
Abstract
Osteoma is a benign bone forming tumor predominantly arising on the surface of craniofacial bones. While the vast majority of osteomas develops sporadically, a small subset of cases is associated with Gardner syndrome, a phenotypic variant of familial adenomatous polyposis caused by mutations in the APC gene resulting in aberrant activation of WNT/β-catenin signaling. In a sequencing analysis on a cohort of sporadic, non-syndromal osteomas, we identified hotspot mutations in the CTNNB1 gene (encoding β-catenin) in 22 of 36 cases (61.1%), harbouring allelic frequencies ranging from 0.04 to 0.53, with the known S45P variant representing the most frequent alteration. Based on NanoString multiplex expression profiling performed in a subset of cases, CTNNB1-mutated osteomas segregated in a defined “WNT-cluster”, substantiating functionality of CTNNB1 mutations which are associated with β-catenin stabilization. Our findings for the first time convincingly show that osteomas represent genetically-driven neoplasms and provide evidence that aberrant WNT/β-catenin signaling plays a fundamental role in their pathogenesis, in line with the well-known function of WNT/β-catenin in osteogenesis. Our study contributes to a better understanding of the molecular pathogenesis underlying osteoma development and establishes a helpful diagnostic molecular marker for morphologically challenging cases.
Similar content being viewed by others


Introduction
Osteoma is a benign bone forming tumor arising predominantly on the surface of bones formed by membranous ossification, i.e., the craniofacial, and jaw bones [1]. While their prevalence has been reported to reach 6.4% in selected series [2], the true incidence of osteomas is unknown, and it seems likely that many lesions remain clinically undetected due to the lack of specific symptoms. In advanced cases, osteomas can lead to deformation of the affected bone and compression of adjacent structures, including obstruction of the paranasal sinuses, associated with headache and paraesthesia [1]. Histologically, osteomas are predominantly composed of mature lamellar bone and can be divided into compact and spongious subtypes which most frequently show low cellularity and inconspicuous osteoblastic activity. However, in sinuorbital tumors, prominent osteoblastic and osteoclastic activity may be present and mimic features of osteoblastoma [3, 4]. Different hypotheses on the aetiology of osteomas have been proposed, including trauma, secondary changes of paranasal polyps, metaplasia, and dysontogenic or hereditary factors, respectively [1]. Osteoma is rarely associated with Gardner syndrome (representing a phenotypic variant of familial adenomatous polyposis (FAP)), characterized by APC mutations and aberrant activation of WNT/β-catenin signaling [5, 6]. The central key mediator of the canonical WNT/β-catenin signaling pathway is β-catenin, which in the pathway´s inactive state is degraded through a multiprotein complex including axis inhibition protein (AXIN), adenomatous polyposis coli (APC), casein-kinase-1 (CK1) and glycogen synthase kinase-3beta (GSK-3β) via sequential phosphorylation, ubiquitination, and proteasomal degradation. Binding of WNT ligands to frizzled receptors and co-receptors of the LRP family leads to inactivation of the ubiquitination/degradation function of the multiprotein destruction complex and nuclear accumulation of β-catenin, where it interacts with transcription factors of the T-cell factor and lymphoid enhancer factor family and induces transcriptional programs [7]. Given well-known examples of neoplasms arising in the setting of FAP (e.g., desmoid fibromatosis, hepatoblastoma) that occur independently from APC mutations in the sporadic setting and the established role of WNT/β-catenin signaling in osteogenesis (reviewed in [8, 9]), we set out to analyze a cohort of craniofacial osteomas for the presence of CTNNB1 gene mutations.
Materials and methods
Patients and samples
Cases of interest with sporadic osteoma were identified by searching the histopathology archives at the Basel Bone Tumor Reference Centre (Basel, Switzerland) and the Gerhard-Domagk-Institute of Pathology (Münster, Germany). 19 tumors were classified as compact, 14 as spongious, and three as osteoblastoma-like osteomas. Thorough investigation of the clinical history and radiological findings was performed to assess a syndromal predisposition with no evidence found in the selected cohort. The study was approved by the Ethical committees in Münster (reference 2020-886fS) and Basel (reference 274/12).
Next-generation sequencing (NGS)
Two customized NGS gene panels were applied to sequence the complete exonic region of human CTNNB1 (encoding β-catenin). The first sequencing approach was conducted using a hybrid capture-based hot spot panel (Illumina, covering 0.013 Mb, selected exons of 19 genes), including exon 3 of the CTNNB1 gene (RefSeq: NM_001098210; Ensembl transcript ID: ENST00000396183; UniProt/Swiss-Prot: P35222). All CTNNB1 exon 3 wild type samples were additionally analyzed employing a second NGS gene panel (Qiagen, covering 0.0638 Mb, 27 genes) to assess exons 2-15 of the CTNNB1 gene. Target enrichment was processed by means of the Nextera DNA Flex Pre-Enrichment Library Prep and Enrichment Reagents kit (Illumina) and the GeneRead DNAseq Panel PCR V2 kit (Qiagen). All purification and size selection steps were performed employing Agencourt AMPure XP magnetic beads (Beckman Coulter). IDT for Nextera DNA Unique Dual Indexes (Illumina) and NEXTflex-96 DNA barcodes (Bioo Scientific) were used. Sequencing was performed on the Illumina MiniSeq system (MiniSeq High Output Reagent Kit; 300-cycles). The Quantitative Multiplex FFPE Reference Standard (Horizon Discovery, ref. no. HD200) was applied as isogenic quality control (11 somatic mutations verified at 0.8–24.5% allelic frequency in genomic DNA) for routine performance monitoring and evaluation of NGS workflow integrity (pre-analytical DNA extraction, NGS workflow and post-analytical bioinformatics). NGS data analysis was performed by means of the CLC Genomics Workbench 20 software (CLC bio, Qiagen).
Immunohistochemistry (IHC)
Immunohistochemical stainings were performed on cases with acceptable preservation of morphology after decalcification using a β-catenin antibody (monoclonal mouse, clone 14, CellMarque, Rocklin, CA, USA) on a BenchMark ULTRA Autostainer (VENTANA/Roche, Basel, Switzerland) using 3 μm tissue sections. In brief, the staining procedure included heat-induced epitope retrieval pretreatment employing CC1 buffer (95 °C; 24 min) followed by incubation with the primary antibody for 32 min and signal detection using the OptiView DAB IHC Detection Kit (VENTANA/Roche, Basel, Switzerland) according to the manufacturer’s instructions. Positive and negative control stainings using an appropriate IgG subtype (DCS) were included. Due to the limited quality of the tissue, nuclear immunoreactivity was assessed as negative or positive, independent from the staining intensity.
Multiplex gene expression profiling
Formalin‐fixed and paraffin‐embedded (FFPE)-derived RNA samples were analyzed employing the NanoString nCounter FLEX gene expression analysis system with DX enablement according to the manufacturer´s instructions. This system was chosen with respect to the limited quality of decalcified tissue as it does not require enzymatic reverse transcription nor amplification, but yields information on the relative abundance of each mRNA transcript of interest through multiplexed hybridization and digital readout of fluorescent barcoded probes hybridized to each transcript [10]. The human PanCancer Pathways CodeSet (XT-CSO-PATH1-1) was used, covering 770 genes including 40 internal reference controls/housekeeping genes from 13 essential cancer hallmark-associated canonical pathways (MAPK, STAT, PI3K, RAS, Cell Cycle, Apoptosis, Hedgehog, WNT, Notch, DNA Damage Control, Transcriptional Regulation, Chromatin Modification, and TGF-β). The CodeSet containing capture and reporter probes was hybridized to 180 ng of total RNA for 18 h at 65 °C. Hybridized samples were loaded into the nCounter Prep Station for post‐hybridization processing. Expression of target mRNA was assessed with the nCounter Digital Analyzer. The NanoString nCounter nSolver software (version 4.0), with an additional NanoString Advanced Analysis Module (version 2.0.134) was used to perform quality control assessment, normalization, differential expression analysis, and pathway analyses. Data quality was assessed by default parameters according to the NanoString gene expression data analysis guidelines. No background subtraction or thresholding parameters were selected. The geometric means were used to compute normalization factors for the mRNA content of 6 internal positive control probes and 40 internal housekeeping genes. In the fold-change estimation settings, the ratio building option was selected and samples were partitioned based on their respective CTNNB1 mutational status. Housekeeping gene normalization was computed with an open-source R statistical package using the geNORM algorithm [11, 12]. Normalized log2 transformed mRNA counts were generated according to the gene expression normalization factor based on the geometric mean of the most stable housekeeping genes and Z score transformation gene expression values were used for subsequent data analysis. P-value adjustments were performed using the Benjamini–Yekutieli method [13]. Expression data is shown in Supplementary Table S1.
Results
We analyzed 36 samples from 36 individuals with histologically confirmed, sporadic craniofacial osteoma. The patient cohort included 19 females (52.8%) and 17 males (47.2%) ranging from 12 to 71 years of age (median 38.5 years). Twenty-five lesions (69.4%) involved the paranasal sinuses, nine (25%) were located in the jaws, and two (5.6%) involved the orbital and mastoid processes, respectively. Radiologically, all cases presented with characteristic imaging features, i.e., well-demarcated round, dense, well-defined ivory-like tumors attached to the underlying bone without cortical invasion. Thorough assessment of the clinical files revealed no evidence of multiple osteomas or accompanying skin lesions, congenital hypertrophy of the retinal pigmented epithelium, desmoid tumors or polyposis coli as indicators of a syndromal association. In 19 out of 36 cases (52.8%), classic (functionally activating) mutations affecting S45 in exon 3 of the CTNNB1 gene were detected, with S45P representing the most frequent alteration (Fig. 1A, B). One tumor (2.8%) displayed the well-known T41A CTNNB1 gene mutation. Two further osteomas were detected to carry rarer CTNNB1 mutations: (i) W383S, lying within Amardillo repeat 6 of β-catenin (Fig. 1B); though W383S has not been functionally described yet, the related W383R alteration has been shown to confer increased function to β-catenin as indicated by enhanced transcriptional activity compared to wild type β-catenin in cultured cells [14], and (ii) an A20V mutation that has been described before in anaplastic thyroid carcinoma where it was found not to be associated with nuclear accumulation of β-catenin, thus being of questionable functional relevance [15]. No significant difference regarding the CTNNB1 mutation frequency was detected between the histological subtypes of osteomas (Fig. 1A). As a significant number of cases was incidentally detected and aggressive acid decalcification (accompanied by protein denaturation) is regularly employed in tissue work-up of bone-containing specimens derived from routine surgery of the paranasal sinuses submitted without clinically suspected atypia or malignancy, immunohistochemical characterization was not informative, i.e., technically unfeasible, in a larger subset of the lesions (15 out of 36 cases). In 14 out of 16 CTNNB1-mutated cases, nuclear β-catenin immunoreactivity was detected, however, considerable variability with regard to the percentage of stained nuclei and staining intensity was observed. In selected cases, β-catenin was consistently detected in the nuclei of osteoblasts rimming lesional bone (Fig. 2). Contrarily, in 7 out of 7 CTNNB1-wildtype cases, no nuclear β-catenin staining was discernible. Given the methodical limitation of immunohistochemistry in acid-decalcified tissue, prediction of CTNNB1 mutational status through evaluation of nuclear β-catenin accumulation appeared not sufficiently reliable. In order to get deeper insights into the biological differences of osteomas according to the CTNNB1 mutational status, we performed multiplex gene expression profiling employing the NanoString nCounter system. This system was used as it does not require enzymatic reverse transcription nor amplification steps, but yields information on the relative abundance of each mRNA transcript of interest through multiplexed hybridization. After RNA extraction and quality control, based on an mRNA expression analysis covering 770 genes from 13 essential cancer hallmark-associated canonical pathways, eight CTNNB1 mutated and four CTNNB1 wild type osteomas were comparatively analyzed. Though the extractable information on individual genes is naturally limited, this approach provides evidence of distinct gene sets being expressed and biological pathways being activated in osteomas with and without CTNNB1 mutation, respectively (Fig. 3A, B). Pathway scores derived from this approach indicated an activation of the Notch, TGF-beta, and WNT signaling pathways in the CTNNB1 mutated group as compared to the CTNNB1 wild type group (Fig. 3B). The distribution of mRNA expression (fold change) of genes derived from the comparison of the group of CTNNB1 mutated to the group of CTNNB1 wild type cases is depicted in Fig. 3C. The relative expression of WNT/β-catenin-associated genes is summarized in Supplementary Fig. 1.

A Clustered mutational profile of osteomas (n = 36). Alterations in CTNNB1 (rows) are indicated for each sample (columns). Clinicopathological information is summarized according to the legend. Cases with indicated sample IDs (framed squares) were included in the NanoString analysis. AF allelic frequency. B Schematic overview of β-catenin indicating relevant protein domains, phosphorylation sites, interaction partners and associated cellular functions. Detected mutations in the N-terminal domain and the Armadillo repeat region are indicated in red along with their frequency. NTD N-terminal domain, CTD C-terminal domain.

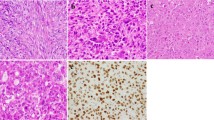
A CT scan and (B, C) H&E stained aspects of an osteoma with osteoblastoma-like features arising in the ethmoidal sinus involving the nasal cavity. Conspicuous osteoblastic and osteoclastic activity is observed against the background of a well-vascularized and moderately cellular and fibrous stroma in the marrow spaces (B, original magnification 10x; C, original magnification 20x). D Osteoid matrix-rimming osteoblasts display nuclear β-catenin positivity (original magnification 20x).

A Exploratory heatmap of the normalized expression data generated via unsupervised clustering clearly distinguishes CTNNB1 mutated and CTNNB1 wild type osteomas according to their mRNA expression profile (red indicates high expression; green indicates low expression). B Heatmap of pathway scores presenting an overview of 13 essential hallmark-associated canonical pathways (MAPK, STAT, PI3K, RAS, Cell Cycle, Apoptosis, Hedgehog, WNT, Notch, DNA Damage Control, Transcriptional Regulation, Chromatin Modification, and TGF-β), indicating that CTNNB1 mutated osteomas exhibit, among others, similar WNT/β-catenin pathway activation profiles, as compared to the CTNNB1 wild type group (red indicates high pathway scores; green indicates low scores). C Volcano plot displaying each gene’s –log10 (p value) and log2 fold change as calculated from the differential expression profile in the set of CTNNB1 mutated osteomas vs. baseline expression in the set of CTNNB1 wild type cases. Statistically significantly regulated genes are shown in the upper part of the plot above the horizontal line, differentially expressed genes fall to either side. Genes related to WNT/β-catenin pathway activation profile are highlighted in red.
Discussion
The aetiology of osteomas is an ongoing matter of debate. Some authors favour trauma, secondary changes of paranasal polyps, or inflammatory processes as the initiating trigger, therefore implying a reactive character for at least a subset of lesions fulfilling the diagnostic criteria of osteoma [1]. On the contrary, rare cases of (multiple) osteomas arising in a context of genetic syndromes, particularly FAP, point to a true neoplastic origin of these lesions [5, 6]. The current WHO classification of soft tissue and bone tumors classifies them as benign tumors, stating that the pathogenesis of sporadic osteomas is unknown [16]. In this focused analysis of a cohort of sporadic, non-syndromal osteomas, we have identified hotspot mutations in the CTNNB1 gene (encoding β-catenin) in 22 of 36 cases (61.1%). The β-catenin T41 and S45 mutations primarily detected here represent major regulatory sites for interaction with GSK-3β (phosphorylation at T41, S33 and S37) and CK1 (S45). These variants are known to prohibit β-catenin ubiquitination and proteasomal degradation, eventually leading to dysregulated WNT/β-catenin signaling [17,18,19].
According to the clinical data available, none of the patients (including those with a CTNNB1 wild type osteoma), had multiple osteomas or stigmata implying a syndromal association and thus potential involvement of the APC gene. This is in line with the results of our NanoString mRNA expression approach, providing evidence of distinct gene sets being expressed in osteomas with and without CTNNB1 mutation, respectively. While in CTNNB1 mutated osteomas a strong WNT pathway activation was discernible, CTNNB1 wild type osteomas did not show a relevant WNT signature activation. This makes alternative hits leading to WNT activation (including APC mutations) unlikely, given the findings of Crago and colleagues in desmoid-type fibromatosis in which CTNNB1 mutated and wild type cases were indiscernible in unsupervised clustering of U133A-derived gene expression profiles; in their analysis, the CTNNB1 wild type group of desmoid-type fibromatoses was shown to comprise cases with APC losses [20]. Alternatively, a mechanism of “selective apoptosis” of β-catenin mutated cells, similar to the supposed mechanism of “vanishing” GNAS mutations in long-standing fibrous dysplasia might be postulated to explain the CTNNB1 wild type status of a subset of osteomas. Eventually, a number of lesions that appear clinically and histologically similar to osteomas may indeed represent reactive compact new bone formations resulting from a possible inflammatory stimulus. Assessing the anatomic distribution of CTNNB1 wild type cases, it is noticeable that mandibular lesions are overrepresented in this group and that the only detectable CTNNB1 variant was the A20V mutation lacking evidence of functional relevance [15]. This might point to mandibular lesions being biologically different, but the size of the cohort is too limited for statistical analyses. Nonetheless, the NanoString mRNA expression analysis convincingly documenting distinct expression profiles according to CTNNB1 status can be regarded as a strong argument for the assumption that CTNNB1 wild type and CTNNB1 mutated cases are biologically distinct. Independent from localization, morphological re-evaluation of CTNNB1 wild type and CTNNB1 mutated cases revealed no significantly different histological patterns.
The frequency of classic CTNNB1 mutations in sporadic osteomas opposed to a well-known syndromal FAP-association of rare cases is reminiscent to other diseases rarely arising in the context of FAP, i.e., desmoid fibromatosis and hepatoblastoma [21, 22]. Desmoid fibromatosis has been reported to carry CTNNB1 mutations in up to 89% of sporadic cases [20, 23,24,25,26,27], while APC mutations have only rarely been detected in the sporadic context [20, 28]. In hepatoblastoma, CTNNB1 mutations have been found in up to 67% of cases, all affecting exon 3 which encodes the degradation targeting box of β-catenin, resulting in nuclear β-catenin accumulation [29,30,31]. The latter observation is of particular interest as WNT/β-catenin signaling is known to play a pivotal role in developmental hepatoblast proliferation, liver organogenesis and regeneration (reviewed in [32]). A comparably essential role of canonical WNT/β-catenin signaling is well established also in osteogenesis (reviewed in [8, 9]). Clinical evidence is derived from the finding that loss of function mutations in the LRP5 gene, encoding a coreceptor of canonical WNT signaling, is linked to a decrease of bone density, associated with defects in osteoblast differentiation and proliferation [33]. Consistently, Lrp5−/− mice display a reduction in bone mass and a disruption of osteoblast proliferation and bone matrix deposition [34]. On the contrary, gain of function mutations in LRP5 are associated with an increase in bone mass (reviewed in [35]), and mice expressing the LRP5 G171V mutation in osteoblasts display increased bone density and elevated numbers of active osteoblasts [36]. Mice deficient in Axin2, a negative regulator of canonical WNT signaling, exhibit craniosynostosis due to an increased proliferation of osteoblastic progenitors in the skull sutures [37], and haploid deficiency of β-catenin was shown to alleviate craniofacial bone defects caused by Axin2 deficiency [38]. Elaborate experiments examining the conditional inactivation of β-catenin in skeletal progenitors revealed that β-catenin activity is crucial for the differentiation of mature osteoblasts and thus for both endochondral and membranous ossification, the latter being particularly important for the development of the craniofacial bones [39, 40].
Our finding of recurrent CTNNB1 mutations in osteomas contributes to the concept of an outstanding role of WNT/β-catenin in bone biology and disease. We show osteoma to represent the first bone tumor associated with the relatively “simple” and recurrent genetic background of single nucleotide variants in the CTNNB1 gene and thus provide evidence for a genetically defined, true neoplasm. Beyond this conceptual aspect, our findings establish CTNNB1 mutations as a helpful diagnostic marker which may be of use in morphologically challenging cases. Given the limitations of immunohistochemistry in larger subsets of such lesions in which, however, tissue quality after decalcification may still be sufficient for highly sensitive NGS approaches, molecular confirmation of CTNNB1 mutations appears to be the most suitable tool to be employed.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. All original mRNA expression data are available (Supplementary Table S1).
References
Larrea-Oyarbide, N., Valmaseda-Castellon, E., Berini-Aytes, L. & Gay-Escoda, C. Osteomas of the craniofacial region. Review of 106 cases. J. Oral. Pathol. Med. 37, 38–42 (2008).
Lee, D. H. et al. Characteristics of paranasal sinus osteoma and treatment outcomes. Acta Otolaryngol. 135, 602–607 (2015).
McCann, J. M., Tyler, D. Jr. & Foss, R. D. Sino-orbital osteoma with osteoblastoma-like features. Head Neck Pathol. 9, 503–506 (2015).
McHugh, J. B., Mukherji, S. K. & Lucas, D. R. Sino-orbital osteoma: a clinicopathologic study of 45 surgically treated cases with emphasis on tumors with osteoblastoma-like features. Arch Pathol. Lab. Med. 133, 1587–93 (2009).
Gomez Garcia, E. B. & Knoers, N. V. Gardner’s syndrome (familial adenomatous polyposis): a cilia-related disorder. Lancet Oncol. 10, 727–35 (2009).
Sundaram, M., Falbo, S., McDonald, D. & Janney, C. Surface osteomas of the appendicular skeleton. AJR Am. J. Roentgenol. 167, 1529–33 (1996).
Nusse, R. & Clevers, H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–99 (2017).
Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 127, 469–80 (2006).
Hartmann, C. A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol. 16, 151–158 (2006).
Geiss, G. K. et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 26, 317–25 (2008).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 (2002).
Waggott, D. et al. NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinformatics 28, 1546–1548 (2012).
Benjamini, Y. & Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–88 (2001).
Li, C. M. et al. CTNNB1 mutations and overexpression of Wnt/beta-catenin target genes in WT1-mutant Wilms’ tumors. Am. J. Pathol. 165, 1943–53 (2004).
Garcia-Rostan, G. et al. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 59, 1811–1815 (1999).
Baumhoer, D., Bredella, M.A. & Sumathi, V. P. Osteoma. In: WHO Classification of Tumours Editorial Board (ed). WHO classification of Soft Tissue and Bone Tumours 391–393 (International Agency for Research on Cancer, 2020).
Rubinfeld, B. et al. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 275, 1790–1792 (1997).
Tejpar, S. et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene 18, 6615–20 (1999).
Yost, C. et al. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 10, 1443–54 (1996).
Crago, A. M. et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer 54, 606–15 (2015).
Garber, J. E. et al. Hepatoblastoma and familial adenomatous polyposis. J. Natl Cancer Inst. 80, 1626–1628 (1988).
Gurbuz, A. K. et al. Desmoid tumours in familial adenomatous polyposis. Gut 35, 377–81 (1994).
Amary, M. F. et al. Detection of beta-catenin mutations in paraffin-embedded sporadic desmoid-type fibromatosis by mutation-specific restriction enzyme digestion (MSRED): an ancillary diagnostic tool. Am. J. Surg. Pathol. 31, 1299–309 (2007).
Domont, J. et al. High frequency of beta-catenin heterozygous mutations in extra-abdominal fibromatosis: a potential molecular tool for disease management. Br. J. Cancer 102, 1032–1036 (2010).
Huss, S. et al. Beta-catenin (CTNNB1) mutations and clinicopathological features of mesenteric desmoid-type fibromatosis. Histopathology 62, 294–304 (2013).
Lazar, A. J. et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am. J. Pathol. 173, 1518–27 (2008).
Trautmann, M. et al. Novel pathogenic alterations in pediatric and adult desmoid-type fibromatosis - a systematic analysis of 204 cases. Sci. Rep. 10, 3368 (2020).
Alman, B. A., Li, C., Pajerski, M. E., Diaz-Cano, S. & Wolfe, H. J. Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors). Am. J. Pathol. 151, 329–34 (1997).
Blaker, H. et al. Beta-catenin accumulation and mutation of the CTNNB1 gene in hepatoblastoma. Genes Chromosomes Cancer 25, 399–402 (1999).
Koch, A. et al. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res. 59, 269–73 (1999).
Wei, Y. et al. Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene 19, 498–504 (2000).
Perugorria, M. J. et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 16, 121–36 (2019).
Gong, Y. et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107, 513–23 (2001).
Kato, M. et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 157, 303–14 (2002).
Johnson, M. L., Harnish, K., Nusse, R. & Van Hul, W. LRP5 and Wnt signaling: a union made for bone. J. Bone Min. Res. 19, 1749–57 (2004).
Babij, P. et al. High bone mass in mice expressing a mutant LRP5 gene. J. Bone Min. Res. 18, 960–74 (2003).
Yu, H. M. et al. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development 132, 1995–2005 (2005).
Liu, B., Yu, H. M. & Hsu, W. Craniosynostosis caused by Axin2 deficiency is mediated through distinct functions of beta-catenin in proliferation and differentiation. Dev. Biol. 301, 298–308 (2007).
Day, T. F., Guo, X., Garrett-Beal, L. & Yang, Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–50 (2005).
Hill, T. P., Spater, D., Taketo, M. M., Birchmeier, W. & Hartmann, C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–38 (2005).
Funding
The study was supported by the Swiss National Science Foundation, the foundation of the Basel Bone Tumor Reference Centre, the Gertrude von Meissner Stiftung, the Stiftung für krebskranke Kinder, Regio Basiliensis, the Wilhelm-Sander Stiftung, the Deutsche Krebshilfe and the DFG. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
D.B., M.T., and W.H. conceived and designed the experiments. D.B., I.G., E.W., F.M.T., C.R., V.V., M.T., and W.H. contributed tumor material and data acquisition. R.B., I.I., L.H., B.A., J.S., and M.T. performed DNA extractions and sequencing analyses. M.T. and J.S. analyzed sequencing data and interpreted the results. D.B., M.T., and W.H. prepared the paper with contributions from all other authors. All authors read and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The study was approved by the Ethical committees in Münster (reference 2020-886fS) and Basel (reference 274/12).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baumhoer, D., Berthold, R., Isfort, I. et al. Recurrent CTNNB1 mutations in craniofacial osteomas. Mod Pathol 35, 489–494 (2022). https://doi.org/10.1038/s41379-021-00956-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00956-x
