
Abstract
Natural killer (NK) cells are lymphocytes of the native immune system that play a pivotal role in host defense and immune surveillance. While the conceptual view of NK-neoplasms is evolving, little is known about the rare NK lymphoblastic leukemia (NK-LL), which remains as a provisional entity in the 2016 WHO Classification. The goal of this study is to characterize NK-LL cases and compare with other CD56 co-expressing acute leukemias. We identified 105 cases, diagnosed as NK-LL (6), CD56+ acute undifferentiated leukemia (AUL) (6), CD56+ T-lymphoblastic leukemia (T-LL) (51), and CD56+ acute myeloid leukemia (AML) (42). Compared to AUL patients, NK-LL patients were significantly younger (p = 0.021) and presented with higher white blood cell (WBC) (p = 0.037) and platelet counts (p = 0.041). Flow cytometry showed more frequent expression of cytoplasmic CD3 (cCD3, p = 0.064) and CD33, (p = 0.065), while HLA-DR was significantly absent from NK-LL (p = 0.035) compared to AUL. Compared to T-ALL, NK-LL cases showed less frequent cCD3 (p = 0.002), CD4 (p = 0.051), and CD10 expression (p = 0.06). The frequency of abnormal karyotypes was similar between NK-LL, AUL, and T-ALL. The mutational profile differed in four leukemia groups, with a significance enrichment of NOTCH1 (p = 0.002), ETV6 (p = 0.002) and JAK3 (p = 0.02) mutations in NK-LL as compared to AML. As compared to T-ALL, NK-LL cases showed a higher number of total mutations (p = 0.04) and significantly more frequent ETV6 mutations (p = 0.004). Clinical outcome data showed differences in overall survival between all four groups (p = 0.0175), but no difference in event free survival (p = 0.246). In this largest study to date, we find that that NK-LL shows clinical presentation, immunophenotypic and molecular characteristics distinct from AUL, T-ALL, and AML. Our findings suggest NK-LL is a distinct acute leukemia entity and should be considered in the clinical diagnosis of acute leukemias of ambiguous lineage.
Similar content being viewed by others

Introduction
Natural killer (NK) cells arise from hematopoietic stem cells in the bone marrow and undergo differentiation in secondary lymphoid tissues including peripheral blood, liver, mucosa-associated lymphoid tissues, lymph nodes, and other organs [1]. NK cells mediate immune-surveillance not only via cytotoxic effector functions, but also by serving as regulatory lymphocytes that secrete cytokines and interact with both innate and adaptive immune cells [1,2,3,4]. Mature NK cells have large granular lymphocyte cytology, express T-cell associated antigens including CD2, CD7, CD16, and CD56, and lack both T-cell receptor (TCR) gene rearrangements (i.e., have TCR genes in germline configuration) and a fully assembled TCR-CD3 complex. NK cells can originate from lymphoid as well as myeloid precursors and from precursors with lympho-myeloid potential [5, 6]. Early markers expressed by CD34 positive hematopoietic cells associated with propensity to differentiate into NK cells include CD7, CD10, CD45RA, and CD127 [5,6,7]. The stages of NK cell maturation have been defined both in vitro and in vivo and expression of CD94 and loss of CD117 mark an important step in this process [8]. Leukemias derived from NK cells, including the precursor neoplasm NK lymphoblastic leukemia (NK-LL), are a rare and heterogeneous group of disorders with several recognized phenotypes that lack expression of specific markers of T cells, myeloid cells, or plasmacytoid dendritic cells. As absence of specific markers makes it difficult to clearly establish the origin of these malignancies, the current 2016 World Health Organization (WHO) classification system considers NK-LL as a provisional entity [9]. A diagnosis of NK-LL is considered in cases expressing CD56 along with immature markers and T-cell markers such as CD2 and CD7, but lacking B-cell and myeloid markers. Aggressive NK-cell leukemia is a systemic neoplastic proliferation of NK cells with a similar immunophenotype to NK-LL, but lacking markers of immaturity; this entity is frequently associated with EBV infection [9].
The phenotypic and clinicopathologic spectra of NK-LL can potentially overlap with other CD56-positive hematopoietic neoplasms. In particular, blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a CD56+ neoplasm with blastic morphology, potentially mimicking NK-LL. BPDCN is derived from the precursors of plasmacytoid dendritic cells and presents in the skin with frequent involvement of the bone marrow, peripheral blood, and lymph nodes. In a study of 47 cases of putative NK cell neoplasms with immature morphologic features, Suzuki et al. in 2005 found that 28 (59%) had cutaneous involvement [10]. The cutaneous group showed more frequent mediastinal involvement, thrombocytopenia, CD4 and HLA-DR expression, and absence of CD34 and CD16 expression, as well as better survival compared to the non-cutaneous cases [10]. However, CD123 and other more specific markers to exclude the possibility of BPDCN were not assessed in this study. A series by Bayerl et al. of seven blastic NK-cell lymphoma/leukemias suggested that CD4 positive cases had frequent cutaneous involvement (5/7, 71%) but included BPDCN cases [11]. A case report and review of literature by Dubois et al. suggested that pediatric acute blastic NK leukemia showed distinct clinical characteristics and infrequent skin involvement [12].
In T-ALL/LBL, CD56 expression has been reported in a small subset of cases (7–28%) and these have distinct clinicopathologic findings [13]. In one study, expression of both CD56 and CD16 in T-ALL correlated with older age, higher platelet count, expression of cytotoxic markers, CD34, and CD33, and significantly worse prognosis [14]. In pediatric T-ALL, CD56-positive cases were also characterized by more frequent expression of myeloid and precursor markers, higher rate of early T-cell precursor (ETP) subtype, and shorter event free survival and overall survival [15]. Molecular genetic data showed that the frequency of mutations in NOTCH1, PTEN, and FBXW7 in the CD56+ cases did not differ significantly from the CD56- cases [16]. These findings are suggestive of a common T/NK/myeloid precursor cell as the biological origin of CD56-positive T-ALLs.
CD56 expression is also seen in a subset (14–58%) of AML cases [17]. Its prognostic significance is controversial, but it has been associated with worse prognosis in acute promyelocytic leukemia and in AML with t(8;21) [18]. One study of adult AML patients demonstrated that CD56 expression in AML was associated with extramedullary infiltrates at diagnosis, 11q23 karyotypic abnormalities, low complete remission rate and poor overall survival [19]. Junge et al. found that expression of CD56 and also another NK marker CD16 on blasts in AML was associated with particularly adverse outcome in multivariate analysis [20]. As compared to other AML cases, these CD56+/CD16+ AML cases showed similar rates of FLT3 mutations and overall infrequent complex karyotype. Other studies have previously reported inferior outcome in subsets of AML patients with a NK/T‐cell phenotypes defined as CD7 and CD56 expression or an HLA‐DR−, CD33+, CD56+, and CD16− phenotype [21, 22]. Some investigators have used the term “myeloid/NK acute leukemia” to encompass these cases and have suggested that they are of precursor NK-cell origin and show an immunophenotypic overlap with AML with minimal differentiation [10].
We have recently shown that AML with minimal differentiation shares many features with acute undifferentiated leukemia (AUL), a rare acute leukemia that lacks evidence of differentiation along any lineage, but can show co-expression of CD56 and T-cell markers, similar to NK-LL [23]. The most common mutations in AUL were mostly myeloid type and included PHF6 (also enriched in T-ALL), SRSF2, RUNX1, ASXL1, and BCOR; the mutational profile of NK-LL has not as of yet been described. Given the paucity of reports of NK-LL and its continued provisional status in the most recent WHO classification, we conducted a multi-institutional study in order to characterize its clinicopathologic, cytogenetic, and molecular genetic findings in comparison to other CD56 positive precursor hematologic neoplasms (AUL, T-ALL, and AML).
Methods
Patients
We searched the databases of multiple institutions (Brigham and Women’s Hospital/Dana-Farber Cancer Institute (DFCI), Massachusetts General Hospital, Seattle Children’s, MD Anderson Cancer Center, University of North Carolina, University of Minnesota, University of Utah, Yale, Stanford Health Care, University of Pennsylvania, and Weill Cornell Medical Center) for patients with diagnoses of NK-LL as well as cases of AUL, T-ALL, and AML with CD56 expression. Pathology slides or reports including flow cytometry and ancillary testing results were reviewed by at least one author of this paper at each institution to confirm that each case met the criteria for T-ALL, AUL, and NK-LL based on the 2016 WHO classification. Specifically, cases meeting criteria of BPDCN (with expression of CD123, CD4, and TCL1 and lacking expression of CD34) were excluded from initial analysis. A separate control group of 39 BPDCN cases from MD Anderson Cancer Center was included to assess immunophenotype differences from NK-LL. Clinical information and follow-up were retrieved from the electronic medical records. This study was approved by the Institutional Review Boards of all participating institutions.
Immunophenotyping
Flow cytometric immunophenotyping was performed at each institution using comprehensive panels for acute leukemia work-up. Although the panels varied among different institutions, all panels were adequate to assess the lineages of leukemic blasts. In the case of cytoplasmic or nuclear antigens, samples were also permeabilized during antibody labeling. The total markers assessed in all institutions included CD34, CD117, CD13, CD33, CD15, CD11b, CD64, myeloperoxidase (MPO), HLA-DR, sCD3, cCD3, CD2, CD4, CD5, CD7, CD56, CD1a, CD19, CD20, CD10, CD22, CD79a, TdT, CD38, and MPO. CD16, CD94, and CD123 were assessed in some, but not all institutions.
Cytogenetic and FISH analysis
Conventional chromosomal analysis was performed on G-banded metaphase cells prepared from unstimulated 24- and 48-h bone marrow aspirate cultures at the time of diagnosis using standard techniques and was documented according to the International System for Human Cytogenetic Nomenclature. Fluorescence in situ hybridization was performed as a part of the clinical evaluation in some cases to identify common gene rearrangements/fusions
Targeted next-generation sequencing
Targeted next-generation sequencing (NGS) studies were performed to detect gene mutations that are commonly identified in hematolymphoid malignancies. DNA was prepared from bone marrow or peripheral blood at each participating institution. Specific NGS methodologies varied between institution, but all included an amplicon based library preparation and sequencing and panels ranged from 33 to 103 genes. The following genes were included in every panel: NOTCH1, ETV6, RUNX1, TET2, FLT3, IDH1, IDH2, JAK2, NPM1, NRAS and TP53. In addition, genes present in at least six of the eight panels were ASXL1, BRAF, CBL, DNMT3A, EZH2, KRAS, JAK2, PHF6, PTPN11, SETBP1, SF3B1, U2AF1, WT1, and ZRSR2.
Statistical Analysis
Fisher’s exact test was used to compare categorical variables. Relapse free survival and overall survival (OS) from diagnosis were estimated using the method of Kaplan and Meier. Complete remission was defined according to the criteria of Cheson et al. [24]. A p value of <0.05 was considered to be statistically significant.
Results
Patient cohort
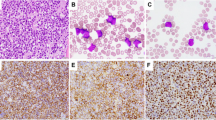
A total of six cases fulfilled immunophenotypic features of NK-LL using the 2016 WHO Classification criteria (Table 1). The remaining CD56 cases control cohort included 6 AUL, 52 T-ALL/LBL patients and 42 AML. The blasts in NK-LL were small to medium in size with scant, agranular cytoplasm and prominent nucleoli. An example of NK-LL case is shown in Fig. 1.

A Blasts show scant, agranular cytoplasm and prominent nucleoli on aspirate smears. B The biopsy is hypercellular with marked predominance of blasts (C) Blasts are diffusely positive for CD56 and also express CD34 (D).
NK-LL patients
Clinical presentation data for all six NK-LL patients is listed in Tables 1 and 2. NK-LL presented with a median age of 13 years (range 4–72 years) (Table 2). Bone marrow evaluation showed hypercellular marrow with a median cellularity of 95% (range 60–95) and median of 88% blasts (range 30–95%). In all six cases, blasts expressed CD56, CD34, and CD7 while 3/6 cases showed cytoplasmic CD3 and TdT. None of the cases had skin involvement or had significant CD4 or CD123 expression. The additional NK markers CD94 and CD16 were performed in three NK-LL cases and were positive in all three NK-LL cases. T-cell receptor gene rearrangement was performed in 3/6 NK-LL cases and was negative in all of these cases. The remaining three cases were presumptive NK LL based on their overall immunophenotype; these three cases all showed expression of CD56, CD16, and CD94 and 2/3 lacked cCD3 expression. Two NK-LL cases had a normal karyotype and the remaining four had an abnormal karyotype, including three with complex karyotype. NGS data were available in five NK-LL cases and showed a median of five mutations (range 0–7). The most common pathogenic mutations found in NK-LL patients were in NOTCH1 (3/5), ETV6 (3/5), and JAK3 (2/5) genes (Fig. 2). Most (5/6) NK-LL patients received ALL-type therapy and 5/6 were alive at last follow up. Median overall follow up time was 36 months (range 2–189, illustrated in Fig. 3). Three of six patients relapsed after therapy and two patients received a stem cell transplant.

Pathogenic mutations, total number of mutations and karyotype abnormalities found in NK-LL, CD56-positive T-ALL, AML, and AUL patients.

Overall survival and event free survival in NK-LL, AUL, T-ALL and AML patients.
NK-LL compared to CD56+ AUL patients
As compared with the six CD56+ AUL patients, the six NK-LL patients were significantly younger (p = 0.021) and presented with higher white blood cell counts (p = 0.037) and platelet count (p = 0.041), but with no difference in hemoglobin, circulating blasts or bone marrow cellularity (Table 2). Flow cytometry data showed that expression of CD34 was seen in all NK-LL cases (6/6) and half of all AUL cases (3/6) (Table 3, p = 0.061). T-cell marker expression was similar in both groups, but cytoplasmic CD3 was present in 3/6 of NK-LL cases and, by definition, absent from all AUL cases (p = 0.064). Myeloid markers were less frequently expressed in AUL cases as compared to NK-LL (CD33, p = 0.064). HLA-DR was present in only 1/6 NK-LL, significantly less frequent than AUL (p = 0.035). CD56 expression intensity was similar in both groups (p = 0.62). Two NK-LL cases had a normal karyotype and the remainder showed an abnormal karyotype, which was not significantly different from AUL (2/6 vs 3/6, p = 0.21). NGS data were available in five AUL cases and the most common mutations were ASXL1 (2/5) and TET2 (2/5) genes. In contrast to the ALL-type therapy given to most NK-LL patients, the majority of AUL patients (4/5) received AML-type therapy; the remaining AUL patient received supportive care only. There was no significant difference in OS and EFS between NK-LL and AUL patients (p > 0.05, Fig. 3).
NK-LL compared to CD56 + T-ALL patients
The control group of CD56 T-ALL patients had a median age of 25 years (range 1–80 years). As compared with NK-LL patients, T-ALL patients presented with similar WBC count, hemoglobin, platelet count, bone marrow cellularity and blast percentage (all p > 0.05). NK-LL showed less frequent cytoplasmic CD3 (p = 0.002), CD4 (p = 0.051) and CD10 expression (p = 0.06) compared to T-ALL. CD16 was assessed in 12 T-ALLs and was negative in all cases. The myeloid marker CD13 was expressed in similar numbers but NK-LL had more frequent CD33 expression (p = 0.038). No difference in TdT, CD123, HLA-DR, or monocytic markers was seen (all p > 0.05).
Molecular analysis was performed in 29 T-ALL patients. The most common mutations included NOTCH1 (9/29), WT1 (7/29), PHF6 (5/29), and IL7R (4/29). As compared with T-ALL, NK-LL cases had higher total median mutation count (5 versus 2, p = 0.04) and were enriched in ETV6 mutations (3/5 as compared with 1/29, p = 0.016). Karyotype was available in 46 cases with 12 cases (26%) showing a normal karyotype. Abnormal karyotypes in T-ALL included deletion of 9p21 in 4/46 (9%) cases and complex karyotype in 9/46 (19%) cases.
All 51 T-ALL patients were treated with T-ALL therapy with varied Children’s Oncology group (COG) or Dana Farber Cancer Institute (DFCI) regiments. Median follow up time for T-ALL patients was 20 months (range 1–132) and median relapse free time was 15 months (range 0–132). Eighteen patients relapsed and 20 ultimately died during the follow up period. There was no significant difference in OS and EFS between NK-LL and T-ALL patients (p > 0.05, Fig. 3).
NK-LL compared to CD56+ ETP-ALL patients
Patients with ETP-ALL were identified on the basis of the following immunophenotypes: CD1a−, CD8−, CD5−(dim), and positivity for 1 or more precursor or myeloid antigens [10]. 14/51 (27%) T-ALL cases fulfilled criteria for ETP-ALL. CD56 + ETP-ALL patients had a median age of 45 years (range 3–80 years). As compared with NK-LL patients, ETP-ALL patients presented with similar WBC count, hemoglobin, platelet count, bone marrow cellularity, and blast percentage and similar rate of abnormal karyotype (all p > 0.05). NK-LL patients showed brighter CD56 expression as compared to ETP-ALL patients (p = 0.015) and less frequent cytoplasmic CD3 (p = 0.039), but there were no significant differences in expression of TdT, CD123, HLA-DR, CD13, CD33, or monocytic markers (all p > 0.05). Mutational analysis was available in eight ETP ALL cases and most frequent mutations included NOTCH1 four cases), DNMT3A (three cases), and ETV6 (two cases). There was no significant difference in OS and EFS between NK-LL and ETP-ALL patients (p > 0.05).
NK-LL compared to CD56+ AML patients
The control group of 42 CD56 AML had a median age of 52 (range 5–82), which was significantly older than NK-LL (p = 0.023). Distribution of AML subtypes included nine AML with myelodysplasia-related changes, 17 AML with mutated NPM1 and 16 patients with AML, not otherwise specified. When comparing NK-LL patients with CD56 positive AML patients, NK-LL patients presented with similar WBC count and hemoglobin but with a significantly higher platelet count (142 vs 38 k/uL p = 0.03). Bone marrow cellularity and bone marrow blast percentages were the same between groups (p > 0.05).
As compared with AML, blasts in NK-LL patients showed less frequent expression of CD117 (p = 0.04) and CD13 (p = 0.002). Monocytic markers, including CD11b (p = 0.033) and CD64 (p = 0.024) were frequently seen in AML. Blasts in NK-LL patients showed significantly more frequent expression of CD7 (p = 0.001), CD5 (p = 0.03) and TdT (p = 0.005).
Molecular analysis was performed on 35 AML patients. The most common mutations included NPM1 (17/35), TET2 (13/35), DNMT3A (12/35), PTPN11 (7/35), ASXL1 (4/35); the median number of total mutations was three (range 0–7). As compared with AML, NK-LL patients show significantly higher frequency of NOTCH1 (p = 0.002), ETV6 (p = 0.002) and JAK3 (p = 0.02) and absence of TET2 mutations (p = 0.05). Cytogenetic analysis showed a normal karyotype in 26 (65%) AML patients, four with abnormalities of chromosome 8, and seven patients with complex karyotype.
Median follow up time was 11 months (range 1–88) and 27 AML patients received standard cytotoxic induction while the remainder were treated with hypomethylating agents. At time of last follow up, 21 patients were deceased. 18 AML patients relapsed with median relapse-free time of 9 months. There was no significant difference in OS and EFS between NK-LL and AML patients (p > 0.05, Fig. 3).
Discussion
NK leukemia is a rare and heterogeneous group of immature disorders with several recognized phenotypes. The first description of this latter entity included myeloid/NK cell acute leukemia which was suggested to be of precursor NK cell origin but has since been shown to have a phenotype that is indistinguishable from AML with minimal differentiation [13]. Currently NK-LL is a provisional entity in the 2016 WHO classification and relatively little is known of its immunophenotypic, cytogenetic and molecular landscape. After an extensive search of numerous large institutions, we identified 6 NK-LL cases and report on the clinical characteristics and outcome in comparison to CD56-positive T-ALL and AML cases. CD56 is a phenotypic marker of natural killer cells but can be expressed by other immune cells, including alpha-beta T cells, gamma-delta T cells, dendritic cells, and monocytes. Common to all these CD56-expressing cell types are strong immunostimulatory effector functions, including T helper 1 cytokine production and an efficient cytotoxic capacity [25].
Early descriptions of NK-LL showed an overlap with BPDCN. In a series of 47 blastic NK cell leukemia/lymphoma cases, Suzuki et al. found cutaneous involvement in 28 patients that was associated with older presenting age [10]. In contrast, our six NK-LL cases occurred in young patients (with a median age of 13) and all of these patients lacked cutaneous involvement. None of our NK-LL cases expressed CD4 and only minimal CD123 was seen in one case, while a group of 39 BPDCN control cases showed significantly more frequent expression of CD123, CD4, CD56 (often partial), and complete absence of CD34 (Table 3). Incorporation of more specific plasmacytoid dendritic cell-associated antigens (CD303, TCF4, TCL1, and CD2AP) and transcription factors (TCF4) can help establish the diagnosis of BPDCN and exclude diseases that present similarly to BPDCN [26]. While there is no dominant cytogenetic lesion described in BPDCN, the most commonly observed mutations include TET2, ASXL1, RAS, and TP53 [19, 26,27,28]. In contrast, the mutational profile of our NK-LL cases included frequent NOTCH1, ETV6, and JAK3 mutations. BPDCN patients generally have a very poor prognosis with most patients relapsing into drug-resistant malignancy with poor overall survival. In this study, NK-LL patients showed a relatively favorable outcome with five of six patients alive at last follow up and median survival of 36 months.
AML is a heterogeneous group of disorders which often present with different morphologic, immunophenotypic and cytogenetic patterns. CD56 can be found across the spectrum of AML subtypes; our CD56 AML cohort cases showed a high frequency of NPM1 mutations and included a subset of cases with complex karyotypes. Although some NK-LL patients also showed a complex karyotype, most presented at a younger age than AML cases and showed a mutational profile more similar to T-ALL with significant enrichment of NOTCH1, ETV6, and JAK3 mutations and a distinctly better outcome as compared to AML (Fig. 2).
A diagnosis of AUL requires a thorough immunophenotypic investigation that reveals none of the lineage-specific markers and less than two myeloid-associated markers CD13, CD33, and/or CD117. In a recent study we found that AUL shows frequent mutations in PHF6, SRSF2, RUNX1, ASXL1, and BCOR with similar clinical presentation to AML with minimal differentiation [23]. As compared to CD56 positive AUL, NK-LL patients were significantly younger and expressed more myeloid markers and cytoplasmic CD3 and also lacked HLA-DR expression. Molecular findings in our NK-LL patients resembled more a T-ALL signature. Not surprisingly NK-LL patients were treated with ALL therapy while AUL patients received AML type therapy.
In T-ALL, CD56 expression has been reported in a small subset of cases and an association with worse outcome has been suggested [29,30,31,32]. Prior studies showed that CD56-positive pediatric and adult T-ALL cases were characterized by a more frequent expression of myeloid and progenitor markers [14, 16], similar to our NK-LL cases. Our CD56+ T-ALL control group included 27% ETP ALL cases, which appears to be higher rate than previously reported the range of 5–16% [33, 34]. Although number of total mutations was significantly higher in NK-LL cases, the overall distribution of mutations was similar with exception of higher ETV6 mutations. NK-LL cases had longer OS compared to T-ALL, but were also characterized by younger age at presentation.
In conclusion, in our multi-institutional study, we confirm the rarity of NK-LL. Recognition of this entity is especially difficult given the nonspecific profile of early NK cell and flow cytometry panels in work up of acute leukemias should consider including CD94, CD56, CD16. CD161, another marker seen in NK cells [35], may also may be helpful and could be explored in future studies, but was not tested in this study and is not widely used in clinical flow cytometry laboratories. The differential diagnosis for this entity includes CD56+ AUL, T-ALL, and AML, as well as BPDCN. Our series shows that NK-LL patients tend to be young and have an overall favorable outcome compared to other CD56+ precursor hematopoietic neoplasms. The molecular profile of NK-LL is similar to T-ALL. Our findings support retention of NK-LL in the classification of acute leukemias and suggest that among other CD56 + precursor neoplasm, it bears most similarity to T-ALL, including frequent NOTCH1 mutations.
References
Agaugué S, Marcenaro E, Ferranti B, Moretta L, Moretta A. Human natural killer cells exposed to IL-2, IL-12, IL-18, or IL-4 differently modulate priming of naive T cells by monocyte-derived dendritic cells. Blood. 2008;112:1776–83.
Vitale M, Della Chiesa M, Carlomagno S, Pende D, Arico M, Moretta L, et al. NK-dependent DC maturation is mediated by TNFalpha and IFNgamma released upon engagement of the NKp30 triggering receptor. Blood. 2005;106:566–71.
Moretta A. The dialogue between human natural killer cells and dendritic cells. Curr Opin Immunol. 2005;17:306–11.
Mattiola I, Pesant M, Tentorio PF, Molgora M, Marcenaro E, Lugli E, et al. Priming of human resting NK cells by autologous M1 macrophages via the engagement of IL-1β, IFN-β, and IL-15 pathways. J Immunol. 2015;195:2818–2828.
Grzywacz B, Kataria N, Kataria N, Blazar BR, Miller JS, Verneris MichaelR. Natural killer-cell differentiation by myeloid progenitors. Blood. 2011;117(Mar):3548–58.
Carotta S, Pang SHM, Nutt SL, Belz GT. Identification of the earliest NK-cell precursor in the mouse BM. Blood. 2011;117:5449–52.
Renoux VM, Zriwil A, Peitzsch C, Michaëlsson J, Friberg D, Soneji S, et al. Identification of a human natural killer cell lineage-restricted progenitor in fetal and adult tissues. Immunity. 2015;43:394–407.
Grzywacz B, Kataria N, Sikora M, Oostendorp RA, Dzierzak EA, Blazar BR, et al. Coordinated acquisition of inhibitory and activating receptors and functional properties by developing human natural killer cells. Blood. 2006;108:3824–33.
Swerdlow SH, Campo E, Harris NL, et al., eds. WHO classification of tumors of haematopoietic and lymphoid tissues. Revised 4th edition ed. Lyon: IARC; 2017.
Suzuki R, Nakamura S, Suzumiya J, Ichimura K, Ichikawa M, Ogata K, et al. Blastic natural killer cell lymphoma/leukemia (CD56-positive blastic tumor): prognostication and categorization according to anatomic sites of involvement. Cancer. 2005;104:1022–31.
Bayerl MG, Rakozy CK, Mohamed AN, Vo DT, Long M, Eilender D, et al. Blastic natural killer cell lymphoma/leukemia: a report of seven cases. Am J Clin Pathol. 2002;117:41–50.
DuBois SG, Etzell JE, Matthay KK, Robbins E, Banerjee A. Pediatric acute blastic natural killer cell leukemia. Leuk Lymphoma. 2002;43:901–6.
Jain N, Lamb AV, O’Brien S, Ravandi F, Konopleva M, Jabbour E, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127:1863–9.
Fischer L, Gökbuget N, Schwartz S, Burmeister T, Rieder H, Brüggemann M, et al. CD56 expression in T-cell acute lymphoblastic leukemia is associated with non-thymic phenotype and resistance to induction therapy but no inferior survival after risk-adapted therapy. Haematologica. 2009;94:224–229.
Dalmazzo LF, Jácomo RH, Marinato AF, Figueiredo-Pontes LL, Cunha RLG, Garcia AB, et al. The presence of CD56/CD16 in T-cell acute lymphoblastic leukaemia correlates with the expression of cytotoxic molecules and is associated with worse response to treatment. Br J Haematol. 2009;144:223–9.
Fuhrmann S, Schabath R, Möricke A, Zimmermann M, Kunz JB, Kulozik AE, et al. Expression of CD56 defines a distinct subgroup in childhood T-ALL with inferior outcome. Results of the ALL-BFM 2000 trial. Br J Haematol. 2018;183:96–103.
Alegretti AP, Matzenbacher Bittar C, Bittencourt R, Kirchner Piccoli A, Schneider L, Silla LM, et al. The expression of CD56 antigen is associated with poor prognosis in patients with acute myeloid leukemia. Rev Bras Hematol Hemoter. 2011;33:202–6.
Ferrara F, Morabito F, Martino B, Specchia G, Liso V, Nobile F, et al. CD56 expression is an indicator of poor clinical outcome in patients with acute promyelocytic leukemia treated with simultaneous all-trans-retinoic acid and chemotherapy. J Clin Oncol. 2000;18:1295–1300.
Chang H, Brandwein J, Yi QL, Chun K, Patterson B, Brien B. Extramedullary infiltrates of AML are associated with CD56 expression, 11q23 abnormalities and inferior clinical outcome. Leuk Res. 2004;28:1007–11.
Junge A, Bacher U, Mueller BU, Keller P, Solenthaler M, Pabst T. Adverse outcome of AML with aberrant CD16 and CD56 NK cell marker expression. Hematol Oncol. 2018;36:576–83.
Suzuki R, Yamamoto K, Seto M, Kagami Y, Ogura M, Yatabe Y, et al. CD7+ and CD56+ myeloid/natural killer cell precursor acute leukemia: a distinct hematolymphoid disease entity. Blood. 1997;90:2417–28.
Scott AA, Head DR, Kopecky KJ, Appelbaum FR, Theil KS, Grever MR, et al. HLA-DR-, CD33+, CD56+, CD16- myeloid/natural killer cell acute leukemia: a previously unrecognized form of acute leukemia potentially misdiagnosed as French-American-British acute myeloid leukemia-M3. Blood. 1994;84:244–55.
Weinberg OK, Hasserjian RP, Baraban E, Ok CY, Geyer JT, Philip JKSS, et al. Clinical, immunophenotypic, and genomic findings of acute undifferentiated leukemia and comparison to acute myeloid leukemia with minimal differentiation: a study from the bone marrow pathology group. Mod Pathol. 2019;32:1373–85.
Cheson BD, Cassileth PA, Head DR, Schiffer CA, Bennett JM, Bloomfield CD, et al. Report of the National Cancer Institute-sponsored workshop on definitions of diagnosis and response in acute myeloid leukemia. J Clin Oncol. 1990;8:813–9.
Van Acker HH, Capsomidis A, Smits EL, Van Tendeloo VF. CD56 in the immune system: more than a marker for cytotoxicity? Front Immunol. 2017;8:892.
Economides MP, Konopleva M, Pemmaraju N. Recent developments in the treatment of blastic plasmacytoid dendritic cell neoplasm. Ther Adv Hematol. 2019;10:2040620719874733.
Montesinos P, Rayón C, Vellenga E, Brunet S, González J, González M, et al. Clinical significance of CD56 expression in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline-based regimens. Blood. 2011;117:1799–805.
Novotny JR, Nückel H, Dührsen U. Correlation between expression of CD56/NCAM and severe leukostasis in hyperleukocytic acute myelomonocytic leukaemia. Eur J Haematol. 2006;76:299–308.
Montero I, Rios E, Parody R, Perez-Hurtado JM, Martin-Noya A, Rodriguez JM. CD56 in T-cell acute lymphoblastic leukaemia: a malignant transformation of an early myeloid-lymphoid progenitor. Haematologica. 2003;88:ELT26.
Onishi Y, Matsuno Y, Tateishi U, Maeshima AM, Kusumoto M, Terauchi T, et al. Two entities of precursor T-cell lymphoblastic leukemia/lymphoma based on radiologic and immunophenotypic findings. Int J Hematol. 2004;80:43–51.
Paietta E, Neuberg D, Richards S, Bennett JM, Han L, Racevskis J, et al. Rare adult acute lymphocytic leukemia with CD56 expression in the ECOG experience shows unexpected phenotypic and genotypic heterogeneity. Am J Hematol. 2001;66:189–96.
Ravandi F, Cortes J, Estrov Z, Thomas D, Giles FJ, Huh YO, et al. CD56 expression predicts occurrence of CNS disease in acute lymphoblastic leukemia. Leuk Res. 2002;26:643–9.
Inukai T, Kiyokawa N, Campana D, Coustan-Smith E, Kikuchi A, Kobayashi M, et al. Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children’s Cancer Study Group Study L99-15. Br J Haematol. 2012;156:358–65.
Ma M, Wang X, Tang J, Xue H, Chen J, Pan C, et al. Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med. 2012;6:416–20.
Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev. 2006;214:56–72.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Weinberg, O.K., Chisholm, K.M., Ok, C.Y. et al. Clinical, immunophenotypic and genomic findings of NK lymphoblastic leukemia: a study from the Bone Marrow Pathology Group. Mod Pathol 34, 1358–1366 (2021). https://doi.org/10.1038/s41379-021-00739-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00739-4
