = 1)16.
Clinical information was obtained from electronic medical records when available. All tumors were confirmed to be mammary in origin based on clinical history, imaging, and pathologic findings.
Capture-based next generation DNA sequencing
Matched tumor and normal tissue were selected from nine pure NEC (6 SCNEC, 1 ANEC, 2 LCNEC) and two mixed NEC/IDC-NST (ANEC2 and LCNEC1) for capture-based NGS (n = 11). For the two mixed NEC/IDC-NST tumors, DNA from NEC and IDC-NST areas was macrodissected and analyzed separately. For patients treated with chemotherapy, only pre-treatment tumor was tested by NGS. Sequencing libraries were prepared from genomic DNA extracted from punch biopsies or macrodissected unstained sections from formalin-fixed paraffin-embedded tissue. Target enrichment was performed by hybrid capture using a custom oligonucleotide library. Capture-based NGS was performed at the UCSF Clinical Cancer Genomics Laboratory, using an assay (UCSF500 panel) that targets the coding regions of 480 cancer-related genes, select introns from ~40 genes, and the TERT promoter with a total sequencing footprint of 2.8 Mb (Supplementary Table S1). Sequencing was performed on a HiSeq 2500 (Illumina, San Diego, CA). Duplicate sequencing reads were removed computationally to allow for accurate allele frequency determination and copy number calling. The analysis was based on the human reference sequence UCSC build hg19 (NCBI build 37), using the following software packages: BWA: 0.7.10-r789, Samtools: 1.1 (using htslib 1.1), Picard tools: 1.97 (1504), GATK: 2014.4-3.3.0-0-ga3711, CNVkit: 0.3.3, Pindel: 0.2.5a7, SATK: 2013.1-10- gd6fa6c3, Annovar: v2015Mar22, Freebayes: 0.9.20 and Delly: 0.5.917,18,19,20,21,22,23,24,25. Only insertions/deletions (indels) up to 100 bp in length were included in the mutational analysis. Somatic single nucleotide variants and indels were visualized and verified using Integrated Genome Viewer (Broad Institute, Cambridge, MA, USA). Tumor mutational burden was quantified, reflecting somatic synonymous and nonsynonymous single nucleotide variants and small indels in coding regions and splice sites. Genome-wide copy number analysis based on on-target and off-target reads was performed by CNVkit and Nexus Copy Number (Biodiscovery, Hawthorne, CA, USA).
Immunohistochemistry and in situ hybridization
The following antibodies were used for immunohistochemistry: synaptophysin (polyclonal, 1:100, Cell Marque, Rocklin, CA, USA), chromogranin (LK2H10, 1:4, Cell Marque), INSM1 (A-8, 1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), NSE (22C9, undiluted, Leica Biosystems), TTF1 (8G7G3/1, 1:500, DAKO, Santa Clara, CA, USA), ER (SP1, undiluted, Ventana, Tucson, AZ, USA), PR (1E2, undiluted, Ventana), HER2 (4B5, undiluted, Ventana), Ki-67 (MIB1, 1:50, DAKO), E-cadherin (HECD-1, 1:100, Invitrogen, Carlsbad, CA, USA), GATA3 (L50-823, 1:50, Biocare Medical, Concord, CA, USA), RB (G3-245, 1:100, BD Biosciences, Franklin Lakes, NJ, USA), p53 (DO7, undiluted, Leica Biosystems), ATRX (polyclonal, 1:100, Sigma, St. Louis, MO, USA), and p16 (E6H4, 1:2, Ventana). RNA in situ hybridization for human papillomavirus (HPV) was HR18 (848568, ACD Bio, Newark, CA, USA). Antigen retrieval was as follows: for synaptophysin, INSM1, NSE, Ki-67, GATA3, Rb, p53, and p16, BOND ER2 (Leica Biosystems); for chromogranin, TTF1, E-cadherin, and ATRX, BOND ER1 (Leica Biosystems); for ER, PR, and HER2, Ventana CC1 (Ventana); and for HPV, RNAprotease.
For ER, PR, and HER2, positive staining was defined according to ASCO/CAP guidelines26,27. HER2 FISH testing was performed using the Abbott Vysis Pathvysion (Des Plaines, IL, USA) FDA-cleared kit, performed per manufacturer recommendations, and scored and interpreted according to ASCO/CAP guidelines27. For synaptophysin, chromogranin, INSM1, NSE, GATA3, and TTF1, positive staining was segregated as ≥90%, 50–89%, and 1–49%. For p16, diffuse (≥90%) strong nuclear and cytoplasmic staining (i.e., “block-positive”) was considered overexpressed, patchy weak to strong cytoplasmic staining was considered normal/wild-type pattern, and lack of staining was considered negative28. For RB, patchy to diffuse nuclear staining was considered intact, and lack of nuclear staining was considered negative. For p53, diffuse (≥90%) moderate to strong nuclear staining was considered positive/aberrant, no nuclear staining was considered negative/null, and patchy heterogeneous nuclear staining was considered wild-type pattern.
Statistical analysis
Statistical analysis was performed using Fisher exact test, using a significance level of p < 0.05. The degree of interobserver agreement was quantified by kappa.
Results
Diagnostic histologic and immunophenotypic features and classification of neuroendocrine carcinomas
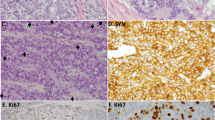
All breast NEC and NEC components of mixed NEC/IDC-NST showed morphologic features typical of poorly-differentiated NEC in the lung and were Nottingham grade 3, with high nuclear grade, high mitotic index, and poor glandular differentiation (Fig. 1 and Table 1). Individual cytomorphologic features including cell size, nuclear, nucleolar, and cytoplasmic features, tumor growth pattern, and the presence of geographic necrosis are listed in Supplementary Table S2. The tumors were independently classified as SCNEC versus LCNEC on H&E slides by two pulmonary pathologists experienced in diagnosis of neuroendocrine carcinomas. Distinction between small and large cell morphology was based primarily on nucleolar features and nuclear:cytoplasmic ratio. Morphologic assessment showed substantial agreement (κ = 0.675): both classified seven cases as SCNEC (SCNEC1-7) and four cases as LCNEC (LCNEC1-4). Disagreement was noted for two cases (ANEC1 and ANEC2), each of which showed high nuclear:cytoplasmic ratio yet variably prominent nucleoli. Although distinction between SCNEC and LCNEC for these two cases was not clear based on morphology, all pathologists agreed that each was NEC based on other histopathologic features (organoid architecture, nuclear features, geographic necrosis, and neuroendocrine marker expression). Two mixed tumors (ANEC2 and LCNEC1) demonstrated a component of high-grade IDC-NST in addition to the NEC. Two tumors (SCNEC5 and LCNEC4) exhibited a single-file growth pattern and aberrant E-cadherin expression, consistent with lobular differentiation (Supplementary Fig. S1).

Neuroendocrine carcinomas exhibited organoid, nested, trabecular, and/or sheet-like growth patterns; some tumors demonstrated circumscribed or expansile borders (A, LCNEC1; B, SCNEC1). C Geographic tumor necrosis was often present (SCNEC3). D Single-cell necrosis and apoptotic bodies were prevalent (SCNEC7). E The majority of cases showed focal in situ carcinoma with neuroendocrine features similar to the invasive tumor (SCNEC4). F Classic cytologic features were used to diagnose small cell neuroendocrine carcinoma, including high nuclear:cytoplasmic ratio, indistinct nucleoli, and nuclear molding (SCNEC4). G Rare cases demonstrated more spindled cytomorphology (SCNEC1). H Features supportive of large cell neuroendocrine carcinoma included abundant cytoplasm, distinct and more frequent nucleoli, and well-defined cellular borders (LCNEC2). I Occasional cases demonstrated scant cytoplasm yet prominent nucleoli; such NEC with mixed features were designated as ambiguous between small cell and large cell neuroendocrine carcinoma (ANEC1). Cases were positive for neuroendocrine markers by immunohistochemistry, including synaptophysin (J), chromogranin (K), and INSM1 (L) (LCNEC2).
By immunohistochemistry, 71% (9/13) of NEC were extensively (≥90% tumor cells) positive for synaptophysin (8/13, 62%) and/or chromogranin (4/13, 31%), with two others (SCNEC5 and ANEC1) showing patchy staining for both neuroendocrine markers (Figs. 1 and 2, Supplementary Table S2). Insulinoma-associated protein 1 (INSM1), an emerging nuclear marker of neuroendocrine differentiation, was expressed in seven NEC, including 4/5 (80%) SCNEC, one ANEC, and 2/4 (50%) LCNEC. By definition, all LCNEC were diffusely positive for synaptophysin (4/4, 100%) and/or chromogranin (2/4, 50%). The majority of SCNEC (4/7, 57%) were diffusely positive for synaptophysin (3/7, 43%) and/or chromogranin (2/7, 29%), with one additional case showing patchy staining for both markers. Of two SCNEC that were synaptophysin and chromogranin negative, both expressed neuron-specific enolase (NSE) diffusely, and one was patchy positive for INSM1. In mixed NEC and IDC-NST tumors, neuroendocrine markers were negative in the IDC-NST component. Nuclear TTF1 was expressed in four tumors (1 SCNEC, 1 ANEC, and 2 LCNEC), all of which had associated ductal carcinoma in situ (DCIS).

Immunohistochemical profiles of neuroendocrine carcinomas.
Diagnostic histologic and immunophenotypic features and classification of grade 3 neuroendocrine tumors
For purposes of comparison to NEC, eight Nottingham grade 3 NEN with diffuse neuroendocrine morphology and extensive (>90%) synaptophysin expression that did not show characteristic cytomorphologic features of either SCNEC or LCNEC of the lung were identified. In contrast to tumors classified as NEC, these tumors had moderate (not marked) nuclear pleomorphism and lacked necrosis, large prominent nucleoli of LCNEC, nuclear molding, and apoptosis/single-cell necrosis, although mitotic activity was high, resulting in overall Nottingham grade 3 (Fig. 3, Supplementary Table S3, and Supplementary Fig. S2). The tumors were classified as grade 3 NET.

Nottingham grade 3 breast carcinomas that were diffusely positive for synaptophysin but did not morphologically resemble SCNEC or LCNEC of the lung were designated as neuroendocrine tumors (A, NET1; B, NET6; C, NET7; D, NET3; E, NET5). These high-grade NET demonstrated intermediate grade nuclei and a high mitotic index. Some exhibited focal glandular architecture (C). Immunohistochemical profiles of NET showed a wild-type p53 pattern (F), intact RB expression (G), and diffusely positive GATA3 (H) (NET5 shown).
Clinicopathologic features of neuroendocrine carcinomas
Clinicopathologic features of the 11 pure NEC and 2 mixed NEC/IDC-NST are shown in Table 1. Patient ages ranged from 38 to 81 years (mean 61 years). The majority of tumors (9/13, 69%) were associated with an in situ component that was generally focal, including eight cases with DCIS and one E-cadherin negative SCNEC with lobular carcinoma in situ (LCIS) (SCNEC5). The in situ carcinoma had neuroendocrine differentiation in six cases (6/8 DCIS), with two DCIS and one LCIS being non-neuroendocrine. Lymphovascular invasion (5/13, 38%) and/or lymph node metastasis (6/11, 55%) were identified at presentation in 54% of cases (7/13). Of patients with clinical follow-up, 60% (6/10) developed distant metastases and 50% (5/10) died of disease (mean follow-up 21 months, range 3–67).
In most cases (7/13, 54%), NEC and NEC components of mixed NEC/IDC-NST were triple negative for ER, PR, and HER2, including 71% (5/7) SCNEC and both ANEC. One LCNEC (LCNEC2) was triple negative by immunohistochemistry but HER2 low amplified by fluorescence in situ hybridization (FISH) (ASCO/CAP group 1, HER2/CEN17 ratio 2.1). Another ER+ PR+ LCNEC (LCNEC3) was HER2 negative before neoadjuvant chemotherapy (ASCO/CAP FISH group 4) but HER2 positive (ASCO/CAP FISH group 3) after treatment. Of the two mixed NEC/IDC-NST, both NEC components were ER- (LCNEC1 was PR+) and HER2-, and both IDC-NST components were ER+ and HER2- (Table 1).
Among patients with available treatment information (12/13), all received chemotherapy (Supplementary Table S4); six of seven patients with SCNEC and one of two patients with ANEC received etoposide, an agent standardly used for small cell lung cancer but not included in guidelines for the management of breast cancer29.
No statistically significant associations were identified between SCNEC and LCNEC with respect to patient age, tumor size, lymphovascular invasion, lymph node metastasis, biomarker status, or outcomes.
Genetics of neuroendocrine carcinomas
Results of targeted DNA sequencing are shown in Fig. 4 and Supplementary Tables S5 and S6. The mean target sequencing coverage was 506 (±219) unique reads per target interval (Supplementary Table S7). Tumor mutational burden ranged from <1 to 18 mutations/megabase (median 4). No pathogenic or likely pathogenic germline alterations were identified in any cases.

Genetic profiles of neuroendocrine carcinomas.
The most frequent pathogenic aberrations were in TP53 and RB1, which were co-altered in 77% (10/13) of NEC. Of the seven SCNEC, six (86%) exhibited co-alteration of TP53 and RB1. The only SCNEC that lacked TP53 and RB1 aberrations (SCNEC7) showed amplifications of MDM2 and MDM4, which are well-known negative regulators of p5330,31,32. Despite the absence of identified RB1 alteration, this tumor was RB negative by immunohistochemistry (see below). TP53 and RB1 co-alteration was also identified in both NEC with ambiguous small versus large cell morphology (ANEC1 and 2) and in 50% (2/4) LCNEC. TP53/RB1 co-alteration was significantly more common in NEC as a group and in SCNEC than in a group of matched grade 3 IDC-NST profiled by UCSF500 assay (7/45, 16%) (p < 0.001 and p = 0.001, respectively). Similar results were found when all NEC or SCNEC were compared to grade 3 IDC-NST in the large publicly available METABRIC dataset, in which TP53/RB1 co-alteration was reported in only 3% (35/1009) tumors (p < 0.001, p < 0.001, and p = 0.007, respectively) (Supplementary Fig. S3).
Pathogenic phosphoinositide (PI)-3 kinase pathway aberrations were identified in 46% (6/13) NEC. These included homozygous PTEN loss in 57% (4/7) SCNEC but not in any LCNEC or ANEC, and activating hotspot PIK3CA mutations in 3 tumors (1 SCNEC, 1 ANEC, 1 LCNEC). ZNF703 amplifications were identified in 43% (6/13) NEC, half of which were triple negative, and included 43% (3/7) SCNEC, 50% (2/4) LCNEC, and 1 ANEC. FGFR1 was co-amplified with ZNF703 in two cases (1 SCNEC and 1 ANEC), with equivocal FGFR1 amplification in a third (SCNEC7) (Supplementary Table S6). One SCNEC and one LCNEC each had a CDH1 truncating mutation with loss of heterozygosity (LOH), consistent with the morphologic and immunophenotypic (E-cadherin loss) impression of lobular differentiation in these tumors (Fig. 4 and Supplementary Fig. S1).
Copy number analysis showed numerous chromosomal gains and losses. Recurrent gains were detected in distal 8q (9/11, 82%) and proximal 1q (7/11, 64%) and recurrent losses were observed in interstitial 22q (10/11, 91%), proximal 13q (8/11, 73%), interstitial 16q (8/11, 73%), distal 8p (8/11, 73%), and proximal 15q (6/11, 55%). No statistically significant copy number differences were identified between SCNEC and LCNEC.
Immunohistochemical expression of RB, p16, p53, and other markers in neuroendocrine carcinomas with comparison to grade 3 neuroendocrine tumors
To further explore RB and p53 pathway inactivation in NEC, immunohistochemical stains for RB, p16, and p53 were performed on tumors with available tissue (Table 2, Figs. 2 and 5). All NEC or NEC components of mixed NEC/IDC-NST harboring RB1 alterations (including frameshift and splice site mutations, deep deletions, and genomic rearrangement) were RB negative by immunohistochemistry, including all SCNEC (6/6) and ANEC (2/2), and 2/4 LCNEC. Notably, the only SCNEC lacking RB1 alteration (SCNEC7) was RB negative by immunohistochemistry. Both LCNEC without RB1 alterations showed intact RB staining. Diffuse p16 overexpression was seen in 82% (9/11) NEC (5/5 SCNEC, 2/2 ANEC, 2/4 LCNEC) and was exclusively associated with RB loss. An aberrant (null type or diffuse positive) p53 staining pattern was seen in 6/10 (60%) NEC, including 3/5 SCNEC, one ANEC, and 2/4 LCNEC. Six of seven (86%) tumors with TP53 alterations demonstrated aberrant p53 expression, whereas none of the three tumors without TP53 alterations had aberrant p53 expression. Negative p53 staining was associated with large deletion, focal homozygous deletion, and splice site mutation in TP53 (three SCNEC), whereas diffuse p53 staining was associated with missense mutations in two tumors and with the p.R342* nonsense mutation (LCNEC1), the latter of which showed nuclear as well as cytoplasmic staining33,34. Of note, TP53 p.G266E mutation has been previously shown to be associated with a non-diffuse staining pattern, as was seen in SCNEC533.

Co-alteration of RB and p53 was frequent in NEC, such as a null p53 staining pattern (A) and negative RB expression (B) in SCNEC1, and diffusely positive (overexpressed) p53 staining pattern (C) with negative RB expression (D) in ANEC1. A subset of LCNEC and all non-NEC exhibited wild-type p53 (E) and intact RB expression (F) (LCNEC4). G-J LCNEC1 demonstrated an unusual aberrant nuclear and cytoplasmic staining pattern for p53 (H) with negative RB expression (I). p16 was diffusely positive in all tumors with RB loss (J).
For comparison to NEC, immunohistochemical expression of RB, p53, and p16 was also assessed in the group of grade 3 NET (n = 8), as well as in two grade 3 IDC-NED and one grade 3 ILC-NED (Fig. 3 and Supplementary Tables S3 and S8). In contrast to the NEC, none of the other grade 3 cancers showed aberrant RB or p53 expression patterns (RB loss in 83% NEC vs 0% grade 3 NET, p = 0.001; aberrant p53 in 60% NEC vs 0% grade 3 NET, p = 0.013), and none were diffusely p16 positive (82% NEC vs 0% grade 3 NET, p = 0.002). The results were also statistically significant when only SCNEC were considered in the analysis (RB loss in 100% SCNEC vs 0% grade 3 NET, p < 0.001; aberrant p53 in 50% SCNEC vs 0% grade 3 NET, p = 0.035; diffuse p16 in 100% SCNEC vs 0% grade 3 NET, p = 0.001). No statistically significant differences in RB, p53, or p16 expression patterns were identified between LCNEC and grade 3 NET (RB loss, aberrant p53, and diffuse p16 in 50% LCNEC vs 0% grade 3 NET, p = 0.091, p = 0.091, and p = 0.109, respectively). NEC and SCNEC were also more frequently RB negative than a control group of 95 grade 3 IDC-NST enriched for triple negative carcinomas (83% NEC and 100% SCNEC vs 38% IDC-NST, p = 0.004 each).
NEC as a group and SCNEC were each more likely than grade 3 NET to be ER negative (69% NEC and 71% SCNEC vs 0% grade 3 NET; p = 0.005 and p = 0.007, respectively) and GATA3 negative (67% NEC and 67% SCNEC vs 0% grade 3 NET, p = 0.013 and p = 0.021, respectively) (Supplementary Tables S3 and S8). No statistically significant differences in ER or GATA3 expression were identified between LCNEC and grade 3 NET (ER negative in 50% LCNEC vs 0% grade 3 NET, p = 0.091; GATA3 negative in 50% LCNEC vs 0% grade 3 NET, p = 0.109). No statistically significant differences in Ki-67 proliferation index were identified between any of the groups (Fig. 2 and Supplementary Table S8). All tested NEC and other grade 3 NEN expressed ATRX (n = 12 NEC, n = 10 other NEN) and were negative for high-risk papillomavirus by in situ hybridization (n = 11 NEC, n = 9 other NEN) (Supplementary Table S8).
Genetic and immunophenotypic analysis of neuroendocrine and invasive ductal carcinoma components of mixed tumors
The neuroendocrine and IDC-NST components of two mixed NEC/IDC-NST tumors (ANEC2 and LCNEC1) were separately analyzed by immunohistochemistry and targeted DNA sequencing (Fig. 6 and Supplementary Fig. S4).

ANEC2 was comprised of mixed components of NEC and IDC-NST (A left, IDC-NST; right, NEC). B Synaptophysin highlights the NEC component. C, D The IDC-NST component shows intact RB expression (D); E, F The NEC component is RB negative (F). G ER is diffusely positive in IDC-NST (left) and negative in the NEC component (right). H GATA3 expression mirrors ER. I Chromosomal copy number plots reveal multiple gains and losses that are shared between the IDC-NST and NEC components, with additional alterations only in NEC (red arrows). J Immunohistochemical and genetic profiles of the separately analyzed components of ANEC2 and LCNEC1, summarizing features that are shared and unique between the components.
In ANEC2, the NEC and IDC-NST components shared hotspot PIK3CA and TP53 mutations and numerous chromosomal copy number changes, consistent with shared clonality between the components. LOH of the TP53 allele and focal homozygous deletion of RB1 exon 1 were exclusive to the NEC component. Consistent with this, RB protein loss and diffuse p16 expression were also restricted to the NEC component. Whereas the IDC-NST component expressed ER and GATA3, these markers were negative in the NEC areas (Fig. 6).
The LCNEC and IDC-NST components of LCNEC1 shared a duplication involving exons 3–25 of RB1 and a TP53 nonsense mutation, as well as numerous chromosomal copy number changes, again consistent with shared clonality between the components. TP53 LOH was detected only in the NEC component. By immunohistochemistry, RB and p53 were aberrant in both components. The IDC-NST component was ER+ PR- and expressed GATA3, whereas the NEC component showed the converse immunoprofile (ER- PR+ and GATA3 negative) (Fig. 6 and Supplementary Fig. S4).
Discussion
Small cell lung carcinoma (SCLC), the prototypical poorly-differentiated NEC, demonstrates near-universal biallelic inactivation of tumor suppressor genes TP53 and RB135, and frequent TP53/RB1 co-alteration has also been identified in small cell/poorly-differentiated NEC of the pancreas, prostate, bladder, and colon/rectum, and in Merkel cell carcinoma36,37,38,39,40,41,42. In contrast to SCLC, LCNEC of the lung is genetically more heterogeneous, with TP53/RB1 co-inactivation only in a subset of tumors, while those lacking these alterations harbor mutations that are more frequently seen in pulmonary adenocarcinomas43,44. In contrast to NEC, TP53 and RB1 alterations are absent or exceedingly rare in extramammary NET (including G3 NET)12,45,46,47,48,49, which instead have mutations in chromatin remodeling genes MEN1 (lung and pancreas), DAXX/ATRX (pancreas), the mTOR pathway (pancreas), and CDNK1B (small intestine)45,46,47,48,49,50,51. In comparison to extramammary NEN, a paucity of data exists for breast tumors with neuroendocrine differentiation.
SCNEC has been recognized as an aggressive type of breast cancer for many years, although most of the literature consists of case reports and small series due to the rarity of these tumors. Histochemical and ultrastructural features of SCNEC were first reported nearly two decades ago in a series of four cases, including the identification of in situ carcinoma supporting primary breast origin52. A subsequent series detailed the morphologic features and expanded immunohistochemical findings of nine breast SCNEC, including four that were mixed with non-NEC components6,9. The largest series to date comprised 19 SCNEC and identified TP53 somatic mutations in 6/8 and PIK3CA mutations in 3/9 cases using a 47-gene NGS panel53. No RB1 mutations were reported, and RB1 copy number and RB immunohistochemistry were not assessed. Although limited by the number of cases, our study offers a comprehensive characterization of the molecular landscape of SCNEC. Akin to SCNEC of the lung and other anatomic sites35,36,37,38,39,40,41,42, we identified for the first time near-universal TP53 and RB1 co-alterations in our cohort of breast SCNEC, with the sole SCNEC that lacked these alterations being RB negative by immunohistochemistry and showing p53 pathway aberrations (MDM2 and MDM4 amplifications) that are known negative regulators of p5330,31,32. These findings suggest that co-inactivation of TP53 and RB1 may be important for the small cell phenotype in the breast, as at other sites35,36,37,38,39,40,41,42. We speculate that methodological differences, notably including the detection of deep deletions, splice site mutations, and a genomic rearrangement, rather than only coding mutations, in our series, may help explain the discrepancy between the mutation prevalence in our study and the prior study that used a small targeted gene panel53.
LCNEC of the breast as defined by the most recent WHO classification is thought to be exceedingly rare and remains largely uncharacterized in terms of molecular features and clinical behavior. Although limited by a small number of cases, we show here that strictly defined LCNEC appear to be genetically heterogeneous, with some but not all harboring TP53/RB1 co-alteration. Two NEC with ambiguous or mixed small versus large cell features (ANEC) in our study also had TP53/RB1 co-alteration. The genetics of breast LCNEC thus appear to mirror the genetic heterogeneity described in pulmonary LCNEC, where some LCNEC are SCNEC-like and others are not44. Whether LCNEC of the breast with and without p53/Rb co-alterations are clinically distinct from one another and/or from other high-grade NEN of the breast will require larger follow-up studies of patients with these rare tumors.
SCNEC with mixed NEC and non-NEC components have been previously reported6. We describe here the genetic and immunohistochemical analysis of a mixed ANEC/IDC-NST and a mixed LCNEC/IDC-NST, both of which were also associated with non-neuroendocrine DCIS. Genetic analysis of separate invasive NEC and IDC-NST areas of these mixed tumors confirms a shared clonality between the components and raises the possibility that NEC could arise as a secondary event from ductal carcinoma in the breast rather than de novo from a committed neuroendocrine progenitor cell. Indeed, a native neuroendocrine progenitor cell has not been identified in the breast11,54,55. We also show that LOH of a shared TP53 mutation was restricted to the NEC components of both mixed tumors, while deep deletion of RB1 was exclusive to the NEC component in one, again suggesting a role for TP53/RB1 co-inactivation in the pathogenesis of the NEC phenotype in the breast, including at least a subset of LCNEC. On the other hand, TP53/RB1 co-inactivation is by no means exclusive to NEC in the breast and is also found in a subset of non-NEC basal-type triple negative and luminal B breast cancers56,57,58,59.
We identified amplification of ZNF703 in 46% of NEC (three SCNEC, one ANEC, and two LCNEC), half of which were triple negative and half of which were ER positive (one LCNEC was triple positive). FGFR1 was co-amplified in two of the triple negative tumors. ZNF703 encodes a zinc finger protein and estrogen-responsive transcriptional cofactor, which appears to be preferentially amplified in luminal B breast cancers and is associated with high proliferation and high histologic grade60,61. FGFR1 amplification is also common in ER-positive carcinomas and has been associated with increased grade and proliferation index62,63. The significance of ZNF703 amplifications in triple negative NEC is not known. However, we note that the IDC-NST components of both mixed NEC/IDC-NST tumors in our series were ER positive, whereas the clonally related NEC components were ER negative. Together, this raises speculation that at least some ER negative SCNEC and LCNEC may be intrinsic luminal B tumors that have lost ER expression during tumor progression, which could explain the enrichment of luminal-type alterations in these tumors. Intrinsic molecular subtyping would help to conclusively address this question in the future.
Inactivating CDH1 mutations were identified in one SCNEC and one LCNEC and corresponded to aberrant E-cadherin expression by immunohistochemistry, supportive of lobular differentiation of these tumors. Although neuroendocrine differentiation has been reported in rare lobular carcinomas, the identification of the lobular phenotype in SCNEC and LCNEC is, to the extent of our knowledge, a novel finding64,65,66.
Most previous studies of breast tumors with neuroendocrine differentiation report an association with luminal subtypes and predominantly include NET and IDC-NED using the current taxonomy; relatively few grade 3 cancers have been studied. Ang et al. identified recurrent PIK3CA (3/15 cases) and FGFR (2/15) alterations in their series, which included only three grade 3 cancers, two with no detected alterations and one which had a pathogenic HRAS mutation64. Marchiò et al. reported recurrent alterations in GATA3, FOXA1, TBX3, ARID1A, PIK3CA, AKT1, and CDH1 in a series of IDC-NED, mucinous, and solid papillary carcinomas, which included only three grade 3 cancers; FOXA1, CDH1, AKT1, and KMT2C alterations were seen in one of the latter65. Neither of these studies appeared to include SCNEC or LCNEC. A study by Lavigne et al. using 2012 (4th edition) WHO terminology included 15 grade 3 cancers, with TP53 mutations identified in two “poorly-differentiated neuroendocrine carcinomas” and one grade 2 NET67. PIK3CA mutations were identified in grade 2 and 3 NET. Wei et al. recently reported a genetic analysis of 11 NEN classified as NEC and found no TP53 or RB1 alterations in any of the tumors, although other genes in the p53 and RB pathways were mutated at 18% and 27%, respectively68. Neither of these latter two studies specified whether the analyzed tumors were SCNEC, LCNEC, or neither. Taken together, the genetics of NEC in our study are clearly distinct from those of the NET and IBC-NED reported in prior studies, supporting the separate classification of NEC. With respect to NEC included in the study by Wei et al., differences in tumor classification are likely to be at least partly responsible for apparent discrepancies from our results.
The 5th edition of the WHO classification of breast tumors defines NET as NEN with >90% neuroendocrine morphology and “extensive” neuroendocrine expression by immunohistochemistry. Defined as such, NET are usually Nottingham grade 1–2, and such tumors can be readily distinguished from NEC, which are high-grade tumors that resemble pulmonary NEC2,5,11. However, NEN with intermediate nuclear grade and high mitotic activity (Nottingham grade 3) that do not resemble pulmonary NEC morphologically are occasionally encountered in practice, although more specific diagnostic criteria to distinguish these tumors from NEC are lacking. Classification of such grade 3 NEN tumors using the WHO system is nebulous. Indeed, they are not excluded from the NET definition and can be best classified as grade 3 NET if adhering strictly to the diagnostic criteria1,11. On the other hand, it is unclear if some authors interpret the WHO classification such that all grade 3 NEN are NEC and all grade 1–2 NEN are NET68,69. In the enteropancreatic system, it has been established that NET with well-differentiated morphology but high mitotic activity and/or Ki-67 index (grade 3 NET) are genetically and clinically distinct from NEC. An analogous dichotomy of grade 3 NEN has not been established in the breast. In this study, we classified a group of uncommon Nottingham grade 3 NEN that did not morphologically resemble NEC of the lung as grade 3 NET and compared them to our cohort of NEC. None of the grade 3 NET showed aberrant RB or p53 staining patterns or diffuse p16 expression, in contrast to the high frequency seen in the NEC, including both SCNEC and LCNEC. We note that immunohistochemical expression of p53 and especially RB reliably correlated with underlying RB1 and TP53 genetic alterations in this study and others33. Strong diffuse overexpression of the cyclin-dependent kinase inhibitor p16 also correlated exclusively with RB alteration, presumably due to the known negative feedback regulation of p16 by RB, and was only seen in NEC70,71. The immunohistochemical findings thus suggest differences in the underlying genetics and biology of NEC compared to tumors classified as grade 3 NET in our study. Significantly higher frequencies of ER and GATA3 expression in grade 3 NET further support an alternate phenotype from NEC. The small number of cases in our series limits any meaningful comparative outcome analysis. However, we note that 60% of patients with NEC developed distant metastases and 50% died of disease (mean follow-up 21 months, range 3–67), whereas no such adverse events were found in patients with grade 3 NET (mean follow-up 32 months, range 3–88). Overall, additional molecular and clinical outcome studies with larger numbers of carefully classified tumors will be essential to determine whether tumors with features of grade 3 NET are biologically distinct from NEC, or whether they should be considered along the spectrum of a heterogeneous group of LCNEC without p53/RB alterations. In this context and given the difficulty in morphologically distinguishing some LCNEC from grade 3 NET, we can speculate that classification of grade 3 NEN based on p53/RB alteration rather than morphology may be clinically relevant.
Our findings raise important questions regarding whether SCNEC of the breast should be managed distinctly from other high-grade breast carcinomas, especially triple-negative carcinomas. We have shown that SCNEC of the breast is convergent genetically, as well as histologically, with SCNEC of the lung, with frequent concurrent loss of p53 and RB. However, we also uncovered clonal origin of mixed NEC/IDC-NST, suggesting that SCNEC may genetically descend from an earlier IDC clone, rather than a distinct progenitor cell. Numerous case reports in the literature reflect the current uncertainty around optimal management of mammary SCNEC, including frequent reports of etoposide-based regimens that are used for SCNEC of the lung and not standardly for breast cancer29,72,73,74,75,76,77. In our series, the majority of patients with SCNEC were treated with etoposide (6/7, 86%), indicating that the diagnosis can influence medical oncologists’ choice of therapy, even in the absence of specific guidelines.
In summary, we have provided a comprehensive genetic characterization of carefully classified NEC of the breast and show that p53 and RB pathway co-alterations are highly prevalent in SCNEC and a subset of LCNEC, similar to their counterparts in the lung35,43,44. Genetic and immunophenotypic analysis of paired NEC and IDC-NST components of mixed tumors confirms their shared clonality and suggests that the NEC phenotype could arise from ductal carcinoma in the breast. Classification of uncommon grade 3 NEN without classic features of NEC is problematic in the most recent WHO classification, and our data suggest that these tumors may be biologically distinct from NEC, or, alternatively, that they fall along a spectrum of heterogeneous LCNEC that lack p53 and RB alterations. The clinical significance of recognizing grade 3 NEN with and without p53/RB alterations awaits further study.
Data availability
Data generated and analyzed during the current study are available from the corresponding author on reasonable request.
Change history
13 June 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41379-022-01099-3
References
Bean GR & Lin CY. Breast neuroendocrine neoplasms: practical applications and continuing challenges in the era of the 5th edition of the WHO classification of breast tumours. Diagn. Histopathol. 27, 139-147 (2021).
Rakha EA, Reis-Filho JS, Sasano H, Wu Y. Neuroendocrine neoplasms: Introduction. In: Allison KH, Brogi E, Ellis IO, Fox SB, Lakhani SR, Lax SF et al., editors. WHO Classification of Tumours Breast Tumours 5th ed, vol 2. Lyon, France: IARC Press, 2019. p. 155.
Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 31, 1770-1786 (2018).
Pareja F & D’Alfonso TM. Neuroendocrine neoplasms of the breast: a review focused on the updated World Health Organization (WHO) 5th edition morphologic classification. Breast J. 26, 1160–1167 (2020).
Rakha EA, Reis-Filho JS, Wu Y. Neuroendocrine carcinoma. In: Allison KH, Brogi E, Ellis IO, Fox SB, Lakhani SR, Lax SF et al., editors. WHO Classification of Tumours Breast Tumours 5th ed, vol 2. Lyon, France: IARC Press, 2019. p. 159–161.
Shin SJ, DeLellis RA, Ying L, Rosen PP. Small cell carcinoma of the breast: a clinicopathologic and immunohistochemical study of nine patients. Am. J. Surg. Pathol. 24, 1231-1238 (2000).
Adegbola T, Connolly CE, Mortimer G. Small cell neuroendocrine carcinoma of the breast: a report of three cases and review of the literature. J. Clin. Pathol. 58, 775-778 (2005).
Ozdirik B, Kayser A, Ullrich A, Savic LJ, Reiss M, Tacke F, et al. Primary neuroendocrine neoplasms of the breast: case series and literature review. Cancers 12, (2020).
Shin SJ, DeLellis RA, Rosen PP. Small cell carcinoma of the breast--additional immunohistochemical studies. Am. J. Surg. Pathol. 25, 831–832 (2001).
Rakha E & Tan PH. Head to head: do neuroendocrine tumours in the breast truly exist? Histopathology https://doi.org/10.1111/his.14627 (2022).
Rakha EA, Reis-Filho JS, Wu Y. Neuroendocrine tumour. In: Allison KH, Brogi E, Ellis IO, Fox SB, Lakhani SR, Lax SF, et al., editors. WHO Classification of Tumours Breast Tumours 5th ed, vol 2. Lyon, France: IARC Press, 2019. p. 156-158.
Tang LH, Untch BR, Reidy DL, O’Reilly E, Dhall D, Jih L, et al. Well-differentiated neuroendocrine tumors with a morphologically apparent high-grade component: a pathway distinct from poorly differentiated neuroendocrine carcinomas. Clin. Cancer Res. 22, 1011-1017 (2016).
Fang JM & Shi J. A clinicopathologic and molecular update of pancreatic neuroendocrine neoplasms with a focus on the new World Health Organization classification. Arch. Pathol. Lab. Med. 143, 1317-1326 (2019).
Tang LH, Basturk O, Sue JJ, Klimstra DS. A practical approach to the classification of WHO grade 3 (G3) well-differentiated neuroendocrine tumor (WD-NET) and poorly differentiated neuroendocrine carcinoma (PD-NEC) of the pancreas. Am. J. Surg. Pathol. 40, 1192-1202 (2016).
Singhi AD & Klimstra DS. Well-differentiated pancreatic neuroendocrine tumours (PanNETs) and poorly differentiated pancreatic neuroendocrine carcinomas (PanNECs): concepts, issues and a practical diagnostic approach to high-grade (G3) cases. Histopathology 72, 168-177 (2018).
Rakha EA, Allison KH, Bu H, Ellis IO, Foschini MP, Horii R, et al. Invasive breast carcinoma of no special type. In: Allison KH, Brogi E, Ellis IO, Fox SB, Lakhani SR, Lax SF, et al., editors. WHO Classification of Tumours Breast Tumours 5th ed, vol 2. Lyon, France: IARC Press, 2019. p. 102-109.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297-1303 (2010).
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491-498 (2011).
Garrison E & Marth G. Haplotype-based variant detection from short-read sequencing. (2012). http://arxiv.org/abs/1207.3907.
Li H & Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589-595 (2010).
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078-2079 (2009).
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333-i339 (2012).
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11 10 11-33 (2013).
Yang H & Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 10, 1556-1566 (2015).
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium-sized insertions from paired-end short reads. Bioinformatics 25, 2865-2871 (2009).
Allison KH, Hammond MEH, Dowsett M, McKernin SE, Carey LA, Fitzgibbons PL, et al. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J. Clin. Oncol. 38, 1346-1366 (2020).
Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. J. Clin. Oncol. 36, 2105-2122 (2018).
Darragh TM, Colgan TJ, Cox JT, Heller DS, Henry MR, Luff RD, et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch. Pathol. Lab. Med. 136, 1266-1297 (2012).
Gradishar WJ, Moran MS, Abraham J, Aft R, Agnese D, Allison KH, et al. NCCN Guidelines Insights: Breast Cancer, Version 4.2021. J. Natl Compr. Canc. Netw. 19, 484-493 (2021).
Oliner JD, Saiki AY, Caenepeel S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb. Perspect. Med. 6, (2016).
Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget 3, 228-235 (2012).
Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell. Biol. 24, 5835-5843 (2004).
Singh N, Piskorz AM, Bosse T, Jimenez-Linan M, Rous B, Brenton JD, et al. p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J. Pathol. 250, 336-345 (2020).
Trostel SY, Sackett DL, Fojo T. Oligomerization of p53 precedes its association with dynein and nuclear accumulation. Cell Cycle 5, 2253-2259 (2006).
George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47-53 (2015).
Shamir ER, Devine WP, Pekmezci M, Umetsu SE, Krings G, Federman S, et al. Identification of high-risk human papillomavirus and RB/E2F pathway genomic alterations in mutually exclusive subsets of colorectal neuroendocrine carcinoma. Mod. Pathol. 32, 290-305 (2019).
Rickman DS, Beltran H, Demichelis F, Rubin MA. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med. 23, 1-10 (2017).
Konukiewitz B, Schlitter AM, Jesinghaus M, Pfister D, Steiger K, Segler A, et al. Somatostatin receptor expression related to TP53 and RB1 alterations in pancreatic and extrapancreatic neuroendocrine neoplasms with a Ki67-index above 20. Mod. Pathol. 30, 587-598 (2017).
Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 22, 298-305 (2016).
Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res. 20, 890-903 (2014).
Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096-1100 (2008).
Shen P, Jing Y, Zhang R, Cai MC, Ma P, Chen H, et al. Comprehensive genomic profiling of neuroendocrine bladder cancer pinpoints molecular origin and potential therapeutics. Oncogene 37, 3039-3044 (2018).
George J, Walter V, Peifer M, Alexandrov LB, Seidel D, Leenders F, et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat. Commun. 9, 1048 (2018).
Rekhtman N, Pietanza MC, Hellmann MD, Naidoo J, Arora A, Won H, et al. Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin. Cancer Res. 22, 3618-3629 (2016).
Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 543, 65-71 (2017).
Clinical Lung Cancer Genome Project & Network Genomic Medicine. A genomics-based classification of human lung tumors. Sci. Transl. Med. 5, 209ra153 (2013).
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199-1203 (2011).
Fernandez-Cuesta L, Peifer M, Lu X, Sun R, Ozretic L, Seidal D, et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat. Commun. 5, 3518 (2014).
Debelenko LV, Brambilla E, Agarwal SK, Swalwell JI, Kester MB, Lubensky IA, et al. Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum. Mol. Genet. 6, 2285-2290 (1997).
Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat. Genet. 45, 1483-1486 (2013).
Banck MS, Kanwar R, Kulkarni AA, Boora GK, Metge F, Kipp BR et al. The genomic landscape of small intestine neuroendocrine tumors. J. Clin. Investig. 123, 2502-2508 (2013).
Papotti M, Gherardi G, Eusebi V, Pagani A, Bussolati G. Primary oat cell (neuroendocrine) carcinoma of the breast. Report of four cases. Virchows Arch. A Pathol. Anat. Histopathol. 420, 103-108 (1992).
McCullar B, Pandey M, Yaghmour G, Hare F, Patel K, Stein M, et al. Genomic landscape of small cell carcinoma of the breast contrasted to small cell carcinoma of the lung. Breast Cancer Res. Treat. 158, 195-202 (2016).
Wachter DL, Hartmann A, Beckmann MW, Fasching PA, Hein A, Bayer CM, et al. Expression of neuroendocrine markers in different molecular subtypes of breast carcinoma. Biomed. Res. Int. 2014, 408459 (2014).
Rakha E, Toss M, Quinn C. Specific cell differentiation in breast cancer: a basis for histological classification. J. Clin. Pathol. https://doi.org/10.1136/jclinpath-2021-207487 (2021).
Freitag CE, Mei P, Wei L, Parwani AV, Li Z. Genetic alterations and their association with clinicopathologic characteristics in advanced breast carcinomas: focusing on clinically actionable genetic alterations. Hum. Pathol. 102, 94-103 (2020).
Zhao SG, Chen WS, Das R, Chang SL, Tomlins SA, Chou J, et al. Clinical and genomic implications of luminal and basal subtypes across carcinomas. Clin. Cancer Res. 25, 2450-2457 (2019).
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 486, 346-352 (2012).
Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 (2016).
Klaestad E, Sawicka JE, Engstrom MJ, Ytterhus B, Valla M, Bofin AM. ZNF703 gene copy number and protein expression in breast cancer; associations with proliferation, prognosis and luminal subtypes. Breast Cancer Res. Treat. 186, 65-77 (2021).
Sircoulomb F, Nicolas N, Ferrari A, Finetti P, Bekhouche I, Rousselet E, et al. ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol. Med. 3, 153-166 (2011).
Bofin AM, Ytterhus B, Klaestad E, Valla M. FGFR1 copy number in breast cancer: associations with proliferation, histopathological grade and molecular subtypes. J. Clin. Pathol. https://doi.org/10.1136/jclinpath-2021-207456 (2021).
Mouron S, Manso L, Caleiras E, Rodriguez-Peralto JL, Rueda OM, Caldas C, et al. FGFR1 amplification or overexpression and hormonal resistance in luminal breast cancer: rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast Cancer Res. 23, 21 (2021).
Ang D, Ballard M, Beadling C, Warrick A, Schilling A, O’Gara R, et al. Novel mutations in neuroendocrine carcinoma of the breast: Possible therapeutic targets. Appl. .Immunohistochem. Mol. Morphol. 23, 97-103 (2015).
Marchio C, Geyer FC, Ng CK, Piscuoglio S, De Filippo MR, Cupo M, et al. The genetic landscape of breast carcinomas with neuroendocrine differentiation. J. Pathol. 241, 405-419 (2017).
Gradinaru SE, Bumbea H, Onisai MC, Stoicea M. Neuroendocrine differentiation in invasive lobular breast carcinoma. Acta Endocrinol. 13, 246-248 (2017).
Lavigne M, Menet E, Tille JC, Lae M, Fuhrmann L, Bonneau C, et al. Comprehensive clinical and molecular analyses of neuroendocrine carcinomas of the breast. Mod. Pathol. 31, 68-82 (2018).
Wei Y, Ke X, Yu J, Jing Q, Bu H, Zeng X, et al. Clinical and genomic analyses of neuroendocrine neoplasms of the breast. Mod. Pathol. https://doi.org/10.1038/s41379-021-00965-w (2021).
Tsang JY & Tse GM. Breast cancer with neuroendocrine differentiation: an update based on the latest WHO classification. Mod. Pathol. 34, 1062–1073 (2021).
Khleif SN, DeGregori J, Yee CL, Otterson GA, Kaye FJ, Nevins JR, et al. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. Proc. Natl Acad. Sci. USA 93, 4350-4354 (1996).
Li Y, Nichols MA, Shay JW, Xiong Y. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res. 54, 6078-6082 (1994).
Atchison L, Hardy T, Mancl T, Quaranta BP, Madan A. Locally advanced primary small cell carcinoma of the breast: a case report and review of current evidence. Case Rep. Oncol. 14, 761–766 (2021).
Raber B, Dao T, Howard E, Bredeweg A. Primary small-cell carcinoma of the breast. Proc. (Bayl. Univ. Med. Cent.) 30, 200–202 (2017).
Abou Dalle I, Abbas J, Boulos F, Salem Z, Assi HI. Primary small cell carcinoma of the breast: a case report. J. Med. Case Rep. 11, 290 (2017).
Boutrid H, Kassem M, Tozbikian G, Morgan E, White J, Shah M, et al. TTF-1 positive primary small cell carcinoma of the breast: a case report and review of the literature. Front. Endocrinol. 11, 228 (2020).
Tremelling A, Samuel S, Murray M. Primary small cell neuroendocrine carcinoma of the breast—a case report and review of the literature. Int. J. Surg. Case Rep. 38, 29-31 (2017).
Nicoletti S, Papi M, Drudi F, Fantini M, Canuti D, Tamburini E, et al. Small cell neuroendocrine tumor of the breast in a 40-year-old woman: a case report. J. Med. Case Rep. 4, 201 (2010).
Acknowledgements
The authors wish to thank Shirley Kwok and Aihui Wang for histology work; Ivy Mangonon, Ellen Gomulia, and Grace Allard for immunohistochemistry; Dr. Jessica Van Ziffle and the UCSF Clinical Cancer Genomic Laboratory for assistance with next-generation sequencing; Dr. Megan L. Troxell for critical reading of the manuscript; and Norm Cyr for help with figures.
Funding
This research was supported and funded by the UCSF Residents’ Teaching and Research Endowments from the UCSF Department of Pathology.
Author information
Authors and Affiliations
Contributions
G.R.B. and G.K. wrote the manuscript; G.R.B., Y.Y.C., and G.K. designed the study; G.R.B., S.N., S.J.S., E.M.H., J.L.C.J., A.U., K.D.J., Y.Y.C., and G.K. acquired and analyzed the data; all authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval and consent to participate
This study was performed in accordance with research policies approved by the Institutional Review Boards of UCSF, Stanford University, and Kaiser Permanente San Francisco.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Due to table and figure errors.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bean, G.R., Najjar, S., Shin, S.J. et al. Genetic and immunohistochemical profiling of small cell and large cell neuroendocrine carcinomas of the breast. Mod Pathol 35, 1349–1361 (2022). https://doi.org/10.1038/s41379-022-01090-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01090-y
This article is cited by
-
Comprehensive analysis of clinicopathologic features and p53 mutation in neuroendocrine neoplasms of the breast: experience from a large academic center
Breast Cancer Research and Treatment (2022)
