
Abstract
Most pathologists are well versed in the diagnosis of lung cancer, given the common nature of the disease. Occasionally more unusual neoplasms are encountered in lung biopsies and resections, which may be difficult to distinguish from “run of the mill” lung cancer cases based on overlapping morphologic and immunophenotypic features. The accurate diagnosis of these rare entities is quite challenging and requires careful morphological examination paired with judicious use of ancillary immunohistochemical and genetic studies. Herein, the clinicopathological and genetic features of five unusual lung tumors will be reviewed, including thoracic SMARCA4-deficient undifferentiated tumor, NUT carcinoma, sclerosing pneumocytoma, primary pulmonary myxoid sarcoma/angiomatoid fibrous histiocytoma, and bronchiolar adenoma/ ciliated muconodular papillary tumor. Since recognition of these entities by pathologists is of increasing importance to guide prognosis and therapy, emphasis will be placed on practical tips to reach these rare diagnoses with confidence.
Similar content being viewed by others
Introduction
Rare neoplasms always pose a particular diagnostic challenge to the pathologist, simply because unusual things are harder to recognize. This has become a much more significant problem over the past 50 years, as tumor classification schemes have become more and more complex. While classical historical pathologic tumor classification systems relied exclusively on morphological features based on H&E examination, the advent of increasingly sophisticated ancillary testing to aid in the subclassification of tumors and the explosion of knowledge surrounding tumor genetics has led to more confident and more complicated tumor subclassification. This makes it difficult for pathologists to keep up with emerging and rare entities. Adding to this challenge is the reality of individualized medicine, whereby the pathologic diagnosis can be critical to determining specific tumor-directed therapy. In this complex setting, misdiagnosis of a rare entity may have clinical implications. However, pathologists armed with basic knowledge about rare entities and proper application of ancillary testing will be able to reach a confident diagnosis in many cases. Louis Pasteur astutely stated “in the fields of observation, chance favors the only prepared mind”1. Indeed, many of these rare lung tumors have quite characteristic morphological features and can be recognized by a pathologist that has a general familiarity with their typical features. To that end, this review will discuss the historical perspective, clinicopathologic features, diagnostic pathology, and genetics of 5 unusual but distinctive pulmonary neoplasms, including thoracic SMARCA4-deficient undifferentiated tumor (SMARCA4-UT), NUT carcinoma, sclerosing pneumocytoma (SP), primary pulmonary myxoid sarcoma/angiomatoid fibrous histiocytoma (PPMS/ AFH), and bronchiolar adenoma/ ciliated muconodular papillary tumor (CMPT).
Thoracic SMARCA4-deficient undifferentiated tumor
Historical perspective and clinicopathologic features
Aggressive thoracic neoplasms with SMARCA4-deficiency were first described by Le Loarer et al. in 20152. The transcriptional profiles of these tumors were felt to be more similar to malignant rhabdoid tumors than to lung nonsmall cell carcinomas with SMARCA4 loss, and thus the term “SMARCA4-deficient thoracic sarcoma” was proposed. Results of this original study as well as several other clinicopathological series have shown that these tumors have a male predominance and tend to occur in younger to middle-aged patients, although the age range is broad2,3,4. Patients often present with symptoms attributable to a large compressive mediastinal mass, and most show at least focal lung involvement. They may also present with symptoms of distant metastasis3. Affected patients are typically smokers, and interestingly, emphysema is often notable even in young patients4. This has led some to postulate that patients at risk for this malignancy may have a particular susceptibility to smoking-related lung damage. Unfortunately, the clinical course is uniformly aggressive, with a high stage at presentation and frequent metastases to the lymph nodes, brain, bone, adrenal glands, and abdominal cavity; death from the disease usually occurs in less than 12 months2,3,4.
Since their original description, it has been noted that thoracic SMARCA4-deficient malignancies do sometimes harbor mutations that are common in lung adenocarcinoma (TP53, STK11, KEAP1, KRAS, etc.), and a recent genetic study has shown that they harbor a genomic smoking signature4,5. They also have a high tumor mutation burden and copy number alterations, similar to nonsmall cell lung carcinoma (NSCLC)5. Rare examples of abrupt SMARCA4-deficiency with typical undifferentiated morphology have also been observed to occur with adjacent areas of recognizable NSCLC5. These genetic and morphological data, along with the frequent association with smoking and the metastatic pattern strikingly similar to NSCLC, have led to the hypothesis that SMARCA4-deficient tumors in the thorax may represented a form of undifferentiated/sarcomatoid lung cancer5. For these reasons, the newly released 2021 thoracic WHO has classified these as “thoracic SMARCA4-deficient undifferentiated tumor” (SMARCA4-UT)6.
Diagnostic pathology
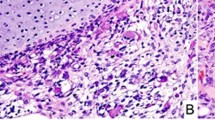
Thoracic SMARCA4-UT has undifferentiated but quite distinctive histomorphologic features. They are composed of sheets of moderately discohesive and relatively monotonous epithelioid cells with indistinct cell borders, moderate eosinophilic cytoplasm, ovoid nuclei, and vesicular chromatin with nucleoli that are often prominent (Fig. 1)2,3,4,7. Mitotic activity and necrosis are common. Characteristically, the tumor shows at least focal areas of rhabdoid morphology, with eccentrically placed nuclei and abundant dense eosinophilic cytoplasm. This undifferentiated morphology may lead to a broad differential diagnosis, with consideration of poorly differentiated carcinoma, melanoma, hematolymphoid neoplasms, or sarcomas that can show rhabdoid morphology such as proximal-type epithelioid sarcoma and malignant rhabdoid tumor.

Thoracic SMARCA4-deficient undifferentiated tumor with sheets of monotonous, moderately dyscohesive epithelioid cells with vesicular chromatin and moderately sized nucleoli (A, H&E, 200x). The tumor demonstrates increased mitotic activity and areas of necrosis (B, H&E, 200x). Focal rhabdoid morphology is observed in an area of lymphovascular invasion (C, H&E, 200x), characterized by abundant eccentric densely eosinophilic cytoplasm. The tumor cells show loss of SMARCA4/BRG1 by immunohistochemistry (D, 200x).
Immunohistochemistry is generally required to establish the diagnosis of thoracic SMARCA4-UT and to exclude other entities in the differential diagnosis. Thoracic SMARCA4-UTs typically show very focal expression of low-molecular weight keratin and EMA, along with characteristic loss of SMARCA4 (also known as BRG1) by immunohistochemistry3,4. While the loss of SMARCA4/BRG1 is typically characterized by complete loss of nuclear expression by immunohistochemistry, rare cases may show a severe global reduction in expression, with barely perceptible weak nuclear staining in tumor cells, which can also be considered a pattern of loss4. As with other stains requiring assessment of abnormal loss of expression, evaluation for strong positive internal control staining in non-tumor cells (inflammatory cells, stromal cells, etc.) is critical to avoid misinterpretation. Thoracic SMARCA4-UT usually also show loss of SMARCA2 (also known as BRM) expression by immunohistochemistry (see genetics section below)2,3,4. S100, desmin, and myogenin are typically negative3, as is claudin 44. Thoracic SMARCA4-UTs frequently express “stem cell markers,” such as CD34, SALL4, and SOX2, and patchy synaptophysin expression is also common3,4,5. The frequent synaptophysin staining can be particularly problematic in the lung, and can lead to misclassification as small cell carcinoma or large cell neuroendocrine carcinoma5. Rare examples may show focal expression of TTF1, p63 or p40, although most SMARCA4-UT are negative for these markers3,5,7.
It is important to remember that SMARCA4/BRG1 loss is not specific to thoracic SMARCA4-UT, and it can be seen in other rhabdoid neoplasms (small cell carcinoma of the ovary-hypercalcemic type, malignant rhabdoid tumor, atypical teratoid/rhabdoid tumor) as well as in carcinomas of the lung, head, and neck, or gastrointestinal tract, and in uterine sarcomas8,9,10,11,12,13. Therefore, the diagnosis should always be made with consideration of the overall immunophenotype, as well as the clinical and radiographic context. The most important consideration in this differential diagnosis is often NSCLC with SMARCA4/BRG1 loss, as this loss may occur in up to 5% of NSCLCs, likely as a passenger mutation8,14,15,16,17. NSCLCs with SMARCA4/BRG1 loss usually show morphology more typical of carcinoma which may include glandular or squamous differentiation, along with diffuse expression of keratin and claudin 4, and retained expression of SMARCA2/BRM17. They are also generally negative for CD34, SALL4, and SOX217. SMARCA4-deficient adenocarcinomas may express Hep-Par, which can lead to confusion with metastatic hepatocellular carcinoma8.
From a practical standpoint, it is a good idea to add immunohistochemistry for SMARCA4/BRG1 when typical morphological features are noted (i.e., undifferentiated monotonous high-grade malignancy with or without focal rhabdoid features), and after immunohistochemistry has been employed to exclude other possible diagnoses which illustrates a compatible immunophenotype (i.e., focal expression of keratin/EMA, possible expression of stem cell markers).
Genetic characteristics
The namesake loss of SMARCA4 is the defining genetic characteristic of these neoplasms. The SMARCA4/BRG1 protein is a critical catalytic subunit of the BAF (BRG1/BRM-associated factor) chromatin remodeling complex18. This chromatin remodeling complex is also frequently called the SWI/SNF complex. Loss of SMARCA4/BRG1 activity may be due to nonsense, frameshift, and/or loss of heterozygosity mutations in SMARCA415,18,19. This complex has tumor suppressor activity through its role in promoting cell differentiation20. The SMARCA2/BRM protein can act as an interchangeable catalytic subunit in the BAF complex in place of SMARCA4/BRG1, and thus thoracic SMARCA4-UT usually shows loss of both proteins7. SMARCB1 (also known as INI1) is also a member of the BAF complex, and thus it is not surprising that SMARCA4-UT shares some morphological characteristics with other tumors driven by SMARCB1 loss (malignant rhabdoid tumor, proximal-type epithelioid sarcoma, atypical teratoid rhabdoid tumor, etc.).
Therapy and Prognosis
Thoracic SMARCA4-UTs have a uniformly aggressive behavior and poor response to conventional chemotherapeutic regiments2,3,4. Given that the tumors are thought to be driven by the loss of BAF complex function, there is a theoretical basis for therapy with bromodomain inhibitors and EZH2 inhibitors21,22, but more research is needed to determine the efficacy of these drugs against thoracic SMARCA4-UT. There have also been individual reports of quite dramatic response to immune checkpoint inhibitors23, making immunotherapy a potentially attractive therapeutic option.
NUT carcinoma
Historical perspective and clinicopathologic features
Rare intrathoracic carcinomas occurring in unusually young patients and showing a characteristic t(15;19) were first described in the early 1990s24,25. This translocation was later shown to lead to BRD-NUT fusion, and resulted in an aggressive form of carcinoma with a proclivity for the midline structures of the body, including the head and neck and mediastinum26. Thus, the name “NUT midline carcinoma” was originally adopted27,28. While first described in young patients, more widespread accessibility of confirmatory testing (genetic testing and/or immunohistochemistry) has revealed that these tumors occur across a wide age range29,30,31. It has also become clear that these tumors do not always occur along the midline, and that they may occur in the lung parenchyma32. Therefore, the WHO classification of tumors now uses the term “NUT carcinoma” for this entity6. Overall, about half of NUT carcinomas occur in the thorax, where they typically present with symptoms of advanced-stage disease including, cough, chest pain, dyspnea, and shortness of breath6,31.
Diagnostic pathology
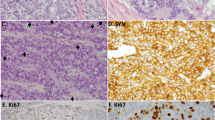
NUT carcinomas can show somewhat variable morphology. Classically, NUT carcinoma is composed of primitive monotonous cohesive cells with scant to moderate cytoplasm (Fig. 2). They may have a basaloid appearance but nuclear molding is usually not present, and glanular differentiation is typically absent6. Nuclei are enlarged and hyperchromatic, and may show prominent nucleoli. Necrosis is common and mitotic activity is brisk. The tumor often shows abrupt squamous differentiation, with discrete nests of cells showing keratinization. However, this classical abrupt keratinization may be present in only a minority of cases, and some tumors are more poorly differentiated without obvious squamous differentiation, bearing resemblance to an undifferentiated NSCLC31,33. Areas of squamous differentiation may also be absent on small biopsy samples, which can make the diagnosis more challenging. The morphologic differential diagnosis may include NSCLC, small cell carcinoma, lymphoma/ leukemia, SMARCA4-UT, or small round cell sarcoma.

NUT carcinoma composed of primitive, monotonous, and cohesive basaloid cells (A, H&E, 200x) showing classical areas of abrupt squamous differentiation with keratin pearls (B, H&E, 400x). This tumor shows the expression of p40 (C, 200x) and NUT (D, 200x) by immunohistochemistry.
NUT carcinomas usually have a squamous phenotype, with the expression of keratin, p63, and p406. There is some evidence that p63 is more sensitive than p40 in this diagnostic setting34. Undifferentiated examples may not show squamous marker expression and may be positive for only keratin, and examples have been described that demonstrate TTF1 and/or neuroendocrine marker expression33. NUT carcinomas are often positive for CD34, which is unusual for carcinoma and can be a helpful diagnostic clue6. The diagnostic NUT immunostain leads to typically dot-like or speckled nuclear staining, and is about 90% sensitive to confirm the diagnosis35. The stain is very specific for neoplasms with NUT rearrangement33, but may also be expressed in germ cell tumors, so consideration of the overall immunophenotype and morphology is important35.
Given the rarity of NUT carcinoma, it may be difficult for a pathologist to decide when to evaluate for this entity, especially if they do not have the immunostain readily available. As a practical approach to thoracic tumors, it is prudent to obtain NUT immunostaining when there is typical morphology (i.e., basaloid-appearing or monotonous squamous carcinoma), for all squamous carcinomas in the mediastinum, and for any lung squamous cell carcinomas that occur outside the expected demographics (i.e., in never smokers or in patients under 45 years old). It has been proposed by some authors that NUT staining is warranted in all primitive-appearing thoracic neoplasms33, which seems to be a reasonable approach once other diagnoses have been excluded.
Genetic characteristics
The t(15;19) translocation that genetically defines NUT carcinoma leads to BRD4-NUT fusion in over three quarters of cases, or the alternative BRD3-NUT fusion in about 15%36. The fusion protein is localized to distinctive nuclear foci corresponding to “megadomains” of hyper-acetylated chromatin, where it binds acetylated histones and drives transcription37. This leads to the promotion of cell growth and inhibition of cell differentiation. The molecular confirmation of NUT rearrangement can be assessed by the florescence in situ hybridization (FISH) for NUT, or by next-generation sequencing33. However, NUT expression by immunohistochemistry is confirmatory in the correct context, so genetic confirmation is not always required35.
Interestingly, NUT rearrangement has been recently demonstrated outside the traditional setting of NUT carcinoma. NUT rearrangement has been observed in tumors with other phenotypes, including round cell sarcoma, spindle cell sarcoma, myxoid sarcoma, and undifferentiated epithelioid/rhabdoid tumors38. These NUT-rearranged sarcomas often demonstrate alternative fusion partners rather than BRD4, including CIC, MGA, and ZNF532, and are usually positive for the NUT immunostain39,40,41. Therefore, it seems likely that the family of tumors with NUT rearrangement is broader than originally recognized, and the classification of this emerging group of tumors is yet to be defined6.
Therapy and prognosis
NUT carcinomas are very aggressive, and resistant to therapy with conventional chemotherapy regiments6. Lymphatic and hematogenous metastases are common, and survival is usually <12 months from the time of diagnosis6,33. There is a hypothetical basis for therapy with bromodomain inhibitors, and there have been some reports of response to this targeted therapy42 but more study is needed regarding therapeutic options for these aggressive tumors.
Sclerosing pneumocytoma
Historical perspective and clinicopathologic features
Unusual neoplasms with frequent blood pools, prominent sclerosis, and mixed growth pattern have long been known to occur in the lung. Due to the often-prominent vascularity, these tumors were originally hypothesized to be of vascular origin, and the name “pulmonary sclerosing hemangioma” was adopted43. However, subsequent immunohistochemical and ultrastructural evidence has shown that these tumors are actually of pneumocyte origin, with TTF1-expression in the neoplastic cells44,45. This realization has led to renaming of this entity as “sclerosing pneumocytoma” (SP)6. SPs usually form small solitary solid peripheral lung masses, which are often incidentally discovered, and are most common in adults with a female predominance44,46.
Diagnostic pathology
SPs typically show a heterogenous growth pattern, which may include varying proportions of papilla, fibrosis, blood pools, and areas of solid growth6. Depending on which components are most represented, this variable morphology can lead to confusion with adenocarcinoma, carcinoid tumor, or vascular tumors. SP is composed of a mixture of surface cells and interstitial round cells, and recognition of these two cell populations based on morphology and immunophenotype will lead to the correct diagnosis (Fig. 3). It is also helpful to note that the cores of the papilla are composed of round cells, and not true fibrovascular cores. The surface cells resemble type 2 pneumocytes both morphologically and immunophenotypically, with the expression of keratin, TTF1, EMA, and napsin44,47. This has led to some controversy as to whether the surface cells are actually a neoplastic component of the tumor, or if they reflect reactive type 2 pneumocytes entrapped within the neoplasm47,48, but the WHO classification currently considers the surface cells to be a neoplastic component6. The interstitial neoplastic cells are bland round to epithelioid cells with small round nuclei, absent or inconspicuous nucleoli, and scant to moderate eosinophilic cytoplasm. They express TTF1 and EMA, may be positive for estrogen and progesterone receptors, and are typically negative for keratin and napsin44,47. Neuroendocrine markers are negative.

Sclerosing pneumocytoma demonstrating blood pools separated by bland interstitial cells (A, H&E, 100x). Papilla lined by luminal type 2 pneumocyte-like cells, which lack true fibrovascular cores and instead have a core composed of bland interstitial round cells (B, H&E, 200x). The interstitial round cells show areas of solid growth (C, H&E, 200x). Both interstitial and round cells are positive for TTF1 (D, 200x).
The diagnosis of sclerosing pneumocytoma can be especially difficult on small biopsies and frozen sections, where it can be mistaken for adenocarcinoma49. From a practical perspective, the diagnosis of SP should be considered and immunohistochemistry should be employed to evaluate for this possibility when the typical mixed growth pattern is observed, or when a diagnosis of adenocarcinoma is considered based on papillary or solid growth, but the cells look unusually bland. Another helpful clue based on immunohistochemistry is the observation that TTF1-positive cells seem to prominently outnumber keratin-positive cells.
Genetic characteristics
SP shows recurrent genetic abnormalities in the AKT1 gene, a serine-threonine kinase in the mTOR pathway, which is an oncogene with a critical role in cell differentiation, proliferation, and survival. AKT1 mutations have been observed in about 80% of SP, and include the common E17K hotspot mutation and other point mutations, as well as internal tandem duplications, insertions, and deletions50,51,52. Uncommon mutations in other genes in the mTOR pathway have also been observed in SP, including PTEN and PIK3R1, providing more evidence that this pathway is key to tumorigenesis in these neoplasms52.
Therapy and prognosis
Most SP are benign and cured by surgical excision6. However, rare patients will experience recurrence, and metastasis to lung and lymph nodes have been reported53,54. Some patients also have multifocal disease52. For the unusual situation where systemic therapy may be required, the genetic underpinnings provide the rationale for targeted therapy with mTOR inhibitors, although more study is needed in this area to determine clinical efficacy52.
Primary pulmonary myxoid sarcoma and pulmonary angiomatoid fibrous histiocytoma
Historical perspective and clinicopathologic features
Primary pulmonary myxoid sarcoma (PPMS) is a very rare mesenchymal neoplasm characterized by EWSR1-CREB1 fusion in 80% of cases, which was first described by Thway et al. in 201155. PPMS usually forms a circumscribed, lobulated mass related to a bronchus with frequent endobronchial growth, and they are most common in middle-aged adults. Due to the endobronchial growth pattern, patients may present with cough and hemoptysis, or signs of bronchial obstruction6,55. In the 2021 thoracic WHO classification, this tumor has been coined “primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion”6. Interestingly, another tumor that can show the exact same fusion gene is angiomatoid fibrous histiocytoma (AFH), which has also been rarely described to occur in the large airways56. Although AFH is most often observed as a pediatric soft tissue tumor, pulmonary tumors are most common in middle aged to older adults56.
Diagnostic pathology
PPMS are composed of spindled, stellate, and polygonal cells deposited with abundant myxoid stroma (Fig. 4). They often have lymphoplasmacytic inflammation associated with the tumor, which may be more prominent at the periphery. A fibrous tumor pseudocapsule may also be present6. The tumor cells are often arranged in strands and cords and show mild atypia with small hyperchromatic nuclei, limited pleomorphism, and a generally low mitotic rate55. Necrosis may be present but is not common. Overall, this appearance may bear a striking resemblance to extraskeletal myxoid chondrosarcoma. PPMS generally has a nonspecific immunophenotype, with focal expression of EMA and negativity for other markers including keratin, S100, desmin, neuroendocrine, and vascular markers55.

Both primary pulmonary myxoid sarcoma (PPMS) and angiomatoid fibrous histiocytoma (AFH) can have prominent fibrous pseudocapsule and lymphoid rim (A). Classical examples of AFH have bland fibrohistiocytic cells with background hemosiderin (B), while PPMS typically has spindled (C) to epithelioid shaped cells (D) in a prominent myxoid background.
Endobronchial AFH generally has a morphology similar to those encountered in soft tissue. AFH classically show large blood lakes, and the tumor is surrounded by a pronounced rim of lymphoplasmacytic infiltrate and a fibrous pseudocapsule57. In the soft tissue, this has often been described as having the low-power appearance of a “bloody lymph node” or a lymph node involved by metastatic disease. However, the classical blood lakes may be lacking in pulmonary examples of AFH56. The neoplastic cells of AFH have a fibrohistiocytic appearance (epithelioid to spindled with indistinct cell borders), and often grow in loose fascicles or whorls. They have relatively monotonous ovoid to slightly irregular nuclei with vesicular chromatin, and nucleoli are usually absent or inconspicuous. Hemosiderin-laden macrophages are common, and there may be other mixed chronic inflammatory cells admixed with the tumor cells. The tumor cells are often focally positive for EMA and desmin57, but they are negative for myogenin, myoD1, S100, and keratin.
Given the overall ambiguous and nonspecific immunophenotype of both the PPMS and AFH, confirmatory genetic testing is often desired (see below). In practice, this testing should be considered for any unusual endobronchial neoplasm with compatible morphology and immunophenotype, that does not show typical features for any of the more common tumors in the differential diagnosis of endobronchial neoplasms, including hamartoma, carcinoid tumor, peripheral nerve sheath tumor, or salivary gland tumors. Of note, the salivary neoplasm hyalinizing clear cell carcinoma can rarely occur in the bronchi and also shows recurrent EWSR1 rearrangements but should not be difficult to distinguish from PPMS and AFH based on its typical trabecular growth, polygonal cell shape, and expression of keratin and p40/p636.
Genetic characteristics
As mentioned above, PPMS have EWSR1-CREB1 fusion in 80%56, and AFH also usually show this fusion (>90%) or the less common EWSR1-ATF157. These fusions can be detected by next-generation sequencing, or by FISH for EWSR1. If FISH is used to support the diagnosis, there must be particular attention to the overall morphology and immunophenotype, since EWSR1 rearrangement can be seen in many different neoplasms. This pair of often interchangeable fusions also occurs in other tumors as well, including clear cell sarcoma, gastrointestinal neuroectodermal tumor (a.k.a. clear cell sarcoma-like tumor of the gastrointestinal tract), and hyalinizing clear cell carcinoma of the salivary gland57,58,59. Interestingly, the alternate EWSR1-ATF1 fusion has recently been described in a typical example of PPMS60. This close genetic kinship between PPMS and AFH has raised the possibility that they are morphological variants of the same entity60,61. Although classical examples of PPMS and AFH are quite morphologically distinct, myxoid examples of AFH certainly show morphological similarities to PPMS. More research is needed to elucidate the exact relationship between these two entities, but the overall similar demographics, behavior, and propensity to occur in the bronchi also lend credence to the idea that they may represent variants of the same entity.
Therapy and prognosis
Limited follow-up data have shown that most examples of PPMS follow an indolent clinical course, without recurrence or metastasis after resection. However, in the original series, one patient died from brain metastasis, and another had renal metastasis but was alive and well at last follow-up55. The tumor in the patient that died from the disease did not have the typical EWSR1-CREB1 fusion and the genetic abnormality driving that tumor was unknown, while the tumor from the patient with renal metastasis did have the classical fusion. Therefore, PPMS is considered a tumor of low-grade malignancy (i.e. rarely metastasizing)6. The limited data on endobronchial AFH indicates a favorable clinical course56. While historically soft tissue AFH had been coined “malignant AFH” due to the possibility of metastasis, long-term follow-up has shown metastasis to be an exceptional occurrence in this tumor type, and even in the setting of metastasis, the disease course is often indolent57. Therefore, AFH is also considered a tumor of low-grade malignancy57.
Bronchiolar adenoma/ciliated muconodular papillary tumor
Historical perspective and clinicopathologic features
Ciliated muconodular papillary tumors (CMPTs) were originally described as an unusual benign pulmonary neoplasm occurring in adult patients of East Asian descent62. Subsequent studies have shown that typical CPMT can also occur in adult Caucasian patients as well63. These tumors are usually located peripherally and found incidentally, and are small (<2 cm). They may show slow growth on serial imaging studies63. A recent study proposed reclassifying CMPTs into the broader diagnostic term “bronchiolar adenoma.” The authors argued that the CMPT name is somewhat misleading, since papilla may be focal or absent, and these tumors may demonstrate a quite variable number of ciliated and mucinous cells, with one cell type lacking entirely64. While the described morphology of bronchiolar adenoma was more variable, all examples had a continuous layer of basal cells and were located in the lung periphery. The 2021 thoracic WHO has combined bronchiolar adenoma and CMPT into a single entity6. There still remains some debate as to the possible relationship of other entities to this group, including mixed glandular and squamous papillomas and sialadenoma papilliferum, which show some morphological similarities and well as similar genetic findings65.
Diagnostic pathology
Bronchiolar adenoma/CMPTs often show a central “naked” artery (i.e., unpaired with an airway), and are typically surrounded by a pool of extracellular mucin (Fig. 5)62,63. There is often an associated chronic inflammatory infiltrate, which may be quite prominent. The tumor often shows a mixture of papillary architecture and flat glandular architecture, but one pattern may predominate. The cells of bronchiolar adenoma/CPMT generally lack cytological atypia. Classical CMPTs are typically characterized by a variable mix of bland ciliated and mucinous cells, and multilayered “squamoid” cells may also be present63. This morphology has also been referred to as “proximal-type” bronchiolar adenoma64. This is in contrast to cases which have a more cuboidal type 2 pneumocyte-like population of cells, which have been described as “distal-type” bronchiolar adenoma64. This distinction is not critical for any clinical purpose but is helpful to understand the spectrum of morphology that can be observed in bronchiolar adenoma/CMPT.

Bronchiolar adenoma/ciliated muconodular papillary tumor, showing mucosatasis and associated chronic inflammation on low power (A, 40x). The tumor is composed of a combination of mucus cells, ciliated cells, and squamoid cells (B, 200x). All tumor components are variably positive for TTF1 (C, 200x) and have a continuous basal layer highlighted by p63 immunohistochemistry (D,200x).
By immunohistochemistry, TTF1 may be variably expressed in the various cellular components. Importantly, bronchiolar adenoma/CMPT shows a continuous layer of basal cells, which are positive for CK 5/6, p40, and p6363,64. This finding is very helpful in differentiating from adenocarcinoma, which is the main differential diagnosis based on the presence of papilla and mucinous cells. This distinction is particularly challenging on frozen section, where cilia and basal cells may be very hard to appreciate. In one study, over three quarters of bronchiolar adenomas/CMPTs analyzed were called adenocarcinoma at the time of frozen section64. From a practical standpoint, it is always prudent to evaluate for ciliated cells before rendering a final diagnosis of mucinous and/or papillary adenocarcinoma, especially in a very small lesion. When it doubt, stains for basal cells will help guide correct classification.
Genetic characteristics
Bronchiolar adenomas/CPMTs have frequent oncogenic hotspot mutations, including BRAF V600E and AKT1 E17K63. Mutations have also been found in EGFR, KRAS, and HRAS64,66. While the clinical course is indolent, the presence of these mutations does indicate a neoplastic etiology.
Therapy and prognosis
Although follow-up data is somewhat limited due to the rare nature of bronchiolar adenoma/CPMT, they are considered benign neoplasms as thus far there have been no convincing reports of recurrence or metastasis62,63,64.
Concluding practical tips
It can be difficult for pathologists to reach the correct diagnosis of a rare neoplastic entity in the lung. However, these distinctions have important implications regarding therapy, patient follow-up, and prognosis. While these diagnoses pose a particular challenge, there are a few practical tips that can help in recognition of these unusual tumor entities:
-
1.
Pathologists should consider staining all undifferentiated high-grade malignancies in the lung and mediastinum for NUT and BRG1/SMARCA4, once other diagnostic possibilities have been excluded.
-
2.
Staining for NUT should always be considered in thoracic squamous cell carcinomas occurring in never smokers, patients <45 years of age, or with a mediastinal location.
-
3.
Evaluation for EWSR1 fusion genes by FISH or next-generation sequencing should be undertaken for unusual bronchial myxoid or fibrohistiocytic neoplasms, once other likely diagnoses have been excluded.
-
4.
Consider unusual pitfalls in the diagnosis of pulmonary adenocarcinoma:
-
a.
Sclerosing pneumocytoma should enter the differential diagnosis if there is a combination of blood lakes, papilla, solid and sclerotic areas, and/or if TTF1 is staining significantly more cells than keratin.
-
b.
Bronchiolar adenoma/ciliated muconodular papillary tumor is a challenging pitfall in the differential diagnosis of adenocarcinoma-always look for ciliated cells before diagnosing adenocarcinoma with mucinous features, and consider markers for basal cells if an unusual mixture of mucinous, ciliated and/or multilayered cells is observed.
These tips used in conjunction with detailed morphological exam and judicious use of ancillary testing will allow for accurate diagnosis to optimize patient care.
Data availability
No primary data was used in the creation of this review.
References
Pasteur, L. Lecture, 1854, University of Lille: Lille (Nord de France).
Le Loarer, F. et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat. Genet. 47, 1200–1205 (2015).
Sauter, J. L. et al. SMARCA4-deficient thoracic sarcoma: a distinctive clinicopathological entity with undifferentiated rhabdoid morphology and aggressive behavior. Mod. Pathol. 30, 1422–1432 (2017).
Yoshida, A. et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities. Mod. Pathol. 30, 797–809 (2017).
Rekhtman, N. et al. SMARCA4-deficient thoracic sarcomatoid tumors represent primarily smoking-related undifferentiated carcinomas rather than primary thoracic sarcomas. J. Thorac. Oncol. 15, 231–247 (2020).
WHO Clsasification of Tumours Editoral Board. in Thoracic Tumours. 5th ed, Vol 5 (eds WHO Classification of Tumours) (IARC Press, 2021).
Perret, R. et al. SMARCA4-deficient thoracic sarcomas: clinicopathologic study of 30 cases with an emphasis on their nosology and differential diagnoses. Am. J. Surg. Pathol. 43, 455–465 (2019).
Agaimy, A. et al. SMARCA4-deficient pulmonary adenocarcinoma: clinicopathological, immunohistochemical, and molecular characteristics of a novel aggressive neoplasm with a consistent TTF1(neg)/CK7(pos)/HepPar-1(pos) immunophenotype. Virchows Arch. 471, 599–609 (2017).
Agaimy, A. et al. SWI/SNF complex-deficient undifferentiated/rhabdoid carcinomas of the gastrointestinal tract: a series of 13 cases highlighting mutually exclusive loss of SMARCA4 and SMARCA2 and frequent co-inactivation of SMARCB1 and SMARCA2. Am. J. Surg. Pathol. 40, 544–553 (2016).
Agaimy, A., Jain, D., Uddin, N., Rooper, L. M. & Bishop, J. A. SMARCA4-deficient sinonasal carcinoma: a series of 10 cases expanding the genetic spectrum of SWI/SNF-driven sinonasal malignancies. Am. J. Surg. Pathol. 44, 703–710 (2020).
Kolin, D. L. et al. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma. Mod. Pathol. 31, 1442–1456 (2018).
Hasselblatt, M. et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am. J. Surg. Pathol. 35, 933–935 (2011).
Karanian-Philippe, M. et al. SMARCA4 (BRG1) loss of expression is a useful marker for the diagnosis of ovarian small cell carcinoma of the hypercalcemic type (ovarian rhabdoid tumor): a comprehensive analysis of 116 rare gynecologic tumors, 9 soft tissue tumors, and 9 melanomas. Am. J. Surg. Pathol. 39, 1197–1205 (2015).
Bell, E. H. et al. SMARCA4/BRG1 is a novel prognostic biomarker predictive of cisplatin-based chemotherapy outcomes in resected non-small cell lung cancer. Clin. Cancer Res. 22, 2396–2404 (2016).
Dagogo-Jack, I. et al. Clinicopathologic characteristics of BRG1-deficient NSCLC. J. Thorac. Oncol. 15, 766–776 (2020).
Naito, T. et al. Non-small cell lung cancer with loss of expression of the SWI/SNF complex is associated with aggressive clinicopathological features, PD-L1-positive status, and high tumor mutation burden. Lung Cancer. 138, 35–42 (2019).
Herpel, E. et al. SMARCA4 and SMARCA2 deficiency in non-small cell lung cancer: immunohistochemical survey of 316 consecutive specimens. Ann. Diagn. Pathol. 26, 47–51 (2017).
Le Quang, M., Ranchère-Vince, D. & Le Loarer F. [Pathological and molecular features of malignancies underlined by BAF complexes inactivation]. Ann. Pathol. 39, 399–413 (2019).
Schoenfeld, A. J. et al. The genomic landscape of SMARCA4 alterations and associations with outcomes in patients with lung cancer. Clin. Cancer Res. 26, 5701–5708 (2020).
Zhang, X. et al. Transcriptional repression by the BRG1-SWI/SNF complex affects the pluripotency of human embryonic stem cells. Stem Cell Rep. 3, 460–474 (2014).
Shorstova, T. et al. SWI/SNF-compromised cancers are susceptible to bromodomain inhibitors. Cancer Res. 79, 2761–2774 (2019).
Yamagishi, M. & Uchimaru, K. Targeting EZH2 in cancer therapy. Curr. Opin. Oncol. 29, 375–381 (2017).
Henon, C. et al. Long lasting major response to pembrolizumab in a thoracic malignant rhabdoid-like SMARCA4-deficient tumor. Ann. Oncol. 30, 1401–1403 (2019).
Kubonishi, I. et al. Novel t(15;19)(q15;p13) chromosome abnormality in a thymic carcinoma. Cancer Res. 51, 3327–3328 (1991).
Kees, U. R., Mulcahy, M. T. & Willoughby, M. L. Intrathoracic carcinoma in an 11-year-old girl showing a translocation t(15;19). Am. J. Pediatr. Hematol. Oncol. 13, 459–464 (1991).
French, C. A. et al. Midline carcinoma of children and young adults with NUT rearrangement. J. Clin. Oncol. 22, 4135–4139 (2004).
French, C. A. NUT midline carcinoma. Cancer Genet. Cytogenet. 203, 16–20 (2010).
French, C. A. et al. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 63, 304–307 (2003).
French, C. A. NUT Carcinoma: clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 68, 583–595 (2018).
Stelow, E. B. et al. NUT rearrangement in undifferentiated carcinomas of the upper aerodigestive tract. Am. J. Surg. Pathol. 32, 828–834 (2008).
Chau, N. G. et al. An anatomical site and genetic-based prognostic model for patients with nuclear protein in testis (NUT) midline carcinoma: analysis of 124 patients. JNCI Cancer Spectr. 4, pkz094 (2020).
Sholl, L. M. et al. Primary pulmonary NUT midline carcinoma: clinical, radiographic, and pathologic characterizations. J. Thorac. Oncol. 10, 951–959 (2015).
Hung, Y. P. et al. Thoracic nuclear protein in testis (NUT) carcinoma: expanded pathological spectrum with expression of thyroid transcription factor-1 and neuroendocrine markers. Histopathology. 78, 896–904 (2021).
Matsuda, K., Kashima, J. & Yatabe, Y. The isoform matters in NUT carcinoma: a diagnostic pitfall of p40 immunohistochemistry. J. Thorac. Oncol. 15, e176–e178 (2020).
Haack, H. et al. Diagnosis of NUT midline carcinoma using a NUT-specific monoclonal antibody. Am. J. Surg. Pathol. 33, 984–991 (2009).
French, C. A. et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 27, 2237–2242 (2008).
Alekseyenko, A. A. et al. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 29, 1507–1523 (2015).
Stevens, T. M. et al. NUTM1-rearranged neoplasia: a multi-institution experience yields novel fusion partners and expands the histologic spectrum. Mod. Pathol. 32, 764–773 (2019).
Goto, T., Arai, Y., Shibata, T., Oyama, T. & Yoshida, A. Sarcoma with MGA-NUTM1 fusion in the lung: an emerging entity. Virchows Arch. 476, 317–322 (2020).
Chien, Y. W. et al. Primary malignant epithelioid and rhabdoid tumor of bone harboring ZNF532-NUTM1 fusion: the expanding NUT cancer family. Genes Chromosomes Cancer. 58, 809–814 (2019).
Schaefer, I. M. et al. CIC-NUTM1 fusion: A case which expands the spectrum of NUT-rearranged epithelioid malignancies. Genes Chromosomes Cancer 57, 446–451 (2018).
Stathis, A. et al. Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Disco. 6, 492–500 (2016).
Huszar, M., Suster, S., Herczeg, E. & Geiger, B. Sclerosing hemangioma of the lung. Immunohistochemical demonstration of mesenchymal origin using antibodies to tissue-specific intermediate filaments. Cancer. 58, 2422–2427 (1986).
Devouassoux-Shisheboran, M., Hayashi, T., Linnoila, R. I., Koss, M. N. & Travis, W. D. A clinicopathologic study of 100 cases of pulmonary sclerosing hemangioma with immunohistochemical studies: TTF-1 is expressed in both round and surface cells, suggesting an origin from primitive respiratory epithelium. Am. J. Surg. Pathol. 24, 906–916 (2000).
Satoh, Y. et al. Pulmonary sclerosing hemangioma of the lung. A type II pneumocytoma by immunohistochemical and immunoelectron microscopic studies. Cancer. 64, 1310–1317 (1989).
Iyoda, A. et al. Clinicopathological analysis of pulmonary sclerosing hemangioma. Ann. Thorac. Surg. 78, 1928–1931 (2004).
Yoo, S. H. et al. Expression patterns of markers for type II pneumocytes in pulmonary sclerosing hemangiomas and fetal lung tissues. Arch. Pathol. Lab. Med. 129, 915–919 (2005).
Wang, Y., Wang, E., Wu, G., Zhang, Z. & Lin, D. Immunohistochemical and ultrastructual study of so-called sclerosing hemangioma of the lung suggests different origins of cells. Zhongguo Fei Ai Za Zhi. 6, 92–96 (2003).
Yang, C. H. & Lee, L. Y. Pulmonary sclerosing pneumocytoma remains a diagnostic challenge using frozen sections: a clinicopathological analysis of 59 cases. Histopathology. 72, 500–508 (2018).
Jung, S. H. et al. Whole-exome sequencing identifies recurrent AKT1 mutations in sclerosing hemangioma of lung. Proc. Natl. Acad. Sci. USA. 113, 10672–10677 (2016).
Yeh, Y. C. et al. AKT1 internal tandem duplications and point mutations are the genetic hallmarks of sclerosing pneumocytoma. Mod. Pathol. 33, 391–403 (2020).
Boland, J. M. et al. Molecular Genetic Landscape of Sclerosing Pneumocytomas. Am. J. Clin. Pathol. 155, 397–404 (2021).
Miyagawa-Hayashino, A., Tazelaar, H. D., Langel, D. J. & Colby, T. V. Pulmonary sclerosing hemangioma with lymph node metastases: report of 4 cases. Arch. Pathol. Lab. Med. 127, 321–325 (2003).
Komatsu, T., Fukuse, T., Wada, H. & Sakurai, T. Pulmonary sclerosing hemangioma with pulmonary metastasis. Thorac. Cardiovasc. Surg. 54, 348–349 (2006).
Thway, K. et al. Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: a new tumor entity. Am. J. Surg. Pathol. 35, 1722–1732 (2011).
Thway, K. et al. Endobronchial pulmonary angiomatoid fibrous histiocytoma: two cases with EWSR1-CREB1 and EWSR1-ATF1 fusions. Am. J. Surg. Pathol. 36, 883–888 (2012).
W. H. O. Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours. 5th ed. WHO Classification of Tumours, 2020, IARC Press: Lyon.
Stockman, D. L. et al. Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am. J. Surg. Pathol. 36, 857–868 (2012).
Antonescu, C. R. et al. EWSR1-ATF1 fusion is a novel and consistent finding in hyalinizing clear-cell carcinoma of salivary gland. Genes Chromosomes Cancer. 50, 559–570 (2011).
Gui, H., Sussman, R. T., Jian, B., Brooks, J. S. & Zhang, P. J. L. Primary Pulmonary myxoid sarcoma and myxoid angiomatoid fibrous histiocytoma: a unifying continuum with shared and distinct features. Am. J. Surg. Pathol. 44, 1535–1540 (2020).
Smith, S. C. et al. At the intersection of primary pulmonary myxoid sarcoma and pulmonary angiomatoid fibrous histiocytoma: observations from three new cases. Histopathology. 65, 144–146 (2014).
Kamata, T. et al. Ciliated muconodular papillary tumors of the lung: a clinicopathologic analysis of 10 cases. Am. J. Surg. Pathol. 39, 753–760 (2015).
Liu, L. et al. Ciliated muconodular papillary tumors of the lung can occur in western patients and show mutations in BRAF and AKT1. Am. J. Surg. Pathol. 40, 1631–1636 (2016).
Chang, J. C. et al. Bronchiolar adenoma: expansion of the concept of ciliated muconodular papillary tumors With roposal for revised terminology based on morphologic, immunophenotypic, and genomic analysis of 25 cases. Am. J. Surg. Pathol. 42, 1010–1026 (2018).
Nakaguro, M. et al. Sialadenoma papilliferum of thebronchus: an unrecognized bronchial counterpart of the salivary gland tumor with frequent BRAF V600E mutations. Am. J. Surg. Pathol. 45, 662–671 (2021).
Kamata, T. et al. Frequent BRAF or EGFR mutations in ciliated muconodular papillary tumors of the lung. J. Thorac. Oncol. 11, 261–265 (2016).
Funding
This manuscript received no funding.
Author information
Authors and Affiliations
Contributions
This manuscript was created and edited by the author (J.M.B.).
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Ethics Approval/Consent to Participate
This is a review paper which does not include any primary data which would require IRB approval or informed consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Boland, J.M. Unusual lung tumors—from morphology to genetics. Mod Pathol 35 (Suppl 1), 57–65 (2022). https://doi.org/10.1038/s41379-021-00914-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00914-7
