
Abstract
Myxoid pleomorphic liposarcoma is a recently defined subtype of liposarcoma, which preferentially involves the mediastinum of young patients and shows mixed histological features of conventional myxoid liposarcoma and pleomorphic liposarcoma. While myxoid pleomorphic liposarcoma is known to lack the EWSR1/FUS-DDIT3 fusions characteristic of the former, additional genetic data are limited. To further understand this tumor type, we extensively examined a series of myxoid pleomorphic liposarcomas by fluorescence in situ hybridization (FISH), shallow whole genome sequencing (sWGS) and genome-wide DNA methylation profiling. The 12 tumors occurred in 6 females and 6 males, ranging from 17 to 58 years of age (mean 33 years, median 35 years), and were located in the mediastinum (n = 5), back, neck, cheek and leg, including thigh. Histologically, all cases consisted of relatively, bland, abundantly myxoid areas with a prominent capillary vasculature, admixed with much more cellular and less myxoid foci containing markedly pleomorphic spindled cells, numerous pleomorphic lipoblasts and elevated mitotic activity. Using sWGS, myxoid pleomorphic liposarcomas were found to have complex chromosomal alterations, including recurrent large chromosomal gains involving chromosomes 1, 6–8, 18–21 and losses involving chromosomes 13, 16 and 17. Losses in chromosome 13, in particular loss in 13q14 (including RB1, RCTB2, DLEU1, and ITM2B genes), were observed in 4 out of 8 cases analyzed. Additional FISH analyses confirmed the presence of a monoallelic RB1 deletion in 8/12 cases. Moreover, nuclear Rb expression was deficient in all studied cases. None showed DDIT3 gene rearrangement or MDM2 gene amplification. Using genome-wide DNA methylation profiling, myxoid pleomorphic liposarcomas and conventional pleomorphic liposarcomas formed a common methylation cluster, which segregated from conventional myxoid liposarcomas. While the morphologic, genetic and epigenetic characteristics of myxoid pleomorphic liposarcoma suggest a link with conventional pleomorphic liposarcoma, its distinctive clinical features support continued separate classification for the time being.
Similar content being viewed by others
Introduction
Myxoid pleomorphic liposarcoma, originally termed “pleomorphic myxoid liposarcoma”, was first described in a large study of liposarcomas in patients younger than 22 years by Alaggio et al. [1]. It is defined in the most recent WHO Classification of soft tissue and bone tumors as an “exceptionally rare, aggressive adipocytic neoplasm, typically occurring in children and adolescents which shows mixed histologic features of conventional myxoid liposarcoma and pleomorphic liposarcoma and lacks the gene fusions and amplifications of myxoid liposarcoma, atypical lipomatous tumor and dedifferentiated liposarcoma” [2, 3].
Although these distinctive tumors were initially thought to most likely represent variants of myxoid liposarcoma, genetic analyses have shown them to be consistently negative for FUS/EWSR1-DDIT3 fusions, the pathognomonic molecular genetic event(s) in myxoid liposarcoma [1, 4]. Furthermore, recent work has shown complex findings by array comparative genomic hybridization and inactivation of RB1, suggesting a possible link to conventional pleomorphic liposarcoma [5, 6]. Thus, it remains unclear whether these lesions are best considered variants of pleomorphic liposarcoma, or a distinct entity altogether [1, 4,5,6,7,8,9,10,11,12].
The purpose of the present study was to analyze the clinicopathologic and molecular genetic features of a well-characterized cohort of myxoid pleomorphic liposarcomas, studied with fluorescence in situ hybridization (FISH), shallow whole genome sequencing (sWGS) and genome-wide DNA methylation profiling, in order to better define their place in the nosology of adipocytic neoplasms.
Material and methods
Following Institutional Review Board approval (approval number/ID B670201938578), cases meeting current criteria for myxoid pleomorphic liposarcoma were collected from the authors’ institutional and consultation archives [3]. Cases of conventional pleomorphic liposarcoma showing “myxofibrosarcoma-like” histology were specifically excluded. The study was performed in accordance with the Code of Conduct of the Federation of Medical Scientific Societies in the United States of America, Belgium, the Netherlands and Germany. Clinical data were provided by the referring pathologists and/or clinicians.
Immunohistochemistry
Immunohistochemistry was performed on 4 µm-thick sections of formalin-fixed, paraffin-embedded material with a Benchmark XT immunostainer (Ventana Medical Systems, Tucson, AZ, USA). Primary monoclonal antibodies were CD34 (1:100; QBEnd10; Dako, Glostrup, Denmark), p16 (ready-to-use; E6H4; CINtec MTM laboratories-Roche, Tucson, AZ, USA), Rb (1:50; G3-245; BD Pharmingen, San Diego, CA, USA) and MDM2 (1:10; IF2; Invitrogen, Carlsbad, CA, USA). Heat-induced epitope retrieval was performed for CD34, p16, Rb and MDM2 using Cell Conditioning 1. Visualization was achieved with the Ultraview Universal DAB Detection kit (Ventana Medical Systems, Tucson, AZ, USA). Appropriate positive and negative controls were used throughout. Rb nuclear immunoreactivity was classified as deficient (<10% of tumor cells with nuclear staining), heterogeneous (10–80% of tumor cells with nuclear staining), or intact (>80% of tumor cells with nuclear staining) [13, 14].
Fluorescence in situ hybridization (FISH)
FISH analysis for the detection of RB1, located on 13q14.2, was performed using a ZytoLight SPEC RB1/13q12 dual-color probe (Zytovision, Bremerhaven, Germany) with standard protocol according to the manufacturer’s instructions. Fifty tumor cells were visually inspected using a fluorescence microscope. The presence or absence of the RB1 gene locus (orange signal) and of the green signal indicating intact 13q12 region was assessed and reported as described previously [15, 16]. A cut-off of >22% was used to define positive FISH results. For MDM2, dual-color FISH was performed, using MDM2 specific probes with a centromere-specific probe for chromosome 12. FISH copy number evaluation was performed by counting MDM2 and chromosome 12 centromere copy numbers in at least 20 nuclei within five tissue areas. An MDM2/CEP12 ratio >2 was considered amplified for the MDM2 gene [17, 18]. For DDIT3 (CHOP) (12q13) analysis, Vysis LSI dual-color break-apart DNA probes (Abbott) were used according to the description by Downs-Kelly et al. [19]. A minimum of 50 cells were scored for the presence of rearranged signals. The interpretation of intact and split signals of DDIT3 was done based on generally accepted guidelines by Vysis. A positive result for DDIT3 was defined as a split of signals observed in more than 10% of tumor cell nuclei.
Shallow whole genome sequencing (sWGS) (copy number profiling)
Sample preparation & sequencing
From eight cases, six to ten sections of FFPE material were used for DNA purification. The zone with the highest percentage of tumor cells was macrodissected. Prior to DNA purification, samples were deparaffinized using Deparaffinization Solution (Qiagen, Hilden, Germany). DNA was purified using the QIAamp DNA FFPE Tissue Kit (Qiagen). DNA quantification was performed using a Qubit Fluorometer and the Qubit dsDNA BR Assay Kit (Life Technologies, Bleiswijk, The Netherlands). Prior to library preparation, DNA quality was determined using the Fragment Analyzer, the High Sensitivity Small DNA Fragment Analysis Kit and PROSize Data Analysis Software according to the manufacturer’s instructions (Advanced Analytical, Ankeny, Iowa, USA). 200 ng of DNA was sheared to 200 bp fragments using Covaris Adaptive Focused Acoustics shearing process and a M220 Focused-ultrasonicator (Covaris, Woburn, Massachusetts, USA). DNA libraries were prepared using the NEXTflex Rapid DNA-Seq Kit and NEXTflex DNA Barcodes (Bio Scientific, Austin, Texas, USA), starting from 128 ng of DNA. Libraries were quantified using a Qubit Fluorometer and the Qubit dsDNA HS Assay Kit (Life Technologies, Bleiswijk, The Netherlands). Sample libraries were equimolarly pooled for multiplexed sequencing, using a 50 bp single-end run on a HiSeq 3000 platform (Illumina, Essex, UK). The minimal number of reads per sample was aimed to be 20 million.
Read mapping & deduplication
Raw reads were mapped by Bowtie 2 against human reference genome CRCh38, using the fast-local flag [20]. Biobambam’s bamsormadup was used to mark duplicate reads and to sort the resulting bam files [21]. The latter documents were indexed using SAMtools [22].
Copy number profiling and aberration calling
WisecondorX was used to reliably deduce normalized log2-ratios from sWGS data [23,24,25]. To enable an optimal technical normalization, the software employed 181 healthy in-house reference samples, prepared similarly as the patient samples. Other than the -binsize argument during the newref phase, set at 100 kb, all parameters remained default. Aberration calling was executed in accordance with Raman et al: [23]. segments, sized two bins or more, were seen as aberrant once they had an absolute Z-score of at least 3, as calculated by WisecondorX, which measures healthy variability based on the provided reference samples, ultimately enabling Z-score calculation [24].
Genome-wide DNA methylation profiling
Samples were subjected to the Illumina Infinium MethylationEPIC BeadChip array (Illumina, San Diego, USA) analysis at the Genomics and Proteomics Core Facility of the German Cancer Research Center (DKFZ) Heidelberg [26,27,28]. Processing of methylation data was performed as described by Koelsche et al [28]. Software packages used for methylation data analysis were R version 3.4.4 (R Development Core Team, 2019), minfi Bioconductor package version 1.24.0, limma package version 3.34.5 and Rtsne (version 0.13). DNA methylation data were normalized by performing background correction and dye bias correction (shifting of negative control probe mean intensity to zero and scaling of normalization control probe mean intensity to 20,000, respectively). Probes targeting sex chromosomes, probes containing multiple single nucleotide polymorphisms and those that could not be uniquely mapped were removed. Human reference genome (hg19) was used for the analysis of multi-site mapping. Probes from the EPIC array were excluded if the predecessor Illumina Infinium 450k BeadChip did not cover them, thereby making data generated by both 450k and EPIC feasible for subsequent analyses. In total, 438,370 probes were kept for analysis. We used t-distributed stochastic neighbor embedding (t-SNE) analysis, a method enabling dimensionality reduction and visualization of complex data to detect methylation clusters. The algorithm was performed using the 20,000 most variable probes with a perplexity of 15 and 3000 iterations. Methylation data of pleomorphic liposarcomas (n = 9) and myxoid liposarcomas (n = 31) was used for comparison.
Results
Clinical data
The 12 tumors occurred in 6 females and 6 males, ranging from 17 to 58 years of age (mean 33 years, median 35 years). The tumors were located in the mediastinum (n = 5), back (n = 1), neck (n = 2), cheek (n = 1) and leg (n = 3), including thigh (n = 2). All the studied specimens were either biopsies or excisional biopsies.
Follow-up information was available for 6 patients with a median duration of 30 months (range: 14–40 months). None of the 12 patients received adjuvant (chemo)therapy. All 6 patients developed distant metastases (lung, bone, and soft tissue) and died of their disease within 40 months. Three of the patients also had multiple local recurrences.
Histology
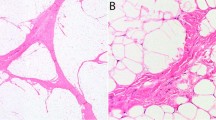
At low power magnification, the tumors showed multinodular, diffusely infiltrative growth into the surrounding soft tissues. All tumors had, at least in part, areas containing abundant myxoid matrix, with a well-developed capillary vasculature and relatively bland round to slightly spindled cells, reminiscent of conventional myxoid liposarcoma (Fig. 1A). However, even in these areas, close inspection disclosed scattered tumor cells showing a greater degree of nuclear enlargement and irregularity than would be expected in conventional myxoid liposarcoma (Fig. 1B). Univacuolated and multivacuolated lipoblasts and floret-like multinucleated cells were present (Fig. 1C and 1D). The mitotic rate in these myxoid liposarcoma-like foci was low in all but one case (Fig. 1D). The predominantly myxoid areas accounted for roughly 30–50% of the tumors. In addition to these low-grade, myxoid areas, all tumors transitioned to much more cellular and pleomorphic sarcoma with easily identified pleomorphic lipoblasts, resembling conventional pleomorphic liposarcoma (Fig. 2). Mitotic activity was greatly elevated in these areas and necrosis was present in three cases. Other morphologic features seen in subsets of cases included prominent fibrous septation (four cases) and ‘lymphangioma-like’ mucin pools (four cases) (Fig. 3A and 3B).

A Abundant myxoid matrix, a well-developed capillary vasculature, lipoblasts and relatively bland round to slightly spindled cells in myxoid liposarcoma-like tumor areas (Hematoxylin and Eosin, original magnification ×40). B Scattered tumor cells showing more atypia and pleomorphism in myxoid liposarcoma-like tumor areas (Hematoxylin and Eosin, original magnification ×100). C Floret-like multinucleated cells (Hematoxylin and Eosin, original magnification ×200). D Multinucleated (floret-like) tumor cells and mitotic activity (Hematoxylin and Eosin, original magnification ×400).

A, B Transition to pleomorphic liposarcoma-like tumor areas. Note the univacuolated and multivacuolated lipoblasts in (A) (Hematoxylin and Eosin, original magnifications ×40). Pleomorphic lipoblasts (C), pleomorphic tumor cells and mitotic activity (D) in pleomorphic liposarcoma-like tumor areas (Hematoxylin and Eosin, original magnifications ×200 and ×400).

A ‘Lymphangioma-like’ mucin pools (Hematoxylin and Eosin, original magnification ×40). B Prominent fibrous septation (Hematoxylin and Eosin, original magnification ×40). Loss of Rb expression in the myxoid liposarcoma-like (C, original magnification ×200) and pleomorphic liposarcoma-like (D, original magnification ×200) tumor areas with intact nuclear expression in the endothelial cells.
Immunohistochemistry
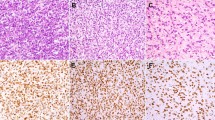
All 12 tumors showed diffuse CD34 and p16 expression, loss of nuclear Rb expression (Fig. 3C and 3D) and were negative for MDM2.
FISH analysis
FISH demonstrated monoallelic deletion of RB1 in 8 of 12 tumors. None showed MDM2 amplification or rearrangement of the DDIT3 gene.
Genome-wide copy number profiling
Genome-wide copy number profiling was performed in eight myxoid pleomorphic liposarcomas. Complex profiles with numerous extensive losses and gains (some with high-level gain/amplifications) were a feature of all cases, with recurrent gains involving chromosomes 1, 6–8, 18–21 and recurrent losses involving chromosomes 13, 16 and 17. Losses in chromosome 13, in particular loss of 13q14 (including RB1, RCTB2, DLEU1 and ITM2B genes), were observed in 4 of the 8 analyzed myxoid pleomorphic liposarcomas (Fig. 4).

A Copy number profile of a representative myxoid pleomorphic liposarcoma tumor. Dots are bins for which copy number is inferred, whereas horizontal graph lines represent segments of predicted equal copy number. The Y-axis indicates the ratio between the observed over the expected number of reads. B Overview of aberrations detected in myxoid pleomorphic liposarcomas. The wave-like figure represents fractions of aberrant samples across the human genome. Patterns above the y = 0 line indicate gains, whilst opposite contours represent losses (e.g., when the top of the wave is at 50%, 50% of samples have a gain at the corresponding locus; when the bottom of the wave is at 10%, 10% of samples have a loss at the corresponding locus). The middle graph line represents mean log2 ratio, as in (A), corresponding to the right-hand axis.
Genome-wide DNA methylation profiling
Unsupervised DNA methylation profiling was performed on 12 tumors and compared with profiles from 9 conventional pleomorphic liposarcomas and 31 conventional myxoid liposarcomas by t-SNE analysis ((t-SNE) analysis) (Fig. 5). The myxoid pleomorphic liposarcomas and conventional pleomorphic liposarcomas formed a common methylation cluster, which segregated from conventional myxoid liposarcomas.

t-Distributed Stochastic Neighbor Embedding analysis. The myxoid pleomorphic liposarcomas and conventional pleomorphic liposarcomas formed a common methylation cluster, which segregated from conventional myxoid liposarcomas.
Discussion
The results of the present study confirm and extend upon those of previous investigations. In agreement with previous studies, the natural history of myxoid pleomorphic liposarcomas appears to be particularly aggressive, with metastases and death from disease within 40 months in all patients with available clinical follow-up. This could be explained at least in part by the unusual mediastinal predilection of this rare liposarcoma subtype, with involvement of this site in 5 of 12 patients. Interestingly, it would seem that myxoid pleomorphic liposarcomas may also occur in older patients than has been previously appreciated, with the patients in the present series having a median age of 35 years, including three patients of >50 years of age.
Previous data on the molecular genetic features of myxoid pleomorphic liposarcoma has been limited to small series and isolated case reports [1, 4,5,6, 29,30,31]. As noted, DDIT3 rearrangements have not been identified in these lesions, arguing against a link to conventional myxoid liposarcoma. Studies of individual cases, by Creytens et al. and Hofvander et al., have found mainly whole chromosome gains of chromosomes 1, 6–8, 19–21 and X and losses of 2–5, 10–17 and 22, including the 13q14 region with absence of RB1 and its flanking genes [5, 6]. Our sWGS data confirms these prior findings, with myxoid pleomorphic liposarcomas showing complex chromosomal alterations, including recurrent large chromosomal gains involving chromosomes 1, 6–8, 18–21 and losses involving chromosomes 13, 16 and 17. Losses in chromosome 13, in particular loss in 13q14 (including RB1, RCTB2, DLEU1, and ITM2B genes), were observed in 4 out of 8 cases analyzed by sWGS. Monoallelic RB1 deletion was confirmed by FISH in 8 of 12 tumors. Moreover, nuclear Rb expression was deficient in all cases, further emphasizing that inactivation of the RB1 tumor suppressor gene is a consistent finding and pathogenetically paramount in this tumor type.
RB1 is one of the best characterized tumor suppressor genes, with loss resulting in extensive changes of chromatin organization, patterns of transcription, metabolic pathways and the proteome [32]. Inactivation of RB1 has been noted in a wide variety of human tumors, and is a feature of certain adipocytic neoplasms including spindle cell/pleomorphic lipoma, atypical spindle cell/pleomorphic lipomatous tumor and conventional pleomorphic liposarcoma [12,13,14, 33,34,35,36,37,38,39]. Based on an initial haploid karyotype in myxoid pleomorphic liposarcoma found by Hofvander et al., it was speculated that early loss of RB1 initiates genomic instability by allowing tumor cells to tolerate dramatic ploidy changes during haploidization and subsequent polyploidization [6]. We postulate that additional losses at 13q14, specifically those found in the flanking genes RCBTB2, ITM2B and DLEU1, also encoding for candidate tumor suppressors, might be important in tumorigenesis of myxoid pleomorphic liposarcoma and may lead to growth advantage [40, 41]. The presence of other complex chromosomal gains and losses likely contributes to the aggressive clinical behavior of these neoplasms.
The differential diagnosis of myxoid pleomorphic liposarcoma includes conventional myxoid liposarcoma, spindle cell/pleomorphic lipoma, atypical spindle cell/pleomorphic lipomatous tumor and conventional pleomorphic liposarcoma, in particular the myxofibrosarcoma-like variant [10]. Although conventional myxoid liposarcoma also often occurs in young adults, and is the most common subtype of pediatric liposarcoma, it lacks the striking pleomorphism that defines myxoid pleomorphic liposarcoma, and invariably harbors DDIT3 rearrangements. Extensively myxoid spindle cell/pleomorphic lipomas and atypical spindle cell/ pleomorphic lipomatous tumors may also mimic myxoid pleomorphic liposarcoma, but typically present as relatively small, more superficial masses with a predilection for the head, neck and shoulder regions, and the limbs and limb girdles respectively, lacking tumor necrosis and the brisk mitotic activity of myxoid pleomorphic liposarcoma. The myxofibrosarcoma-like variant of pleomorphic liposarcoma is perhaps the most difficult to differentiate from myxoid pleomorphic liposarcoma, and there may be situations in which this distinction may be subjective. In general, however, myxofibrosarcoma-like areas within conventional pleomorphic liposarcomas display often an arborizing thick-walled (rather than delicate capillary vasculature), and appear within the overall context of a tumor resembling conventional pleomorphic liposarcoma, either with predominantly undifferentiated pleomorphic sarcoma-like histology, or less often sheets of pleomorphic lipoblasts [42,43,44,45].
Finally, the question remains of whether myxoid pleomorphic liposarcoma should be considered a distinct entity, as currently classified by the WHO, or is perhaps better thought of as a variant of conventional pleomorphic liposarcoma. From a morphologic perspective, one could argue that these two tumors form a continuum, with conventional pleomorphic liposarcoma at the far end. This hypothesis is not, however, entirely satisfying, as myxoid pleomorphic liposarcoma shows distinctive clinical features (e.g., frequent occurrence in pediatric patients, mediastinal predilection) and behaves in a more aggressive fashion, despite having, at least in part, “low-grade” morphology. On the other hand, the genetic and epigenetic features of myxoid pleomorphic liposarcoma and conventional pleomorphic liposarcoma overlap significantly, with both tumors showing similar complex patterns of chromosomal gains and losses and overlapping patterns of genome-wide methylation, suggesting that myxoid pleomorphic liposarcomas and conventional pleomorphic liposarcomas are (epi)genetically closely related. Even here, though, there are some differences, as myxoid pleomorphic liposarcomas lack the genomic complexity of conventional pleomorphic liposarcoma, typically showing focal copy number changes, rather than large/whole chromosomal gains and losses [9, 14, 35]. Conceivably these differences might reflect the younger median age of patients with myxoid pleomorphic liposarcoma.
In conclusion, the results of the present study confirm the distinctive clinicopathologic features of myxoid pleomorphic liposarcoma, including its unusual mediastinal predilection with uniformly aggressive clinical behavior. Our genetic and epigenetic results suggest a possible link with conventional pleomorphic liposarcoma. Additional study of this very rare liposarcoma subtype should help to further refine its place in the overall nosology of adipocytic tumors. For the time being, myxoid pleomorphic liposarcoma is best considered a distinct liposarcoma subtype.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Alaggio R, Coffin CM, Weiss SW, Bridge JA, Issakov J, Oliveira AM, et al. Liposarcomas in young patients. A study of 82 cases occurring patients younger than 22 years of age. Am J Surg Pathol. 2009;33:645–58.
WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. Lyon, France: IARC Press; 2020
Alaggio R, Creytens D. Myxoid pleomorphic liposarcoma. In: WHO Classification of Tumours of Soft tissue and Bone. 5th ed. Lyon, France: IARC; 2020
Boland JM, Colby TV, Folpe AL. Liposarcomas of the mediastinum and thorax: a clinicopathologic and molecular cytogenetic study of 24 cases, emphasizing unusual and diverse histologic features. Am J Surg Pathol. 2012;36:1395–403.
Creytens D, van Gorp J, Ferdinande L, Van Roy N, Libbrecht L. Array-based comparative genomic hybridisation analysis of a pleomorphic myxoid liposarcoma. J Clin Pathol. 2014;67:834–5.
Hofvander J, Jo VY, Ghanei I, Gisselsson D, Martensson E, Mertens F. Comprehensive genetic analysis of paediatric pleomorphic myxoid liposarcoma reveals near-haploidization and loss of RB1 gene. Histopathology. 2016;69:141–7.
Evans HL. Liposarcoma. A study of 55 cases with reassessment of its classification. Am J Surg Pathol. 1979;3:507–23.
Evans HL, Soule EH, Winkelmann RK. Atypical lipoma, atypical intramuscular lipoma, and well differentiated retroperitoneal liposarcoma. A reappraisal of 30 cases formerly classified as well differentiated liposarcoma. Cancer. 1979;43:574–84.
Demicco EG. Molecular updates in adipocytic neoplasms. Semin Diagn Pathol. 2019;36:85–94.
Creytens D. A contemporary review of myxoid adipocytic tumors. Semin Diagn Pathol. 2019;36:129–41.
Putra J, Al-Ibraheemi A. Adipocytic tumors in children: a contemporary review. Semin Diagn Pathol. 2019;36:95–104.
Creytens D. What’s new in adipocytic neoplasia? Virchows Arch. 2020;476:29–39.
Creytens D, van Gorp J, Savola S, Ferdinande L, Mentzel T, Libbrecht L. Atypical spindle cell lipoma: a clinicopathologic, immunohistochemical, and molecular study emphasizing its relationship to classical spindle cell lipoma. Virchows Arch. 2014;465:97–108.
Creytens D, Mentzel T, Ferdinande L, Lecoutere E, van Gorp J, Atanesyan L, et al. ‘Atypical’ pleomorphic lipomatous tumor: a clinicopathologic, immunohistochemical and molecular study of 21 cases, emphasizing its relationship to atypical spindle cell lipomatous tumor and suggesting a morphologic spectrum (atypical spindle cell/pleomorphic lipomatous tumor). Am J Surg Pathol. 2017;41:1443–55.
Agaimy A. Anisometric cell lipoma: insight from a case series and review of the literature on adipocytic neoplasms in survivors of retinoblastoma suggest a role for RB1 loss and possible relationship to fat-predominant (“fat-only”) spindle cell lipoma. Ann Diagn Pathol. 2017;29:52–6.
Agaimy A, Michal M, Giedl J, Hadravsky L, Michal M. Superficial acral fibromyxoma: clinicopathological, immunohistochemical, and molecular study of 11 cases highlighting frequent Rb1 loss/deletions. Hum Pathol. 2017;60:192–98.
Weaver J, Downs-Kelly E, Goldblum JR, Turner S, Kulkarni S, Tubbs RR, et al. Fluorescence in situ hybridization for MDM2 gene amplification as a diagnostic tool in lipomatous neoplasms. Mod Pathol. 2008;21:943–9.
Creytens D, van Gorp J, Ferdinande L, Speel EJ, Libbrecht L. Detection of MDM2/CDK4 amplification in lipomatous soft tissue tumors from formalin-fixed, paraffin-embedded tissue: comparison of multiplex ligation-dependent probe amplification (MLPA) and fluorescence in situ hybridization (FISH). Appl Immunohistochem Mol Morphol. 2015;23:126–33.
Downs-Kelly E, Goldblum JR, Patel RM, Weiss SW, Folpe AL, Mertens F, et al. The utility of fluorescence in situ hybridization (FISH) in the diagnosis of myxoid soft tissue neoplasms. Am J Surg Pathol. 2008;31:8–13.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Tischler G, Leonard S. Biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol Med. 2014;9:13.
Li H, Handsaker B, Wysoker A, Fenell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Raman L, Van der Linden M, De Vriendt C, Van den Broeck B, Muylle K, Deeren D, et al. Shallow-depth sequencing of cell-free DNA for Hodgkin and diffuse large B-cell lymphoma (differential) diagnosis: a standardized approach with underappreciated potential. Haematologica. (2020). https://doi.org/10.3324/haematol.2020.268813
Raman L, Dheedene A, De Smet M, Van Dorpe J, Menten B. WisecondorX: improved copy number detection for routine shallow whole-genome sequencing. Nucleic Acids Res. 2019;47:1605–14.
Van der Linden M, Raman L, Vander Trappen A, Dheedene A, De Smet M, Sante T, et al. Detection of copy number alterations by shallow whole-genome sequencing of formalin-fixed, paraffin-embedded tumor tissue. Arch Pathol Lab Med. (2019). https://doi.org/10.5858/arpa.2019-0010-OA
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–74.
Capper D, Stichel D, Sahm F, Jones DTW, Schrimpf D, Sill M, et al. Practical implementation of DNA methylation and copy‑number‑based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathologica. 2018;136:181–210.
Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F, Reuss DE, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12:498.
Sinclair TJ, Thorson CM, Alvarez E, Tan S, Spunt SL, Chao SD. Pleomorphic myxoid liposarcoma in an adolescent with Li-Fraumeni syndrome. Pediatr Surg Int. 2017;33:631–5.
Francom CR, Leoniak SM, Lovell MA, Herrmann BW. Head and neck pleomorphic myxoid liposarcoma in a child with Li-Fraumeni syndrome. Int J Pediatr Otorhinolaryngol. 2019;123:191–4.
Zare SY, Leivo M, Fadare O. Recurrent pleomorphic myxoid liposarcoma in a patient with Li-Fraumeni syndrome. Int J Surg Pathol. 2020;28:225–8.
Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 2016;30:1492–502.
Dal Cin P, Sciot R, Polito P, Stas M, de Wever I, Cornelis A, et al. Lesions of 13q may occur independently of deletion of 16q in spindle cell/pleomorphic lipomas. Histopathology. 1997;31:222–5.
Schneider-Stock R, Walter H, Radig K, Rys J, Bosse A, Kuhnen C, et al. MDM2 amplification and loss of heterozygosity at RB and p53 genes: no simultaneous alterations in the oncogenesis of liposarcomas. J Cancer Res Clin Oncol. 1998;124:532–40.
Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42:715–21.
Mentzel T, Palmedo G, Kuhnen C. Well-differentiated spindle cell liposarcoma (‘atypical spindle cell lipomatous tumor’) does not belong to the spectrum of atypical lipomatous tumor but has a close relationship to spindle cell lipoma: clinicopathologic, immunohistochemical, and molecular analysis of six cases. Mod Pathol. 2010;23:729–36.
Ghadimi MP, Liu P, Peng T, Bolshakov S, Young ED, Torres KE, et al. Pleomorphic liposarcoma: clinical observations and molecular variables. Cancer. 2011;117:5359–69.
Marino-Enriquez A, Nascimento AF, Ligon AH, Liang C, Fletcher CDM. Atypical spindle cell lipomatous tumor: clinicopathologic characterization of 232 cases demonstrating a morphologic spectrum. Am J Surg Pathol. 2017;41:234–44.
Lecoutere E, Creytens D. Atypical spindle cell/pleomorphic lipomatous tumor. Histol Histopathol. 2020;35:769–78.
Latil A, Chêne L, Mangin P, Fournier G, Berthon P, Cussenot O. Extensive analysis of the 13q14 region in human prostate tumors: DNA analysis and quantitative expression of genes lying in the interval of deletion. Prostate. 2003;57:39–50.
Garding A, Bhattacharya N, Claus R, Ruppel M, Tschuch C, Filarsky K, et al. Epigenetic upregulation of IncRNAs at 13q14.3 in leukemia is linked to the In Cis downregulation of a gene cluster that targets NF-kB. PLoS Genet. 2013;9:e1003373.
Gebhard S, Coindre JM, Michels JJ, Terrier P, Bertrand G, Trassard M, et al. Pleomorphic liposarcoma: clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases. Am J Surg Pathol. 2002;26:601–16.
Hornick JL, Bosenberg MW, Mentzel T, McMenamin ME, Oliveira AM, Fletcher CD. Pleomorphic liposarcoma: clinicopathologic analysis of 57 cases. Am J Surg Pathol. 2004;28:1257–67.
Pedeutour F, Montgomery EA. Pleomorphic liposarcoma. In: WHO Classification of Tumours of Soft tissue and Bone. 5th ed. Lyon, France: IARC; 2020
Anderson WJ, Jo VY. Pleomorphic liposarcoma: updates and current differential diagnosis. Semin Diagn Pathol. 2019;36:122–8.
Acknowledgements
We are very grateful for technical assistance to Isabelle Rottiers and Lynn Supply (Department of Pathology, University Hospital Ghent, Belgium) and Tamara De Clercq (Center for Medical Genetics, Ghent University, Ghent, Belgium). We thank the Microarray unit of the Genomics and Proteomics Core Facility, German Cancer Research Center (DKFZ) for providing excellent DNA methylation services. This work was supported by the German Cancer Aid (Grant no. 70112499).
Funding
The DNA methylation profiling part in this study was supported by the German Cancer Aid (Grant no. 70112499). Other authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
DC performed study concept, design and writing of the paper; JvD and UF performed study concept, design and review of the paper; AF, TM, and KF provided clinical specimens and helped write the paper. LF and JVG helped write the paper; CK, MvdL, LR, BM, and AvD provided acquisition, analysis and interpretation of data, and statistical analysis. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of the Ghent University Hospital/Ghent University (approval number/ID B670201938578). This study was performed in accordance with the Declaration of Helsinki and with the Code of Conduct of the Federation of Medical Scientific Societies in the United States of America, Belgium, the Netherlands, and Germany.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Creytens, D., Folpe, A.L., Koelsche, C. et al. Myxoid pleomorphic liposarcoma—a clinicopathologic, immunohistochemical, molecular genetic and epigenetic study of 12 cases, suggesting a possible relationship with conventional pleomorphic liposarcoma. Mod Pathol 34, 2043–2049 (2021). https://doi.org/10.1038/s41379-021-00862-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00862-2
This article is cited by
-
Myxoid liposarcoma with nuclear pleomorphism: a clinicopathological and molecular study
Virchows Archiv (2024)
-
Systemic Therapy in Advanced Pleomorphic Liposarcoma: a Comprehensive Review
Current Treatment Options in Oncology (2023)
-
Mandibular Myxoid Liposarcoma: a Case Study
Indian Journal of Surgical Oncology (2023)
-
Myxoid pleomorphic liposarcoma is distinguished from other liposarcomas by widespread loss of heterozygosity and significantly worse overall survival: a genomic and clinicopathologic study
Modern Pathology (2022)
-
Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Soft Tissue Tumors
Head and Neck Pathology (2022)
