
Abstract
Merkel cell carcinoma (MCC) is an aggressive cutaneous neuroendocrine carcinoma without a known dysplastic precursor. In some cases, MCC is associated with SCCIS in the overlying epidermis; however, the MCC and SCCIS populations display strikingly different morphologies, and thus far a relationship between these components has not been demonstrated. To better understand the relationship between these distinct tumor cell populations, we evaluated 7 pairs of MCC-SCCIS for overlapping genomic alterations by cancer profiling panel. A subset was further characterized by transcriptional profiling and immunohistochemistry. In 6 of 7 MCC-SCCIS pairs there was highly significant mutational overlap including shared TP53 and/or RB1 mutations. In some cases, oncogenic events previously implicated in MCC (MYCL gain, MDM4 gain, HRAS mutation) were detected in both components. Although FBXW7 mutations were enriched in MCC, no gene mutation was unique to the MCC component across all cases. Transcriptome analysis identified 2736 differentially expressed genes between MCC and SCCIS. Genes upregulated in the MCC component included Polycomb repressive complex targets; downregulated transcripts included epidermal markers, and immune genes such as HLA-A. Immunohistochemical studies revealed increased expression of SOX2 in the MCC component, with diminished H3K27Me3, Rb, and HLA-A expression. In summary, MCC-SCCIS pairs demonstrate clonal relatedness. The shift to neuroendocrine phenotype is associated with loss of Rb protein expression, decrease in global H3K27Me3, and increased expression of Merkel cell genes such as SOX2. Our findings suggest an epidermal origin of MCC in this setting, and to our knowledge provide the first molecular evidence that intraepithelial squamous dysplasia may represent a direct precursor for small cell carcinoma.
Similar content being viewed by others


Introduction
Merkel cell carcinoma (MCC) is an aggressive cutaneous neuroendocrine carcinoma, that classically presents as a rapidly growing nodule on the sun-exposed skin of older individuals1. There is significant risk of metastatic disease, and an estimated 33–46% historic rate of mortality. Surgery and radiotherapy are mainstays of treatment1. Immunotherapy can be effective for advanced disease; however, a significant fraction of patients will progress on immunotherapy, highlighting the need for improved biological understanding and targeted therapies for this malignancy1.
Genomic data indicate that MCC can arise via two distinct pathways: virus-positive MCC (VP-MCC) with genomic integration of the tumorigenic Merkel cell polyomavirus (MCPyV), or virus-negative MCC (VN-MCC) with UV-signature genomic damage1,2,3,4. In VP-MCC, viral oncoproteins inactivate Rb and p53 tumor suppressors, among other activities1,5. By contrast, VN-MCC displays a high tumor mutation burden with UV-mutational signature, similar to melanoma, and harbors near-ubiquitous inactivating mutations of RB1 and TP531,3,4. Oncogene activation events are less frequently observed, and include activating mutation in PIK3CA and HRAS, as well as copy number gain in MYCL or MDM4 genes1,5,6.
Morphologically, MCC has characteristic features of small cell neuroendocrine carcinoma, including minimal cytoplasm, stippled chromatin, nuclear molding, and high mitotic rate1,7. MCC classically presents as a dermal tumor. Although most tumors lack visible connection to an epithelial structure, in a fraction of cases the dermal tumor is accompanied by an intraepidermal component7. There is no known precursor lesion for MCC, and the cell of origin remains unclear8,9,10,11,12. By immunohistochemistry, MCC displays characteristic paranuclear dot patterning of intermediate filaments including keratins 8 and 20 in most cases; neuroendocrine markers; and the master Merkel cell lineage transcription factors SOX2 and ATOH17,13,14. Rb protein expression is low or absent in VN-MCC15, consistent with genomic loss of this tumor suppressor.
In contrast to MCC, cutaneous squamous cell carcinoma (SCC) displays relatively low propensity for metastasis, and has been associated with dysplastic keratinocytic precursor lesions in the epidermis16. Unlike MCC, RB1 mutation is rare in cutaneous invasive SCC, although a subset of SCCIS harbors RB1 mutations17,18,19,20.
In a minority of VN-MCC, SCCIS is noted in the overlying or adjacent epidermis1. This co-occurrence of MCC and SCCIS is a distinct phenomenon from combined MCC-SCC tumors, in which squamous and neuroendocrine components are intimately mingled in the dermis, possibly representing squamous metaplasia7,21. To our knowledge an association between MCC and SCCIS has not been rigorously examined. The potential relatedness between these components has profound implications for models of MCC tumorigenesis.
Here, we compare 7 cases of MCC-associated SCCIS to the matched MCC tumors, using a focused next-generation sequencing (NGS) cancer panel optimized for sensitive detection of mutations and chromosomal copy number variations in small samples. In 6 of 7 cases, we found shared TP53 and/or RB1 mutations between MCC and SCCIS pairs. The 7th sample demonstrated clonal relatedness between SCCIS and spindle cell SCC, but not the adjacent MCC. Gene expression profiling revealed that MCC-associated SCCIS was associated with an intermediate transcriptional profile between MCC and conventional SCCIS, with expression of both epidermal and Merkel cell genes. Transcriptome data and immunohistochemistry demonstrated that the squamous-neuroendocrine shift correlated with decrease in the Polycomb epigenetic mark H3K27 trimethylation (H3K27Me3), loss of Rb1 protein expression, and downregulation of HLA-A. Our findings suggest that in rare cases, cutaneous SCCIS undergoes a shift to a neuroendocrine MCC phenotype accompanied by epigenetic and transcriptional changes, probable decreased immune surveillance, and an aggressive phenotype.
Methods
Case identification and collection
The study was conducted under a protocol approved by the Institutional Review Board of the University of Michigan. Cases of MCC with concurrent SCCIS were identified by database search of the University of Michigan Department of Pathology archives from 1999 to 2020, followed by slide review by a certified dermatopathologist (P.W.H.). Of 45 confirmed cases of MCC-SCCIS, 9 cases had available formalin-fixed, paraffin-embedded (FFPE) tumor blocks and sufficiently distinct large regions of SCCIS and MCC that allowed for dissection of pure areas for each component. Specifically, we targeted areas of SCCIS without intermingled intraepidermal MCC or direct underlying dermal MCC, and areas of dermal MCC avoiding the overlying epidermis (Fig. 1A, Supplementary Figs. S1–S7). For 8 tumors, primary MCC tumor material was available for analysis; for the 9th tumor, only MCC tumor material from a lymph node metastasis could be obtained for analysis alongside the paired SCCIS. For each case, regions of interest were identified on hematoxylin and eosin (H&E) stained slides and designated as SCCIS or MCC by a board-certified dermatopathologist. For one case, an area of invasive SCC with spindle cell features in the same clinical lesion as MCC and SCCIS was also sampled. For one case, multiple areas of MCC and SCCIS were sampled. FFPE blocks were cut to make 4-8 10-µm sections. Areas with high tumor purity were designated using flanking H&E stained sections, and macro-dissected using a scalpel. DNA and RNA from each sample were co-isolated using the Qiagen Allprep FFPE DNA/RNA kit (Qiagen) using manufacturer’s instructions and quantified using the Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA) as previously described22.

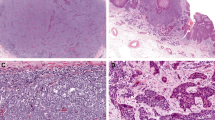
A Diagram showing intraepidermal SCCIS and dermal MCC. Intraepidermal MCC was also present in some cases. Yellow circles represent relative areas of SCCIS and dermal MCC sampled for NGS profiling. B Morphologic features of MCC-SCCIS cases. Some cases had intraepidermal MCC and/or areas of transitional cell morphology in the epidermis. Scale bars: 200 microns (SCCIS), 100 microns (others).
Multiplex PCR-based next-generation sequencing (NGS)
Targeted multiplex PCR-based next-generation DNA sequencing (DNAseq) was performed essentially as described previously23,24. Briefly, up to 20 ng of FFPE-extracted DNA from each tumor component was utilized to generate NGS libraries with the Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific, Waltham, MA) and a custom AmpliSeq panel (Thermo Fisher Scientific) targeting 91 cancer-related genes (Supplementary Table S1). Pairs with adequate DNA in both components were also evaluated by the Ion AmpliSeq Comprehensive Cancer Panel (CCP; Thermo Fisher Scientific), a larger panel targeting the full coding regions of 409 cancer-related genes (Supplementary Table S1). Multiplex PCR-based whole-transcriptome next-generation RNA sequencing (RNAseq) was performed as described previously using up to 20 ng of FFPE-extracted RNA and the Ion AmpliSeq Transcriptome Human Gene Expression Kit (Thermo Fisher Scientific)20,25. Barcoded NGS libraries were templated using the Ion Chef System (Thermo Fisher Scientific) and sequenced using the Ion GeneStudioTM S5 Prime System (Thermo Fisher Scientific). NGS reads were processed and aligned using the Torrent Suite software (Thermo Fisher Scientific) prior to analysis with in-house bioinformatics pipelines. Variant filtering and prioritization, copy number analysis, and differential gene expression analysis was performed essentially as described previously22,25, using a variant allele frequency threshold of 0.10 for most samples (with a lower threshold of 0.05 for the SCCIS and SCC components of Pair 12, due to lower tumor purity).
For 2 of the 9 MCC-SCCIS pairs, targeted DNAseq was unsuccessful due to insufficient quantity and/or quality of FFPE-extracted DNA from either the MCC or SCCIS component. These cases were excluded from subsequent analyses (with the exception of random pair comparisons for similarity index calculation as described below), resulting in a final total of 7 paired MCC-SCCIS cases.
Immunohistochemistry, immunofluorescence, and slide digitization
Antibodies, staining conditions, and samples analyzed are listed in Supplementary Table S2. Immunohistochemistry protocols for MCPyV large T antigen and CK20 have been previously reported3,26. Immunohistochemical stains were visualized by brown chromogen (DAB). Immunofluorescence fluorophores are listed in Supplementary Table S2. Whole slide images (WSIs) were acquired by Vectra Polaris scanner at 20x magnification (fluorescence) or 40× magnification (brightfield).
Digital scoring was conducted on WSIs using QuPath software. A board-certified dermatopathologist (P.W.H.) predesignated pure regions of MCC and SCCIS on flanking H&E stained sections, and then outlined those regions on the corresponding WSIs to include the greatest possible scoreable area for each lesion. Following cell segmentation, regions were analyzed using the positive cell detection feature for H-scoring (chromogenic immunohistochemistry)26, or fluorescence intensity values (immunofluorescence). Fold changes in protein expression were calculated for each pair by dividing by the H-score of the SCCIS component for that pair.
Statistical analysis
GraphPad Prism software was used for statistical testing and data visualization. Genetic relatedness between MCC and SCCIS pairs was calculated using the similarity index approach for specific nucleotide variations, as previously described23,27,28. Briefly, similarity index quantitates similarity on a scale of 0 (no shared changes) to 1 (exact similarity), based upon the number of shared mutations divided by the total mutations across both samples in the pair. Mutation signature analysis (based upon previously described patterns29) was performed using MuSiCa (http://bioinfo.ciberehd.org:3838/MuSiCa)30 to analyze the shared mutation calls combined from all tumor pairs profiled by CCP. Phylogeny comparisons were generated based upon the number of shared or unique mutations in each component, with a relaxed variant allele frequency threshold (VAF < 0.1 if the same variant was present in the other component at VAF > 0.1) to allow for more sensitive detection of subclonal populations. Gene expression heatmaps and 3-dimensional principal components analysis were generated using Qlucore software. Mutation and copy number heatmaps were generated by Oncoprinter31,32. Statistical comparisons for IHC were performed by Student’s t test, with p < 0.05 defined as significant.
Results
Clinicopathologic features of MCC and associated SCCIS
Our study cohort included 7 cases of MCC with associated SCCIS suitable for analyses Clinicopathologic features and representative microscopic images are provided in Supplementary Table S3 and Supplementary Figs. S1–7). Six tumors lacked evidence of MCPyV by IHC and were classified as VN-MCC (Supplementary Table S3); the remaining tumor (Pair 10) lacked sufficient material for MCPyV testing following sequencing. As a requirement for inclusion, all cases displayed dermal MCC with SCCIS in the overlying or adjacent epidermis (Fig. 1, Supplementary Figs. S1–S7). Additional microscopic findings were present in some cases (Fig. 1B): intraepidermal MCC (Pairs 8, 9, 12; Fig. 1B, bottom left), and potential intraepidermal transition zones in which cells displayed intermediate morphology between neuroendocrine (small round cells with stippled chromatin) and squamous (polygonal cells with abundant cytoplasm) features (Pairs 4, 8, and 9; Fig. 1B, bottom right).
Most cases lacked evidence of squamous differentiation within the dermal component of MCC (Supplementary Figs. S1–S6). In one case (Pair 12), the clinical lesion was composed of multiple adjacent components: an area of intraepidermal MCC overlying dermal MCC with foci of squamous differentiation, adjacent to SCCIS and invasive spindled SCC (Supplementary Fig. S7). The spindle cell SCC in this case labeled with pan-keratin in a cytoplasmic pattern, but displayed loss of other epidermal markers including K5/6 and p63.
Diagnostic immunohistochemistry for keratin 20 (K20) was positive in all but one case (MCC tumor for pair 5); this K20-negative case displayed a paranuclear dot pattern for pan-keratin expression, and the patient had a negative chest CT at the time of diagnosis (Supplementary Table S3). When present in sections immunostained at diagnosis, the associated SCCIS was negative for K20 and neuroendocrine markers.
By immunofluorescence, we observed that the SCCIS and MCC components displayed predicted expression patterns of epidermal and MCC markers. Keratin 8 expression was absent in SCCIS, and expressed in a paranuclear dot pattern in most MCC cases (Supplementary Fig. S8A). Keratin 5 was expressed at a high level in SCCIS, and showed paranuclear dot expression in a minority of MCC (Supplementary Fig. S8B). SOX2 displayed low expression in SCCIS, although significantly higher than background epidermis (Supplementary Fig. S8B). By contrast, SOX2 expression in MCC was elevated above background epidermis in every case, and above SCCIS in all but one case (Supplementary Fig. S8C–E). Some SCCIS cases displayed a bimodal distribution of relatively stronger and weaker SOX2-expressing populations, whereas MCC tumors tended to display a more uniformly elevated level of expression (Supplementary Fig. S8E).
Genomic relatedness between MCC tumors and associated SCCIS
To compare genomic features of MCC and associated SCCIS, we used a focused NGS panel of 91 cancer-related genes (Supplementary Table S1) optimized to sensitively detect mutations and copy number changes in small FFPE tumor samples. By this approach, in 6 of 7 cases, there was a high rate of mutational similarity between the MCC and paired SCCIS (Fig. 2A, Supplementary Table S4). The average mutational similarity between paired MCC-SCCIS tumors significantly exceeded the rate of coincidental similarity in random pairings, and was similar to the relatedness seen in paired samples from different regions of a single MCC tumor (Pair 4), as well as a paired SCC and invasive spindle cell SCC (Pair 12) discussed below (Fig. 2A, Supplementary Figs. S1, S7, and S9). Phylogeny comparisons supported a common clonal origin, followed by genetic drift upon or after divergence of the populations (Fig. 2B).

A Mutational similarity in matched MCC-SCCIS pairs is similar to that observed in a SCC-SCCIS pair and multiple samplings within the same MCC tumor (MCC intratumor), and significantly higher than observed for random pairings. One MCC-SCCIS pair lacked evidence of genetic relatedness. B Phylogenetic comparison of MCC-SCCIS pairs, highlighting selected shared and distinct mutation events in each component. S-SCC: spindled SCC. C Specific shared mutations and copy number alterations in matched MCC-SCCIS pairs. Genes displaying identical nucleotide variations or copy number alterations are outlined in purple. All pairs of MCC-SCCIS with genetic similarity display common TP53 and/or RB1 mutation. In contrast, an unrelated MCC-SCCIS pair displayed distinct TP53 mutations. D Relative frequencies of aberrancy (mutation or copy number alteration) in major cancer-related genes in MCC and SCCIS. RB1 and TP53 mutations are predominantly shared. Oncogenic copy gains (MYCL, MDM4) were shared in some cases, or restricted to the MCC component in others. FBXW7 mutations were restricted to the MCC component, whereas CDKN2A alterations were concentrated in SCCIS components.
With regard to specific mutations, related MCC-SCCIS pairs consistently displayed shared mutations identical at the nucleotide level in TP53, RB1, or both (Fig. 2C). A smaller fraction of cases displayed shared aberrations in oncogenes previously nominated to be drivers in MCC such as MYCL copy gain, MDM4 copy gain, and HRAS G12D mutation. CDKN2A mutation or copy loss was observed exclusively in the SCCIS component for 3 cases, and exclusively in the MCC component for 1 case (Fig. 2D). By contrast, FBXW7 mutation or copy loss was restricted to the MCC component in 4 cases, and was not detected in SCCIS for any case (Fig. 2D). In 3 cases, an oncogene activation event previously implicated as a MCC driver was observed in the MCC but not SCCIS component, including MYCL gain (2 cases) and PIK3CA E545K activating mutation (1 case) (Fig. 2C, D).
The sample that lacked similarity between the MCC and SCCIS components was the clinical lesion with combined elements of MCC, SCC, and spindle cell SCC (Supplementary Figs. S7 and S9). In this lesion, the SCCIS and spindle cell SCC components displayed high relatedness, with an additional PTEN nonsense mutation restricted to the spindle cell SCC component (Fig. 2B, Supplementary Fig. S9).
To evaluate genomic relatedness at greater depth, we employed more comprehensive NGS analysis across 400 cancer-related genes (Supplementary Table S1) on four pairs with adequate DNA in both components. By this approach, we again identified a high degree of mutational overlap between paired MCC and SCCIS samples, that significantly exceeded random chance (p < 0.01) (Supplementary Fig. S10A, Supplementary Table S5). For some pairs, we identified shared mutations in additional tumor suppressor genes recurrently mutated in MCC that were not evaluated by the initial focused panel; these included NOTCH2, KMT2C, and KMT2D (Supplementary Fig. S10B). SMARCA4 mutations were restricted to the MCC component (50% of cases; Supplementary Fig. S10C). By mutation signature analysis, mutations shared between MCC and SCCIS displayed strong signature 7 pattern consistent with UV mutations (Supplementary Fig. S10D).
Transcriptional landscape of MCC and associated SCCIS
Four primary MCC-SCCIS pairs had adequate RNA for whole-transcriptome analysis. These gene expression profiles were compared to previously sequenced SCCIS without MCC20. Because the presence of RB1 mutation has previously been associated with transcriptome differences in SCCIS, we included previously sequenced cases of SCCIS with RB1 mutation (n = 4) and without RB1 mutation (n = 5). By principal component analysis (PCA), MCC displayed clear separation from all SCCIS (Fig. 3A). SCCIS associated with MCC formed a cluster that was adjacent but non-overlapping with other SCCIS, and oriented toward the matching MCC tumors. Comparison to pre-defined gene sets revealed that MCC tumors displayed high expression of genes related to Atonal or Merkel cell differentiation, and low expression of epidermal markers (Fig. 3B). SCCIS unrelated to MCC displayed the converse pattern. SCCIS associated with MCC displayed an intermediate profile, combining epidermal marker expression with weak but increased expression of Merkel cell markers (Fig. 3B), despite having displayed absence of neuroendocrine marker expression by immunohistochemistry.

A 3-dimensional principal components analysis of global transcriptome profiles demonstrates that SCCIS associated with MCC form a distinct cluster oriented toward the MCC counterpart tumors. B MCC displays strong expression of Merkel cell genes, and downregulation of epidermal markers. SCCIS not associated with MCC demonstrates the converse pattern. SCCIS associated with MCC demonstrates a mixed profile, with epidermal marker expression accompanied by intermediate-level expression of Merkel cell genes. C Volcano plot of genes with significant differential expression in MCC-SCCIS pairs. D Gene set enrichment analysis identifies increased expression of Polycomb Repressive Complex targets in MCC, with decreased expression of immune associated transcripts (including HLA-A). E Immunofluorescence (IF) demonstrates strong HLA-A expression in SCCIS, with loss in paired dermal MCC tumor. SOX2 expression is shown to distinguish SCCIS from MCC. Scale bar: 40 microns (SCCIS), 50 microns (MCC). F Quantitation of IF expression of HLA-A in MCC tumors relative to related SCCIS pair, displaying significant downregulation in MCC component in 3 of 4 pairs. Asterisk: p < 0.05. G Representative IF intensities of SOX2 and HLA-A in related SCCIS-MCC pair. Each point represents a single cell. Regions of multispectral whole slide images were designated SCCIS or MCC based upon matched H&E stained section. Cells in the SCCIS region display a SOX2-low/HLA-high phenotype, whereas cells in the MCC region are predominantly SOX2-high with relatively lower HLA-A.
We further examined the distinctive transcriptional profile of MCC-associated SCCIS by directly comparing these to SCCIS unrelated to MCC. PCA including only SCCIS confirmed distinct clustering of MCC-associated SCCIS from other SCCIS, with an adjacent cluster of RB1-mutant SCCIS, and more heterogeneous distribution of other SCCIS (Supplementary Fig. S11). We found 203 differentially expressed genes between MCC-associated and RB1-mutant SCCIS groups (Supplementary Fig. S11B, Supplementary Table S6), with enrichment of gene sets including Polycomb Repressive Complex targets, neuronal, and embryonic genes in MCC-associated SCCIS (Supplementary Fig. S11C). Upregulated transcripts associated with MCC-associated SCCIS included NCAM1, KRT20, and PIEZ02. By contrast, there was no significant difference in epidermal markers KRT5, KRT6A, KRT6B, or GATA3. These transcriptional profiling results suggest that SCCIS associated with MCC might represent a distinct molecular subtype of SCCIS.
We then compared gene expression patterns in MCC-associated SCCIS to the paired MCC tumors, revealing 2,736 differentially expressed genes (Supplementary Table S7, Fig. 3C). Unsupervised gene-set enrichment analysis demonstrated upregulation of neural genes, and further upregulation of targets of the Polycomb Repressive Complex (Fig. 3D). By contrast, the SCCIS component was relatively enriched for expression of immune and inflammatory gene sets (Fig. 3D).
The squamous-to-neuroendocrine transition correlates with loss of HLA-A, Rb, and H3K27Me3
Of immune-related genes, MCC tumors displayed significant downregulation of HLA-A transcript, which has been previously shown to be a target of epigenetic silencing in MCC. To validate this observation, we examined protein expression of HLA-A by immunofluorescence in 4 related pairs. HLA-A displayed fold downregulation in 3 of 4 pairs (Fig. 3E, F). We then digitally quantitated co-expression of HLA-A and SOX2 in a multispectral WSI of a representative case. By this analysis, cells in regions of SCCIS displayed strong HLA-A expression and weak SOX2 expression, whereas cells in regions of MCC formed a distinct cluster with the converse pattern of expression (Fig. 3G).
Based upon the strong geneset enrichment signature for upregulation of Polycomb-targets in MCC, we evaluated H3K27Me3 staining. We observed a consistent fold decrease in global H3K27Me3 in MCC relative to the paired SCCIS (average 2.9 fold decrease, p = 0.01) (Fig. 4A, B).

A High H3K27Me3 in SCCIS with loss of labeling in MCC. B Consistent decrease in H3K27Me3 in the MCC component relative to the SCCIS component in paired tumors. C Variably high expression of Rb in SCCIS, with diffusely low expression in MCC. Black arrow denotes endothelial cells. D Consistent decrease in Rb expression in the MCC component relative to the SCCIS component in paired tumors. E Model for progression of SCCIS to MCC. Scale bar: 100 microns.
Given the central role of Rb function in MCC tumorigenesis, we next examined Rb expression patterns, and observed a consistent fold decrease in Rb in MCC relative to the paired SCCIS (average 9.2 fold decrease, p < 0.001) (Fig. 4C, D).
Discussion
Cutaneous carcinomas display diverse phenotypes and clinical behaviors, ranging from tumors that resemble keratinocytes and have relatively low metastatic potential (SCC), to poorly-differentiated neuroendocrine tumors with a high rate of metastasis (MCC). Here, we demonstrate evidence of clonal relatedness between VN-MCC and associated SCCIS, and potential morphologic transition zones were identified in some cases. Tumor suppressor inactivation events in TP53 and RB1 characteristic of VN-MCC were also detected in the SCCIS component, supporting these to be early driver events in the neoplastic process. The SCCIS and MCC components did not show consistent differences in genomic profiles that might explain the phenotypic switch. However, the shift from squamous to neuroendocrine morphology was accompanied by distinctive transcriptional profiles, decreased global H3K27Me3, and loss of Rb protein expression (Fig. 4E).
In all but one of the related cases in our series, the SCCIS component harbored RB1 mutation or copy loss. Evidence suggests that Rb inactivation (by mutation or oncogenic virus) plays a critical role in the formation of small cell neuroendocrine carcinomas in the skin and other body sites1,33. By contrast, cutaneous SCC is more consistently associated with inactivation of TP53 and CDKN2A tumor suppressors, rather than Rb17,18,34. However, we recently identified RB1 mutations in a subset of SCCIS20. This subset of SCCIS may have a stronger predisposition for neuroendocrine shift; however, the relatively common frequency of RB1 mutation in SCCIS (estimated 27% of all SCCIS)20 suggests that this event alone is unlikely to be sufficient for progression to MCC.
In addition to RB1 mutations, we identified further shared changes between SCCIS and MCC components, most consistently TP53, as well as oncogenic events. Mutations unique to the MCC and/or SCCIS component were also identified, compatible with continued clonal drift following divergence of the two populations. With regard to specific mutations, we observed FBXW7 copy loss or mutation restricted to the MCC component in a subset of cases, that might play a functional role analogous to proposed FBXW7 inhibition by the MCPyV small T antigen35. SMARCA4 mutations were also identified in the MCC component. Loss of this tumor suppressor component of the SWI/SNF complex has been described in some non-cutaneous neuroendocrine tumors, and might initiate transition from epithelial to round cell phenotype36,37,38,39. However, in previous studies of MCC, SMARCA4 mutation was not a frequent event, and protein expression was retained3,4,36. Thus, the role for SMARCA4 mutation in our cases remains unclear. Overall, we found no single genomic event that was consistently restricted to the MCC component in every case. Hence our analysis did not nominate a universal driver mutation for the squamous to neuroendocrine transition. Moreover, the overall pattern of changes suggests that fundamental drivers are established within the SCCIS prior to MCC formation.
These observations raise the possibility of a role for epigenetics in the SCCIS-MCC shift. In support of this, unsupervised analysis revealed upregulation of Polycomb-regulated gene transcripts in the MCC component. This observation was further supported by significantly weaker labeling for H3K27Me3 in MCC compared to SCCIS. H3K27Me3 is an inhibitory histone modification mediated by the Polycomb Repressive Complex (PRC) 2, that contains components including EZH2. Roles of Polycomb-mediated silencing in tumor biology are complex and context-dependent. Epigenetic changes, including Polycomb-mediated gene silencing, have been proposed to mediate the shift to neuroendocrine phenotype following RB1 inactivation in prostate carcinoma40,41. The role of EZH2 and Polycomb-mediated silencing in MCC biology is less clear42,43. However, inhibition of PRC2 activity can de-repress expression of SOX2 and promote Merkel cell differentiation44. Furthermore, globally decreased H3K27Me3 labeling has been described in MCC, although studies have shown conflicting results regarding specific patterns of this change45,46. Rb is an essential cofactor for PRC in some contexts47, hence the loss of Rb expression we observed could result in reduced PRC function. Thus, our observations are compatible with a model wherein diminished PRC activity in the precursor SCCIS predisposes to initiation of the Merkel cell differentiation pathway. However, the mechanism by which PRC2 activity is altered remains unclear. Although we noted an overall decrease in H3K27Me3, we did not directly examine methylation for specific promoters. Similarly, we cannot exclude the possibility that the decrease in global H3K27Me3 is a consequence of Merkel cell differentiation rather than an initiating event. Finally, our approach does not account for altered DNA methylation, which has been implicated in establishing the neuroendocrine phenotype in prostate carcinoma40 and displays distinct global patterning between MCC and SCC48. Of note, epigenetic therapies have shown promise in MCC48,49,50,51, and this treatment can be associated with differential expression of genes inhibiting neurogenesis48,49.
The most extensively characterized target of epigenetic silencing in MCC is the HLA-A gene and its associated antigen presentation machinery, which likely contributes to immune evasion by the tumor48,50,52. We found that HLA downregulation was prominent in MCC, but not SCCIS, component of tumors in our study. Proposed mechanisms for epigenetic silencing of HLA-A include histone hypo-acetylation and/or DNA methylation;48,50 although we did not directly examine these epigenetic modifiers, our findings suggest that silencing of this immune recognition gene may be an intrinsic feature of MCC epigenetic programming.
Identification of the cell of origin for a tumor type has profound implications for basic science investigations such as animal modeling, and potential clinical implications for therapeutic sensitivities. Due to the dermal location and unusual immunophenotype of MCC, diverse candidates have been proposed for the MCC cell of origin, including epidermal, follicular epithelial, lymphoid, fibroblasts, and mesenchymal stem cells. Benign Merkel cells are post-mitotic and display distinct patterns of anatomic and histologic distribution from MCC, and thus are considered less likely precursors. Several lines of evidence support an origin for MCC in epidermal or hair follicle populations. In transgenic mouse models, expression of MCPyV oncoproteins in epidermal progenitors gives rise to squamous dysplasia analogous to SCCIS9. Reprogramming the same cell population to neuroendocrine lineage by ATOH1 expression results in the formation of MCC-like proliferations8. As additional evidence for an epithelial origin, a combined tumor with components of VP-MCC and benign follicular tumor (trichoblastoma) displayed genetic relatedness between these morphologic populations53. MCC has also been rarely observed in association with other epithelial tumors such as poroma and basal cell carcinoma, although genetic relatedness has not been directly demonstrated in such cases.
Our results agree with evidence supporting an epithelial lineage for neuroendocrine carcinoma arising at other sites. Tumors combining small cell carcinoma morphology with other epithelial components (especially SCC or adenocarcinoma) have been described at sites including the larynx, lung, urinary bladder, and uterine cervix54,55. A shift to neuroendocrine phenotype heralds progression to more aggressive disease in prostate adenocarcinoma40. In contrast, MCC does not typically present as progression from another carcinoma type, and the most frequently associated second neoplasm is SCCIS7. Cases of small cell carcinoma accompanied by SCCIS have also been described in the esophagus, anal canal, and uterine cervix55,56,57, as well as urothelial carcinoma in situ in the urinary bladder58. Therefore, the same intraepithelial squamous-to-neuroendocrine transition we observe for MCC might also occur at other anatomic sites. However, this is likely not the sole pathway for neuroendocrine carcinoma formation, given that small cell carcinomas also arise in tissues or tumors that lack squamous epithelium.
Our study significantly expands upon previous studies examining the relationship between MCC and SCC. A case report of MCC overlapping with SCC identified differing chromosomal copy number profiles by array comparative genomic hybridization, consistent with collision of unrelated tumors59. A larger study of combined MCC tumors (with squamous differentiation in the dermis) compared the matched neuroendocrine and squamous components in two of the tumors, and demonstrated similarity in patterns of copy number alteration and gene-level mutations between the components21. However, unlike our study, rigorous statistical comparisons of nucleotide-level genomic similarity between the components were not performed to exclude the possibility of coincidental overlap. The areas of dermal squamous differentiation in combined tumors (as evaluated in that study) might represent squamous metaplasia; in contrast, in our study we evaluate SCCIS, which is substantially more likely to represent a precursor lesion. Of note, with only one exception, tumors in our study were “pure” MCC lacking overt squamous differentiation within the dermal component.
By coincidence, our study included another example of neoplastic progression with morphologic shift: SCCIS to invasive spindle cell SCC. To our knowledge, this is the first comparison of genomic changes in human SCCIS with associated spindle cell SCC. The spindle cell SCC demonstrated an inactivating PTEN mutation not detected in the SCCIS component. Of note, PTEN mutations appear to be infrequent in cutaneous spindle cell SCC60. Thus, investigation of additional SCCIS/spindle-cell SCC cases is necessary to provide additional evidence for a role for PTEN loss in this form of SCCIS progression.
Our study has several limitations. Because our sequencing approach is based upon a targeted cancer panel optimized for small FFPE samples, rather than whole exome sequencing, we cannot exclude the possibility that significant genomic events might not be captured by our panels. However, our approach encompasses oncogenes and tumor suppressors previously established by whole exome sequencing approaches to be recurrently altered in MCC, thus it is relatively unlikely that major, highly recurrent events are undetected by our approach. Similarly, our approach provides limited information on fine clonal substructure within the tumors. Lack of comparison against matched normal limits our sensitivity for detecting some variants. Although we sequenced in high depth to allow for sensitive detection of mutations, we cannot exclude the possibility that lower tumor purity in some SCCIS samples (relative to background keratinocytes and other normal cell types) limited sensitivity, especially for detecting chromosomal copy number alterations. Although we hypothesize that MCC-associated SCCIS has an inherently distinct transcriptome profile, we acknowledge that this profile might be influenced by proximity to MCC and/or its tumor microenvironment61. Furthermore, this study type carries inherent risk of cross-contamination between tumor components during collection or processing; however, we took extensive precautions to avoid this (Supplementary Figs. S1–7), and the variant allele frequencies of shared mutations (Supplementary Table S4) indicate that these are not likely due to cross-contamination. Finally, given the small size of our case series, we cannot rigorously predict what fraction of MCC-SCCIS are clonally related, rather than collision phenomena of unrelated tumors (as was the case for Pair 12).
Many important questions remain regarding the SCCIS-MCC transition, in addition to those mentioned above. Functional studies are needed to determine whether any changes we identified might be responsible for initiating the neuroendocrine shift in SCCIS. In addition, it is unclear whether the squamous-neuroendocrine shift is a rare stochastic event independent of the tumor microenvironment, or if immune surveillance might exert selection pressure for an immune-evasive neuroendocrine phenotype. Finally, the precise tumorigenic pathway for this subset of MCC may not be generalizable to other MCC groups, especially VP-MCC that likely arise in a more photo-protected niche1. Because most VN-MCC are not associated with SCCIS, an epidermal origin would imply that an intraepidermal precursor must be inconspicuous or regressed in such tumors.
In summary, in this case series we establish clonal relatedness between MCC and associated SCCIS in the overlying epidermis, suggesting an epidermal origin for MCC in these cases. Inactivation of TP53 and/or RB1 tumor suppressor genes occurs in the precursor squamous lesion. The shift to neuroendocrine phenotype is associated with global loss of H3K27Me3 and decreased Rb protein expression. Our findings raise the possibility of an epidermal origin for this subset of MCC, and to our knowledge represent the first genomic evidence suggesting that intraepithelial dysplasia can represent a direct precursor of small cell carcinoma.
Data availability
All nucleotide variants and differentially expressed genes meeting statistical thresholds are listed in supplemental materials. Additional data related to this manuscript can be provided upon request.
References
Harms, P. W. et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat. Rev. Clin. Oncol. 15, 763–776 (2018).
Feng, H., Shuda, M., Chang, Y. & Moore, P. S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–1100 (2008).
Harms, P. W. et al. The distinctive mutational spectra of polyomavirus-negative merkel cell carcinoma. Cancer Res. 75, 3720–3727 (2015).
Wong, S. Q. et al. UV-associated mutations underlie the etiology of MCV-negative merkel cell carcinomas. Cancer Res. 75, 5228–5234 (2015).
Park, D. E. et al. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 116, 1027–1032 (2019).
Paulson, K. G. et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J. Invest. Dermatol. 129, 1547–1555 (2009).
Tetzlaff, M. T. & Harms, P. W. Danger is only skin deep: aggressive epidermal carcinomas. An overview of the diagnosis, demographics, molecular-genetics, staging, prognostic biomarkers, and therapeutic advances in Merkel cell carcinoma. Mod. Pathol. 33, 42–55 (2020).
Verhaegen, M. E. et al. Merkel cell polyomavirus small T antigen initiates merkel cell carcinoma-like tumor development in mice. Cancer Res. 77, 3151–3157 (2017).
Verhaegen, M. E., et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Invest. Dermatol. https://doi.org/10.1038/jid.2014.446 (2014).
Zur Hausen, A., Rennspiess, D., Winnepenninckx, V., Speel, E. J. & Kurz, A. K. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res. 73, 4982–4987 (2013).
Liu, W. et al. Identifying the target cells and mechanisms of merkel cell polyomavirus infection. Cell Host Microbe 19, 775–787 (2016).
Spurgeon, M. E., Cheng, J., Bronson, R. T., Lambert, P. F. & DeCaprio, J. A. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 75, 1068–1079 (2015).
Laga, A. C. et al. Expression of the embryonic stem cell transcription factor SOX2 in human skin: relevance to melanocyte and merkel cell biology. Am. J. Pathol. 176, 903–913 (2010).
Leonard, J. H. et al. Proneural and proneuroendocrine transcription factor expression in cutaneous mechanoreceptor (Merkel) cells and Merkel cell carcinoma. Int. J. Cancer 101, 103–110 (2002).
Sihto, H. et al. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin. Cancer Res. 17, 4806–4813 (2011).
International Agency for Research on Cancer. WHO Classification of Skin Tumors. 4th edn, (International Agency for Research on Cancer: Lyon, France, 2018).
Pickering, C. R. et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 20, 6582–6592 (2014).
Li, Y. Y. et al. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 21, 1447–1456 (2015).
Inman, G. J. et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 9, 3667 (2018).
Lazo de la Vega, L. et al. Invasive squamous cell carcinomas and precursor lesions on UV-exposed epithelia demonstrate concordant genomic complexity in driver genes. Mod. Pathol. 33, 2280–2294 (2020).
Carter, M. D. et al. Genetic profiles of different subsets of Merkel cell carcinoma show links between combined and pure MCPyV-negative tumors. Hum. Pathol. 71, 117–125 (2018).
Chan, M. P. et al. Next-generation sequencing implicates oncogenic roles for p53 and JAK/STAT signaling in microcystic adnexal carcinomas. Mod. Pathol. 33, 1092–1103 (2020).
Harms, K. L. et al. Molecular profiling of multiple primary merkel cell carcinoma to distinguish genetically distinct tumors from clonally related metastases. JAMA Dermatol. 153, 505–512 (2017).
Faisal, F. A. et al. CDKN1B deletions are associated with metastasis in African American men with clinically localized, surgically treated prostate cancer. Clin. Cancer Res. 26, 2595–2602 (2020).
Lazo de la Vega, L. et al. Multiclonality and marked branched evolution of low-grade endometrioid endometrial carcinoma. Mol. Cancer Res. 17, 731–740 (2019).
Harms, K. L., et al. Virus-positive merkel cell carcinoma is an independent prognostic group with distinct predictive biomarkers. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-20-0864 (2021).
Harms, P. W. et al. Cutaneous basal cell carcinosarcomas: evidence of clonality and recurrent chromosomal losses. Hum. Pathol. 46, 690–697 (2015).
Nemes, S. et al. A diagnostic algorithm to identify paired tumors with clonal origin. Genes Chromosomes Cancer 52, 1007–1016 (2013).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Diaz-Gay, M. et al. Mutational Signatures in Cancer (MuSiCa): a web application to implement mutational signatures analysis in cancer samples. BMC Bioinform. 19, 224 (2018).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco 2, 401–404 (2012).
Sung, S., Shirazi, M., Shu, C. A., Crapanzano, J. P. & Saqi, A. Pulmonary small cell carcinoma: review, common and uncommon differentials, genomics and management. Diagn. Cytopathol. 48, 790–803 (2020).
Zilberg, C. et al. Analysis of clinically relevant somatic mutations in high-risk head and neck cutaneous squamous cell carcinoma. Mod. Pathol. 31, 275–287 (2018).
Kwun, H. J. et al. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 14, 125–135 (2013).
Gandhi, J. S., Alnoor, F., Sadiq, Q., Solares, J. & Gradowski, J. F. SMARCA4 (BRG1) and SMARCB1 (INI1) expression in TTF-1 negative neuroendocrine carcinomas including merkel cell carcinoma. Pathol. Res. Pr. 219, 153341 (2021).
Orlando, K. A., et al. Re-expression of SMARCA4/BRG1 in small cell carcinoma of ovary, hypercalcemic type (SCCOHT) promotes an epithelial-like gene signature through an AP-1-dependent mechanism. Elife 9, e59073 (2020).
Huang, S. C. et al. The clinicopathological and molecular analysis of gastric cancer with altered SMARCA4 expression. Histopathology 77, 250–261 (2020).
Vanacker, L. et al. Mixed adenoneuroendocrine carcinoma of the colon: molecular pathogenesis and treatment. Anticancer Res. 34, 5517–5521 (2014).
Davies, A., Zoubeidi, A. & Selth, L. A. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr. Relat. Cancer 27, R35–R50 (2020).
Clermont, P. L. et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenetics 7, 40 (2015).
Harms, P. W. et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod. Pathol. 29, 240–248 (2016).
Harms, K. L. et al. Increased expression of EZH2 in Merkel cell carcinoma is associated with disease progression and poorer prognosis. Hum. Pathol. 67, 78–84 (2017).
Perdigoto, C. N. et al. Polycomb-mediated repression and sonic hedgehog signaling interact to regulate merkel cell specification during skin development. PLoS Genet 12, e1006151 (2016).
Matsushita, M. et al. Decreased H3K27me3 expression is associated with merkel cell polyomavirus-negative merkel cell carcinoma, especially combined with cutaneous squamous cell carcinoma. Anticancer Res. 39, 5573–5579 (2019).
Busam, K. J. et al. Reduced H3K27me3 expression in Merkel cell polyoma virus-positive tumors. Mod. Pathol. 30, 877–883 (2017).
Dahiya, A., Wong, S., Gonzalo, S., Gavin, M. & Dean, D. C. Linking the Rb and polycomb pathways. Mol. Cell 8, 557–569 (2001).
Harms, P. W., et al. Viral status predicts patterns of genome methylation and decitabine response in merkel cell carcinoma. J. Invest. Dermatol. Electronically published Aug 30, 2021 (2021).
Song, L. et al. The HDAC inhibitor domatinostat promotes cell-cycle arrest, induces apoptosis, and increases immunogenicity of merkel cell carcinoma cells. J. Invest. Dermatol. 141, 903–912 e904 (2021).
Ritter, C. et al. Reversal of epigenetic silencing of MHC class I chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci. Rep. 6, 21678 (2016).
Ugurel, S. et al. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: a case series. Cancer Immunol. Immunother. 68, 983–990 (2019).
Paulson, K. G. et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2, 1071–1079 (2014).
Kervarrec, T. et al. Polyomavirus-positive merkel cell carcinoma derived from a trichoblastoma suggests an epithelial origin of this merkel cell carcinoma. J. Invest. Dermatol. 140, 976–985 (2020).
La Rosa, S., Sessa, F. & Uccella, S. Mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs): unifying the concept of a heterogeneous group of neoplasms. Endocr. Pathol. 27, 284–311 (2016).
Li, P. et al. Cervical small cell carcinoma frequently presented in multiple high risk HPV infection and often associated with other type of epithelial tumors. Diagn. Pathol. 13, 31 (2018).
Marcus, D. M., Edgar, M. A., Hawk, N. N., Sullivan, P. S. & Stapleford, L. J. Small cell carcinoma of the anus in the setting of prior squamous dysplasia and carcinoma in situ. J. Gastrointest. Oncol. 4, E1–E4 (2013).
Yamamoto, J., Ohshima, K., Ikeda, S., Iwashita, A. & Kikuchi, M. Primary esophageal small cell carcinoma with concomitant invasive squamous cell carcinoma or carcinoma in situ. Hum. Pathol. 34, 1108–1115 (2003).
Wang, G. et al. Small cell carcinoma of the urinary bladder: a clinicopathological and immunohistochemical analysis of 81 cases. Hum. Pathol. 79, 57–65 (2018).
Falto Aizpurua, L. A. et al. A case of combined Merkel cell carcinoma and squamous cell carcinoma: Molecular insights and diagnostic pitfalls. JAAD Case Rep. 4, 996–999 (2018).
Miller, T. I. et al. A genomic survey of sarcomas on sun-exposed skin reveals distinctive candidate drivers and potentially targetable mutations. Hum. Pathol. 102, 60–69 (2020).
Aran, D. et al. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 8, 1077 (2017).
Acknowledgements
We thank the Rogel Cancer Center Tissue and Molecular Pathology Shared Resource (supported by NIH P30 CA04659229) for assistance with tissue processing. We thank Aniket Dagar for bioinformatics assistance. We thank Sharon Kerr, Threase Nickerson, and Karen Myers for administrative support.
Funding
This work was supported by NIH grants CA189352 (A.A.D. and M.E.V.), CA241947 (A.A.D., M.E.V., and P.W.H.), CA046592 (University of Michigan Rogel Cancer Center Support Grant to A.A.D. and P.W.H.), AR075043 (University of Michigan Skin Biology and Diseases Resource-based Center to A.A.D. and P.W.H.), and the AP Project Funding Committee, Department of Pathology, University of Michigan.
Author information
Authors and Affiliations
Contributions
Conceptualization: P.W.H., A.M., A.A.D.; Data Curation: K.H., A.M.U.; Formal Analysis: P.W.H., K.H., A.M.U.; Funding Acquisition: P.W.H., M.E.V., M.P.C., A.U.M., A.A.D.; Investigation: M.E.V., K.H., S.M.H., R.M.P., M.P.C., A.M.U., P.W.H.; Methodology: M.E.V., K.H., S.M.H., M.P.C., A.M.U., A.A.D.; Project Administration: C.L., A.M.U., A.A.D.; Resources: M.P.C., R.M.P., A.M.U., A.A.D.; Software: K.H., A.M.U.; Supervision: C.L., M.P.C., A.M.U., A.A.D.; Validation: P.W.H., M.E.V., K.H., S.M.H.; Visualization: P.W.H., M.E.V.; Writing - Original Draft Preparation: P.W.H., M.E.V., A.M.U., A.A.D.; Writing - Review and Editing: P.W.H., M.E.V., K.H., S.M.H., M.P.C., C.L., M.G., R.M.P., A.M.U., A.A.D.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was conducted under a protocol approved by the Institutional Review Board at the University of Michigan (HUM00045834).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Harms, P.W., Verhaegen, M.E., Hu, K. et al. Genomic evidence suggests that cutaneous neuroendocrine carcinomas can arise from squamous dysplastic precursors. Mod Pathol 35, 506–514 (2022). https://doi.org/10.1038/s41379-021-00928-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00928-1
This article is cited by
-
RB1-deficient squamous cell carcinoma: the proposed source of combined Merkel cell carcinoma
Modern Pathology (2022)
