
Abstract
Gene fusions constitute pivotal driver mutations often encoding aberrant chimeric transcription factors. However, an increasing number of gene fusion events have been shown not to be histotype specific and shared among different tumor types, otherwise completely unrelated clinically or phenotypically. One such remarkable example of chromosomal translocation promiscuity is represented by fusions between EWSR1 or FUS with genes encoding for CREB-transcription factors family (ATF1, CREB1, and CREM), driving the pathogenesis of various tumor types spanning mesenchymal, neuroectodermal, and epithelial lineages. In this study, we investigate a group of 13 previously unclassified malignant epithelioid neoplasms, frequently showing an epithelial immunophenotype and marked predilection for the peritoneal cavity, defined by EWSR1/FUS–CREB fusions. There were seven females and six males, with a mean age of 36 (range 9–63). All except three cases occurred intra-abdominally, including one each involving the pleural cavity, upper, and lower limb soft tissue. All tumors showed a predominantly epithelioid morphology associated with cystic or microcystic changes and variable lymphoid cuffing either intermixed or at the periphery. All except one case expressed EMA and/or CK, five were positive for WT1, while being negative for melanocytic and other mesothelioma markers. Nine cases were confirmed by various RNA-sequencing platforms, while in the remaining four cases the gene rearrangements were detected by FISH. Eleven cases showed the presence of CREM-related fusions (EWSR1–CREM, 7; FUS–CREM, 4), while the remaining two harbored EWSR1–ATF1 fusion. Clinically, seven patients presented with and/or developed metastases, confirming a malignant biologic potential. Our findings expand the spectrum of tumors associated with CREB-related fusions, defining a novel malignant epithelioid neoplasm with an immunophenotype suggesting epithelial differentiation. This entity appears to display hybrid features between angiomatoid fibrous histiocytoma (cystic growth and lymphoid cuffing) and mesothelioma (peritoneal/pleural involvement, epithelioid phenotype, and cytokeratin and WT1 co-expression).
Similar content being viewed by others
Introduction
Recurrent gene fusions involving CREB family of transcription factors with genes encoding FET family RNA-binding proteins, such as EWSR1 and FUS, have been implicated in driving the oncogenesis of a diverse group of neoplasms, including a variety of benign and highly malignant soft tissue tumors, in addition to a subset of carcinomas and mesotheliomas [1, 2]. EWSR1–CREB fusions represent the genetic hallmark of angiomatoid fibrous histiocytoma (AFH), soft tissue and gastrointestinal clear cell sarcoma (CCS), primary pulmonary myxoid sarcoma (PPMS), hyalinizing clear cell carcinoma of salivary gland, and a subset of malignant mesotheliomas occurring in young adults [2,3,4,5,6,7,8]. ATF1 and CREB1 partners are the most prevalent, while cAMP responsive element modulator (CREM)-related fusions being less common and documented in fewer pathologic entities to date [9,10,11]. Although CREB1 and ATF1 are interchangeable gene partners, there is a striking propensity for EWSR1–CREB1 to occur in AFH and for EWSR1–ATF1 in soft tissue CCS and in hyalinizing clear cell carcinoma [3, 8], while only EWSR1–CREB1 was reported in PPMS [7]. In addition, an EWSR1–CREM fusion appears to define—or at least be most prevalent in—a unique myxoid mesenchymal neoplasm with an intracranial predilection [11].
In this study, we further expand the morphologic spectrum of tumors characterized by EWSR1/FUS–CREB fusions. Herein, we describe a series of distinctive malignant epithelioid neoplasms that often show cytokeratin or EMA positivity and predilection for peritoneal cavity. These tumors do not appear to fit in any previously described neoplastic category and likely represent a novel pathologic entity.
Materials and methods
Patients and cases
We selected 13 previously unclassified cases from our consultation files (PA and CRA) defined by a malignant epithelioid phenotype and harboring EWSR1/FUS–CREB fusions. In each case, H&E stained slides and paraffin tissue blocks or unstained slides were available for immunohistochemistry, RNA sequencing, and/or fluorescence in situ hybridization (FISH) as described below. Cases were assessed for microscopic features, including cell type (epithelioid and spindle), cytoplasmic appearance, and nuclear shape and degree of pleomorphism, mitotic activity, and necrosis. The study was approved by the Institutional Review Board at our institutions.
Immunohistochemistry
Immunohistochemical labeling was performed on the Benchmark XT autostainer (Ventana Medical Systems Inc, Tucson, AZ) using the I-View detection kit. The standard antibodies used, vendors, pretreatments, and dilutions included: cytokeratin AE1/3 (Chemicon, steam, 1:4000), EMA (Ventana, 760-4259, steam, prediluted), WT1 (Cell Marque, clone 6F-H2, Steam, Prediluted), Calretinin (Biocare, Steam, Prediluted polyclonal), desmin (Dako M0760, clone D33, steam, 1:100), CD99 (Leica, Clone 12E7, steam, prediluted), S100 protein (Ventana, 760-2914, stream, prediluted), BAP1 (Santa Cruz, clone C-4, steam, 1:200), and INI1 (BD Transduction Laboratories, clone 25/BAF47, steam, 1:100).
RNA sequencing
RNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA) in all except one case tested for RNA sequencing. Fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). In two cases, RNA-sequencing libraries were prepared using 20–100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA), as previously described [12]. Each sample was subjected to targeted RNA sequencing on an Illumina MiSeq at eight samples per flow cell (∼3 million reads per sample). All reads were independently aligned with STAR (version 2.3) and BowTie2 against the human reference genome (hg19) for Manta-Fusion and TopHat-Fusion analysis, respectively.
Four cases were tested by Anchored Multiplex RNA-sequencing assay using the Archer FusionPlex Solid tumor Kit (Archer, Boulder, CO) [13]. Anchored Multiplex polymerase chain reaction amplicons were sequenced on Illumina Miseq, and the data was analyzed using the Archer software. In two cases, the molecular reports were available from FoundationOne®CDX. In one case, frozen tissue was available for whole transcriptome analysis as previously described [11].
Fluorescence in situ hybridization (FISH)
FISH was performed as previously described [11]. Briefly, FISH on interphase nuclei from paraffin-embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking the EWSR1, FUS, ATF1, and CREM genes. BAC clones were chosen according to USCS genome browser (http://genome.ucsc.edu) [11]. The BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (https://bacpacresources.org/). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution. The genomic location of each BAC was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems). A positive score was interpreted when at least 20% of the nuclei showed a break apart signal. Nuclei with incomplete set of signals were omitted from the score.
Results
Clinical features
The study included 13 patients, 6 males and 7 females, ranging in age from 9 to 63 years, with a mean and median of 36 years (Table 1). Ten cases occurred intra-abdominally, often involving or spreading along the peritoneal surface, omentum or mesentery, with five cases arising in the mesocolon or peri-rectal/rectovaginal pouch. Two of the three pediatric patients presented with intra-abdominal tumors. Four cases further involved to various extents abdominal viscera, such as stomach, cecum, adrenal, and kidney. Two cases occurred in the soft tissues of the extremities, one of them presenting as a 15 cm forearm mass surrounding ulna and radius, being associated with bone erosion. The second case occurred in the deep soft tissue of the thigh, involving the periosteum and displaying grossly a cystic hemorrhagic appearance simulating a soft tissue aneurysmal bone cyst. One patient presented with a large, solid-cystic, pleural-based mass at the level of the right lower lung zone, causing mediastinal shift to the left. Three patients showed lymph node involvement, either as the initial presentation in two cases or as locoregional metastasis 6 years after diagnosis. Sizes ranged from 2 to 15 cm (mean 8 cm).
The submitted diagnoses varied significantly, also related to the wide anatomic distribution of the lesions, including Ewing sarcoma (three cases), sex cord stromal tumor (two cases), localized malignant mesothelioma (two cases), and in one case each: myoepithelial carcinoma and epithelioid inflammatory myofibroblastic tumor.
Morphologic features
All cases had at least focally an epithelioid phenotype, which predominated in eight cases. In the remaining five cases, the tumors showed either a mixed epithelioid and spindle morphology (two cases), or a mixed epithelioid and round cell phenotype (three cases)(Figs. 1–3). The epithelioid areas contained predominantly solid growth, with cells arranged in relatively cohesive sheets, often in a syncytial pattern (Fig. 1). A cystic or microcystic pattern was identified in all cases, either as a conspicuous finding or as a focal feature (Figs. 1–3). The microcystic spaces often contained pink serous fluid and were lined by the epithelioid neoplastic cells, which formed a flat community border. In four cases the cystic spaces were grossly observed and microscopically were associated with hemorrhage and hemosiderin deposition (Fig. 3). Most cases appeared well circumscribed and surrounded by a thick fibrous capsule, with prominent pericapsular lymphoid aggregates (Figs. 1, 2).

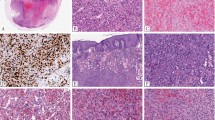
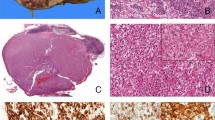
a–d (Case 1, 54/F, mesocolonic mass) Well-circumscribed neoplasm surrounded by a thick fibrous capsule associated with a dense lymphoid cuffing (a). The predominant architecture was solid (a, bottom). However, both macro- and microcysts were noted, the smaller cysts containing serous fluid (b). At higher power, the epithelioid cells had ill-defined borders, with eosinophilic cytoplasm, relatively round but slightly irregular nuclear membranes, vesicular chromatin, prominent nucleoli, and rare mitotes (b). The neoplastic cells showed diffuse immunoreactive for cytokeratin AE1:AE3 (c) and nuclear labeling for WT1 (d). e–h (Case 2, 10/F, rectovaginal pouch). This solid and cystic neoplasm was composed of epithelioid cells arranged in sheet-like pattern (e) or forming small tubular structures surrounding serous fluid (f). The cells were diffusely immunoreactivity for cytokeratin (g) and showed nuclear labeling for WT1 (h). i–l (Case 4, 9/M, adrenal) At low power, this epithelioid neoplasm was surrounded by a fibrous capsule and associated with a prominent lymphoplasmacytic cuff and abundant dystrophic calcification (i). At high power, the epithelioid cells showed eosinophilic cytoplasm, with forming focal microcysts containing serous fluid (j) and tubular structures within a hyalinized matrix (k). The neoplastic cells demonstrated patchy cytokeratin labeling (l) and nuclear labeling for WT1 (not shown).

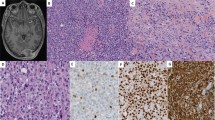
a, b (Case 6, 25/M, intra-abdominal lesion) Cystic metastasis to the liver (a), showing at high power a mixture of primitive round cells with areas of epithelioid cells arranged in solid sheets (b). c–d (Case 11, 14/F, thigh) Solid and cystic soft tissue mass showing epithelioid cells with clear cytoplasm arranged in sheets (c); tumor was diffusely positive for CD99, being misinterpreted as an Ewing sarcoma (d). e, f (Case 12, 29/M) Cystic and solid renal tumor (e) composed of predominantly round cells with focal areas of epithelioid appearance (f); which by immunohistochemistry showed multifocal cytokeratin (g) and diffuse CD99 positivity (h). i, j (Case 8, 44/F) Large pleural-based mass showing round and epithelioid cell morphology arranged in solid and pseudopapillary architecture. k, l (Case 5, 47/F, mesocolic) Multinodular mass surrounded by a fibrous capsule with lymphoid cuffing (k). At high power an abrupt transition between spindle fascicular growth to epithelioid areas arranged in nests and solid sheets (l).

a–c (Case 3, 63/M) predominantly solid omental mass associated with an abundant lymphoid infiltrate, resembling an involved lymph node (a), which at higher power showed sheets of monomorphic epithelioid cells with eccentric round nuclei and somewhat rhabdoid appearance (b). Immunohistochemical stain for Cam5.2 cytokeratin showed diffuse positivity (c). d–f (Case 9, 62/M, peripancreatic) Cystic lesion associated with prominent lymphoid aggregates and focal hemorrhagic changes (d), which at higher power showed a mixture of epithelioid and spindle cells with mild to moderate nuclear atypia (e). Tumor showed diffuse positivity for ALK (f). g–i (Case 13, 36/F, rectovaginal pouch) A similar cystic and hemorrhagic lesion (g), which at high power was composed of epithelioid and rhabdoid cells with densely eosinophilic cytoplasm, and was diffusely positive for cytokeratins and ultrastructurally showed abundant intracytoplasmic tonofilaments but lacking mesothelial differentiation (i).
Among the eight cases with predominant epithelioid morphology, five also contained focal rhabdoid morphology (Fig. 3). In nine cases the tumor cells showed a moderate amount of pale eosinophilic cytoplasm, while in the remaining four there was a mixture of cells with both clear and eosinophilic cytoplasm (Fig. 2). The nuclei were round and predominantly monomorphic, with smooth nuclear contours, and vesicular chromatin. Only one case showed some scattered moderate nuclear pleomorphism and enlarged, irregular nuclei (Fig. 3).
In two cases, the epithelioid component showed a mixed spindle and epithelioid phenotype, with an abrupt transition between the two cellular components in one of the cases (Fig. 2). The spindle cells were arranged in intersecting fascicles and showed delicate cell processes and uniform fusiform nuclei (Fig. 2). In three cases a round cell phenotype was noted (Fig. 2), blending in with areas of epithelioid cells that displayed more abundant clear to pale eosinophilic cytoplasm (Fig. 2). In one of the three cases, the round cell component predominated (Case #12, Fig. 2), with only focal interspersed areas of epithelioid cells.
All except three cases showed an associated chronic inflammatory infiltrate consisting of lymphoid aggregates, which in five cases was prominent, obscuring the neoplastic component or mimicking a lymph node (Figs. 1, 3). In two of the cases, the prominence of the lymphoid infiltrate prompted additional immunohistochemical stains to exclude a concurrent low-grade lymphoma. In the remaining three cases, the combination of cystic or cystic hemorrhagic changes, with the rich lymphoid infiltrate, mimicked an AFH at low power. However, at a closer examination, the cells showed malignant epithelioid features and expressed epithelial markers which excluded that diagnosis. In the remaining cases the lymphoid infiltrate was less conspicuous, either seen at the periphery around the fibrous pseudocapsule surrounding the lesion or interspersed within the lesion (Figs. 1, 3).
Other less common features included: prominent calcifications (two cases, Fig. 1), pseudopapillary growth pattern (two cases, Fig. 2), myxoid stromal component (one case), and thick collagen bundles (two cases).
Most cases had relatively low mitotic activity, with nine cases showing 1–2 MF/10 HPFs. Two cases showed an intermediate mitotic count of 5–6 MF/10 HPFs, with one of them also showing rare atypical mitoses, patchy necrosis, and scattered moderate nuclear pleomorphism (case 9, Fig. 3). There was only one outlier case, showing a predominantly small blue round cell histology, with areas of necrosis and a brisk mitotic rate of 20 MF/10 HPFs (case 12, Fig. 2). One additional case showed focal punctate necrosis (case 1), but otherwise had a low mitotic activity and lacked nuclear pleomorphism.
Immunohistochemical profile
By immunohistochemistry, all except one case showed evidence of epithelial differentiation. Five of the cases (including two of the pediatric tumors) showed diffuse immunoreactivity for cytokeratin (AE1:AE3 and/or Cam5.2) and positivity for EMA (four diffuse and 1 focal) (Fig. 1), with four of them also co-expressing WT1 (three diffuse and one focal) (Fig. 1) (Table 1). The remaining seven cases showed focal positivity for both CK and EMA (three cases), or only CK (two cases) or EMA (two cases). Only one additional case from this latter group showed rare WT1 staining, while the remaining cases were negative. One case (case 7, forearm) was negative for EMA, CK, and a large battery of immunstains, including WT1, showing only nonspecific staining for calponin. All cases tested were negative for calretinin and retained BAP1 and INI1 expression. Other results included four cases with variable CD99 reactivity (including one case with diffuse pattern, case 11, thigh, resulting in an erroneous diagnosis of Ewing sarcoma) and three cases showing focal desmin positivity. Two cases showed reactivity for inhibin, which in one case was focal. Other pertinent negative stains included P40, P63, SF1, and S100. One case showed diffuse and strong expression for ALK (case 9, Fig. 3).
In one case (case 13, rectovaginal pouch) electron microscopy was performed in the clinical work-up using paraffin-embedded tissue, showing abundant intracytoplasmic tonofilaments but no evidence to support mesothelial differentiation, i.e., long, thin microvilli (Fig. 3). Additional stains are summarized in Supplementary Table 1.
Molecular pathology
Seven cases harbored a EWSR1–CREM fusion, while four revealed a FUS–CREM fusion (Table 1). The breakpoints were available in six cases studied by various RNA-sequencing platforms. In two of the cases with FUS–CREM fusion, exon 8 of FUS was fused to either exon 5 or exon 7 of CREM. In four cases with EWSR1–CREM fusion the exonic composition showed CREM exon 7 was fused to EWSR1 exons 7, 13, 14, or 15. The two remaining cases showed an EWSR1–ATF1 fusion, which in one case showed ATF1 exon 5 fused to either EWSR1 exon 7 or exon 14. The rest of the cases were tested by FISH and showed gene rearrangements for EWSR1, FUS, and CREM genes.
In addition, we have interrogated the gene expression of 2 of the study cases positive for EWSR1–CREM fusion (cases#5 &12), both tested on the TruSight RNA Fusion Panel and compared to a large number of various neoplasms available on the same platform, including other tumors with EWSR1–CREB fusions: three CCSs, two GI CCSs, five AFH, one myxoid mesenchymal tumor, and one hyalinizing clear cell carcinoma (Fig. 4). In addition, three cases of fusion-positive mesotheliomas studied on the same platform were available for analysis, including two pediatric patients with EWSR1–ATF1 and one adult patient with EWSR1–YY1 fusion. The two study cases clustered together (red lines) and separate from all the other tumors with similar gene fusions or fusion-positive mesotheliomas.

Two study cases positive for EWSR1–CREM fusion (cases#5 & 12, red lines), clustered together, being away from 12 other tumors with EWSR1–CREB fusions: 3 clear cell sarcomas (2 with EWSR1–ATF1, 1 EWSR1–CREB1, green), 2 GI clear cell sarcomas (EWSR1–ATF1 fusion, brown), 5 AFH (2 with EWSR1–ATF1, 3 EWSR1–CREB1, blue), 1 myxoid mesenchymal tumor (EWSR1–CREM, purple) and 1 hyalinizing clear cell carcinoma (EWSR1–ATF1, orange). The study group cases did not cluster close to the 3 fusion-positive mesotheliomas (2 EWSR1–ATF1, 1 EWSR1–YY1, pink). A large number of various sarcoma types available on the same platform shown in gray lines.
Clinical follow-up
Clinical follow-up was available in nine patients, as remaining were either very recent cases or lost to follow-up. Despite this limitation, the information available clearly demonstrated its malignant potential and propensity for both peritoneal, lymph node, and distant spread (Table 1). Six patients developed peritoneal or pleural local recurrence/metastatic implants, despite extensive resection and/or chemotherapy in four patients. One of the patients developed three recurrences over a 10-year period and is currently alive with no evidence of diseases at 17 years follow-up. Three patients developed locoregional lymph node metastases, two at presentation and one 8 years from diagnosis. One patient developed liver metastases one year after diagnosis.
Discussion
We report a series of previously unrecognized malignant epithelioid neoplasms demonstrating unique clinical, morphologic and molecular features. Among the 13 patients included, 11 demonstrated involvement of a mesothelial-lined cavity, often forming masses within the abdominal cavity with or without visceral extension. Most lesions occurred in the omentum, mesocolon or rectovaginal pouch, with one patient presenting with pleural-based disease. Two patients presented with large lesions within the deep soft tissues of the extremities, being associated in one case with bone erosion. Microscopically, in addition to the predominant epithelioid phenotype, the tumors exhibited distinctive cystic changes and a brisk lymphocytic infiltrate, in the form of lymphoid aggregates either intermixed with or at the periphery of the lesion. Immunohistochemically, all except one case showed convincing evidence of epithelial differentiation, with positivity for cytokeratin, EMA or both. In one case, the available ultrastructural analysis showed abundant cytoplasmic filaments but no evidence of mesothelial differentiation. Although WT1 nuclear labeling was present in a third of cases, other mesothelial markers, such as calretinin were negative and BAP1 expression was retained. Moreover, all cases lacked the typical tubulo-papillary architecture as seen in classic variants of epithelioid mesotheliomas.
The morphologic findings of this novel subset reveal certain overlap with two other pathologic entities that have been shown to harbor EWSR1/FUS–CREB-related fusions, specifically AFH and mesothelioma in young adults. The combination of cystic changes, occasionally associated with hemorrhage and hemosiderin deposition, and the brisk lymphocytic cuffing and lymphoid aggregates was highly reminiscent at low magnification to AFH in a small subset of cases. However, the lesional cells had a predominant epithelioid morphology arranged in cohesive sheets, admixed with variable rhabdoid, tubular, spindle, and round cell components. All except one case showed positivity for epithelial markers, including strong and diffuse cytokeratin staining, and occasionally for WT1, which are not in keeping with a diagnosis of AFH. Moreover, the malignant phenotype and the pattern of metastasis (lymph node and liver) is also highly unusual for AFH. In contrast, AFH is a rarely metastasizing mesenchymal neoplasm, with predilection for the superficial soft tissues of extremities of children and young adults, being characterized by a constellation of microscopic findings, such as nodules of monomorphic ovoid or histiocytoid cells arranged in solid sheets, intermixed with blood-filled cavernous spaces, surrounded by a dense lymphocytic infiltrate. Most AFH show variable positivity for desmin, EMA, and CD99 in about half of the cases, but are consistently negative for cytokeratins. AFH harbor mostly EWSR1–CREB1 fusions, though rare cases with EWSR1–ATF1 or FUS–ATF1 fusion have been reported [3].
On the other hand, the overlap of our cohort with epithelioid mesothelioma is even more striking, in particular due to its predilection for peritoneal or pleural surface involvement and the immunoreactivity for cytokeratin and/or EMA, with co-expression of WT1 in a third of the cases. However, the lymphoid cuffing and localized nature of these abdominal neoplasms argue against mesothelioma, as does the complete absence of calretinin immunoreactivity, retained BAP1 expression and ultrastructural findings in one case studied. Moreover, our group has reported recently on a small subset of epithelioid mesothelioma occurring in young adults harboring EWSR1/FUS–ATF1 fusions [2]. Interestingly, this molecular subset of mesotheliomas also shows a predisposition for abdominal cavity and retains BAP1 expression, but microscopically are indistinguishable from conventional epithelioid mesotheliomas, showing at least focally papillary architecture, and positivity for WT1 and calretinin. However, two of our study group cases did not cluster together with any of the three cases of fusion-positive mesotheliomas, either EWSR1–ATF1 or EWSR1–YY1, investigated on the same RNA sequencing platform.
The differential diagnosis also included sex cord stromal tumors, due to WT1 nuclear reactivity and the immunoreactivity for inhibin in two of the cases. However, the diffuse immunoreactivity for cytokeratin and EMA in these cases, along with the absence of SF1 and Melan A labeling, argues against a sex cord stromal tumor diagnosis. Myoepithelial carcinomas were also considered, based on the EMA and/or cytokeratin expression, but the absence of S100 protein in all cases is not consistent with this diagnosis. Only one case of myoepithelial tumor of soft tissue was so far reported with an EWSR1–ATF1 fusion; however, that case had a well-documented myoepithelial immunophenotype [14].
Another consideration was the so-called myxoid mesenchymal spindle cell tumor that has a predilection for intracranial location that similarly harbors EWSR1–CREM fusions [11, 15]. Although some have suggested that this lesion represents a myxoid variant of AFH rather than a separate pathologic entity [16, 17], it clearly shows distinctive morphologic features, such as extensive myxoid stroma, frequent amianthoid fibers and lacks lymphoid aggregates and hemorrhagic pseudoangiomatous spaces. However, none of the reported cases to date showed convincing epithelial differentiation with cytokeratin positivity or WT1 labeling.
The common intra-abdominal location and occasional gastric or bowel involvement also raises the possibility of a gastrointestinal CCS, which are characterized by EWSR1–CREB1 or ATF1 fusions [6]. However, these tumors consistently show positivity for S100 protein, while none of the cases in the present cohort showed expression for this marker.
Interestingly, one of the intra-abdominal lesions that involved the pancreas and resembled an AFH at low magnification with cystic hemorrhagic changes and abundant lymphoid aggregates, showed strong and diffuse ALK positivity and was thought to represent an epithelioid inflammatory myofibroblastic sarcoma. However, no ALK fusions were detected, and instead Archer FusionPlex showed the presence of an EWSR1–ATF1 fusion. Indeed, ALK overexpression unrelated to an ALK gene rearrangement has been recently described in other sarcomas with recurrent gene fusions, such as the intra-osseous rhabdomyosarcoma with EWSR1/FUS–TFCP2 fusion [18, 19]. Our result adds tumors with EWSR1–CREB fusion to this group of lesions with associated upregulation of ALK protein through alternative mechanisms unrelated to ALK fusions.
CREM, together with ATF1 and CREB1, belong to the CREB family of basic leucine zipper (bZIP) transcription factors, which share a high degree of homology in the C-terminal bZIP domain. The bZIP domain binds to target CRE present in the regulatory regions of over a hundred putative target genes [20, 21], reflecting the functional diversity of the CREB family of transcription factors, including neuronal development, synaptic plasticity, glucose homeostasis, spermatogenesis, and cytokine regulation [20,21,22,23,24].
Similar to fusions encompassing other CREB family members, CREM-related fusions are evolving as promiscuous abnormalities, spanning the pathogenesis of a number of tumor entities, of different cell lineages. EWSR1–CREM represents the main genetic alteration for the intracranial myxoid mesenchymal tumor [11]. However, a recent report has described three cases of myxoid AFH harboring EWSR1–CREM fusions [10]. This observation further raises the debate regarding the pathogenesis of these two lesions, one occurring with predilection intracranially and being predominantly myxoid with frequent amianthoid fibers, and the other showing features of AFH in addition to myxoid changes [16, 17]. Moreover, the EWSR1–CREM fusion was also recently described in a single case of CCS of soft tissue [10], as well as in three cases of hyalinizing clear cell carcinoma of salivary gland, as an alternative to the more common EWSR1–ATF1 fusions in these tumor types [9]. Finally, Yoshida et al. recently reported two unclassified neoplasms with EWSR1–CREM fusions [10]. One was an abdominal cavity spindle cell neoplasm that was cytokeratin positive in a 15-year-old male, while the other was a chest wall round cell sarcoma in a 63-year-old female. It is possible that one or both of these cases may be related to the current study group.
In conclusion, our study describes a unique group of malignant epithelioid neoplasms, with a striking predilection for mesothelial-lined cavities and EWSR1–CREM fusions. A significant proportion of the cases displayed epithelial differentiation by immunohistochemistry, either by cytokeratin, EMA or both in all except one case. All lesions were previously unclassified and did not correspond to any known tumor category, suggesting a novel pathologic entity with distinctive microscopic features. Morphologically, these cases combined features of other tumors harboring EWSR1–CREB fusions, specifically AFH and malignant mesothelioma. As the current series displays a variegated phenotype, with a small subset morphologically resembling AFH, while other cases displaying a pure epithelioid appearance, the possibility of a heterogeneous group of tumors driven by an EWSR1/FUS–CREM fusion pleiotropy cannot be entirely excluded. Despite a broad histologic spectrum, most cases share a significant core of microscopic and immunohistochemical features in keeping with a single entity. Specifically, all tumors showed some degree of cystic changes and most of the cases revealed lymphoid cuffing or lymphoid aggregates. In fact, a subset of cases occurring intra-abdominally displayed a homogeneous phenotype, composed of a pure epithelioid population, being diffusely positive for cytokeratin and often WT1, but lacking other convincing mesothelial differentiation. At the other ends of the spectrum were lesions with a more prominent spindle or round cell component intermixed with the epithelioid areas and a more focal expression of epithelial markers and complete lack of WT1 expression. It is tempting to speculate, that this novel tumor entity shows hybrid morphologic features of two completely different diseases, of distinct histogenesis (i.e., AFH and malignant mesothelioma), as a result of their common EWSR1–CREB gene fusion pathogenesis. At the gene expression level, two of the cases with EWSR1–CREM fusion, including one with mixed epithelioid and spindle phenotype and the other with predominant round cell features, clustered together and separate from other EWSR1–CREB fusion-positive tumors, such as AFH and CCSs, adding support for a distinct entity. Further studies with larger number of cases and investigated by alternative genomic platforms, including methylation classifiers, are needed to draw more definitive conclusions regarding the relationship of this group of tumor with other lesions defined by EWSR1–CREB fusions.
References
Thway K, Fisher C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: the current status. Am J Surg Pathol. 2012;36:e1–e11.
Desmeules P, Joubert P, Zhang L, Al-Ahmadie HA, Fletcher CD, et al. A subset of malignant mesotheliomas in young adults are associated with recurrent EWSR1/FUS-ATF1 fusions. Am J Surg Pathol. 2017;41:980–8.
Antonescu CR, Dal Cin P, Nafa K, Teot LA, Surti U, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosom Cancer. 2007;46:1051–60.
Antonescu CR, Tschernyavsky SJ, Woodruff JM, Jungbluth AA, Brennan MF, et al. Molecular diagnosis of clear cell sarcoma: detection of EWS-ATF1 and MITF-M transcripts and histopathological and ultrastructural analysis of 12 cases. J Mol Diagn. 2002;4:44–52.
Wang WL, Mayordomo E, Zhang W, Hernandez VS, Tuvin D, et al. Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcripts in clear cell sarcoma (melanoma of soft parts). Mod Pathol. 2009;22:1201–9.
Antonescu CR, Nafa K, Segal NH, Dal Cin P, Ladanyi M. EWS-CREB1: a recurrent variant fusion in clear cell sarcoma-association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res. 2006;12:5356–62.
Thway K, Nicholson AG, Lawson K, Gonzalez D, Rice A, et al. Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: a new tumor entity. Am J Surg Pathol. 2011;35:1722–32.
Antonescu CR, Katabi N, Zhang L, Sung YS, Seethala RR, et al. EWSR1-ATF1 fusion is a novel and consistent finding in hyalinizing clear-cell carcinoma of salivary gland. Genes Chromosom Cancer. 2011;50:559–70.
Chapman E, Skalova A, Ptakova N, Martinek P, Goytain A, et al. Molecular profiling of hyalinizing clear cell carcinomas revealed a subset of tumors harboring a novel EWSR1-CREM fusion: report of 3 cases. Am J Surg Pathol. 2018;42:1182–9.
Yoshida A, Wakai S, Ryo E, Miyata K, Miyazawa M, et al. Expanding the phenotypic spectrum of mesenchymal tumors harboring the EWSR1-CREM fusion. Am J Surg Pathol. 2019;43:1622–30.
Kao YC, Sung YS, Zhang L, Chen CL, Vaiyapuri S, et al. EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol. 2017;41:482–90.
Suurmeijer AJH, Dickson BC, Swanson D, Zhang L, Sung YS, et al. A morphologic and molecular reappraisal of myoepithelial tumors of soft tissue, bone, and viscera with EWSR1 and FUS gene rearrangements. Genes Chromosom Cancer. 2020;59:348–56.
Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84.
Flucke U, Mentzel T, Verdijk MA, Slootweg PJ, Creytens DH, et al. EWSR1-ATF1 chimeric transcript in a myoepithelial tumor of soft tissue: a case report. Hum Pathol. 2012;43:764–8.
Sciot R, Jacobs S, Calenbergh FV, Demaerel P, Wozniak A, et al. Primary myxoid mesenchymal tumour with intracranial location: report of a case with a EWSR1-ATF1 fusion. Histopathology. 2018;72:880–3.
Gareton A, Pierron G, Mokhtari K, Tran S, Tauziede-Espariat A, et al. ESWR1-CREM fusion in an intracranial myxoid angiomatoid fibrous histiocytoma-like tumor: a case report and literature review. J Neuropathol Exp Neurol. 2018;77:537–41.
Bale TA, Oviedo A, Kozakewich H, Giannini C, Davineni PK, et al. Intracranial myxoid mesenchymal tumors with EWSR1-CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol. 2018;28:183–91.
Le Loarer F, Cleven AHG, Bouvier C, Castex MP, Romagosa C, et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol. 2020;33:404–19.
Agaram NP, Zhang L, Sung YS, Cavalcanti MS, Torrence D, et al. Expanding the spectrum of intraosseous rhabdomyosarcoma: correlation between 2 distinct gene fusions and phenotype. Am J Surg Pathol. 2019;43:695–702.
Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol cell Biol. 2001;2:599–609.
Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–23.
Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–61.
Don J, Stelzer G. The expanding family of CREB/CREM transcription factors that are involved with spermatogenesis. Mol Cell Endocrinol. 2002;187:115–24.
Rauen T, Hedrich CM, Tenbrock K, Tsokos GC. cAMP responsive element modulator: a critical regulator of cytokine production. Trends Mol Med. 2013;19:262–9.
Acknowledgements
We thank Norman Barker and Bruce Crilly for expert photographic assistance.
Funding
This study was supported in part by: P50 CA217694 (CRA), P50 CA140146 (CRA), P30 CA008748 (CRA), Cycle for Survival (CRA), Sara’s Cure (CRA), Kristin Ann Carr Foundation (CRA), Dahan Translocation Carcinoma Fund (PA), Joey’s Wings (PA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Argani, P., Harvey, I., Nielsen, G.P. et al. EWSR1/FUS–CREB fusions define a distinctive malignant epithelioid neoplasm with predilection for mesothelial-lined cavities. Mod Pathol 33, 2233–2243 (2020). https://doi.org/10.1038/s41379-020-0646-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0646-5
This article is cited by
-
CREB fusion–associated epithelioid mesenchymal neoplasms of the female adnexa: three cases documenting a novel location of an emerging entity and further highlighting an ambiguous misleading immunophenotype
Virchows Archiv (2023)
-
Advances in the management of peritoneal malignancies
Nature Reviews Clinical Oncology (2022)
-
Comprehensive genomic profiling of EWSR1/FUS::CREB translocation-associated tumors uncovers prognostically significant recurrent genetic alterations and methylation-transcriptional correlates
Modern Pathology (2022)
-
Intra-abdominal EWSR1/FUS-CREM-rearranged malignant epithelioid neoplasms: two cases of an emerging aggressive entity with emphasis on misleading immunophenotype
Virchows Archiv (2022)
-
Emerging glioneuronal and neuronal tumors: case-based review
Brain Tumor Pathology (2022)
