
Abstract
Anaplastic thyroid carcinomas (ATC) are rare, but represent the most lethal malignancy of the thyroid. Selective molecular markers and drivers distinguishing ATC from other thyroid carcinomas of follicular origin remain largely unknown, limiting advances in diagnosis and treatment. In a retrospective study, we analyzed gene expression in 36 ATC, 18 poorly differentiated, 132 papillary, and 55 follicular thyroid carcinoma, as well as 124 paired and unpaired normal thyroid tissues in three independent cohorts by RNA-sequencing and immunohistochemistry. RNA-sequencing data in the test cohort suggested selective ATC protein biomarkers. Evaluation of these revealed that ATCs are characterized by the de novo expression of various testis antigens, including melanoma-associated antigen A3 (MAGEA3), but most importantly the oncofetal IGF2 mRNA binding protein 1 (IGF2BP1). Shallow whole genome sequencing essentially excluded that IGF2BP1 upregulation results from gene copy number alterations. Immunohistochemical analyses in all three tumor cohorts confirmed the selective de novo expression of IGF2BP1 protein in ATC. In sum, 75% (27/36) of all tested ATC and 0.5% (1/204) of poorly and well-differentiated thyroid carcinoma tissue samples were positive for IGF2BP1 protein. This indicates that IGF2BP1 protein expression identifies ATC with a diagnostic odds ratio of 612 (95% CI: 74.6–5021). In addition, we found that MAGEA3 is exclusively, although less consistently upregulated in ATC, presenting with an odds ratio of 411 (95% CI: 23.8–7098.7). Importantly, we provide confirmatory evidence that IGF2BP1 and MAGEA3 expression distinguishes ATC from poorly differentiated thyroid carcinoma. IGF2BP1 furthermore identified ATC foci within low-grade follicular thyroid carcinoma. In conclusion, IGF2BP1 represents the most promising single-gene marker available for ATC, followed by MAGEA3, improving on current techniques. Robust markers are essential to help distinguish this high-grade malignancy from other thyroid carcinomas, to guide surgical decision making, therapy and post-resection/therapy monitoring strategies.
Similar content being viewed by others


Introduction
Thyroid cancer of follicular origin is the most common endocrine malignancy, with significantly increasing incidence. The majority of tumors are further classified in well-differentiated carcinomas (WDTCs) including papillary thyroid carcinomas (PTC; incidence, 80–90%) and follicular thyroid carcinomas (FTC; incidence, 10–15%). These are distinguished from poorly differentiated (PDTC; incidence, 1–6%) and considerably rarer anaplastic thyroid carcinomas (ATC; incidence, 1–2%) [1, 2]. The majority of WDTCs have a good prognosis, and PDTC an intermediate prognosis. ATC, however, present a nearly uniformly fatal disease, accounting for the majority of thyroid cancer-associated deaths [3, 4].
Diagnosis of an ATC, either arising de novo or as aggravation of a WDTC or PDTC, demands for urgent and radical surgical interventions due to the local aggressive behavior and early metastasis formation, but still therapy options remain palliative. Therefore, the possible accurate identification, even of already microscopic foci of ATC within lower-grade thyroid cancer, would bring a benefit to any patient in sufficient time [1, 3, 5].
Although, histological classification of thyroid cancer remains the gold standard for diagnosis, massive-parallel sequencing results are increasingly being considered. Mutations in BRAF, (H/K/N) RAS, and other genes are found to a certain degree in all thyroid cancers of follicle epithelium, including ATC. However, these insights have failed to greatly improve patient survival [3, 6, 7]. An FDA-approved therapy option targeting BRAF-mutated ATC by applying dabrafenib and trametinib [8] changed the therapeutic landscape of the disease, but continued dismal outcomes highlight the need to identify selective markers and reveal targetable genes expressed in ATC. To further point at specific marker-driven approaches for ATC therapy and diagnosis improvement, the investigation of the ATC transcriptome should be instrumental. To date, microarrays and RNA-sequencing (RNA-seq) datasets on thyroid carcinomas are available [6, 9], but did not elucidate factors exclusive to ATC. Notably, ATCs are not included in TCGA transcriptomic datasets limiting avenues for biomarker and drug development.
The oncofetal IGF2 mRNA binding protein 1 (IGF2BP1) is a bona fide oncofetal protein, upregulated in some advanced solid cancers. The protein promotes the expression of oncogenes like MYC, LIN28B, and SRF by impairing their mRNA decay [10,11,12]. Consistent with its role supporting oncogene expression, IGF2BP1 was reported as a posttranscriptional driver of tumor cell proliferation, migration, metastatic potential, and therapy resistance [13, 14]. In that respect, it represents a prognostic marker for low survival probability in ovarian and neuroblastic cancers [15, 16].
Here, we reveal that IGF2BP1 and MAGEA3 are the first reliable protein and RNA markers of ATC, specifically distinguishing this malignancy from any other thyroid cancer of follicular origin, including PDTC. Besides their diagnostic value, therapeutic targeting of both may provide a promising future perspective for the treatment of ATC, independent of mutational status.
Methods
Patient samples
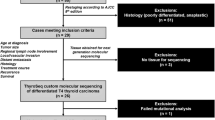
For the test cohort ten human primary ATC, six PTC and six FTC samples were collected from 1999 to 2012 at the University clinic of Halle, Germany. In addition, six nonmalignant thyroid tissue samples (NT) samples served as healthy controls. Specimens were formalin-fixed and paraffinized for immunohistochemistry, or snap frozen in liquid nitrogen and stored at −80 °C. All samples were re-evaluated histologically and with review of patient records by two pathologists (NP and CW).
An independent in-house tissue microarray (TMA I) contained 147 primary thyroid cancer samples (20 ATC, 18 PDTC, 82 PTC, and 29 FTC) and 108 paired normal thyroid tissue samples of all entities. A commercial microarray (TMA II, TH8010a, Biomax) contained 6 primary ATC, 44 PTC, 20 FTC, and 10 unpaired normal tissue samples.
The clinical characteristics of the tumor cohorts are summarized in Table 1.
Immunohistochemistry
Immunohistochemistry was performed on 3 µm thick, consecutive sections of formalin-fixed, paraffin-embedded samples with the Bond Polymer refine detection Kit (Leica, DS9800), according to the manufacturer’s instructions on a fully automated immunohistochemistry stainer (Leica Bond). Sections were imaged with an Olympus BX50/51 microscope. Two pathologists (US and MB), independently and blinded to the clinical data, scored all samples by using a Histoscore, as described previously [17]. In brief, the relative amount of tumor cells being positively stained (%) was multiplied by their intensity from 0 (negative), 1 (weak), 2 (moderate), to 3 (intense). Expression classified into absent (0), low (1–100), intermediate (101–200), or strong (201–300) overall expression. Antibodies are summarized in Supplementary Table S2.
Western blotting
For Western blotting, cells were lysed in lysis buffer (50 mM Tris-HCl (pH 7.4), 50 mM NaCl, 2 mM MgCl2, 1% SDS). Protein expression was analyzed by Western blotting with indicated antibodies (Supplementary Table 2), by an infrared scanner (LICOR).
Databases
For Kaplan–Meier analysis, patient survival was analyzed by using the cBioPortal platform (http://cbioportal.org), combining patient data from the TCGA for PTC and FTC and from the MSKCC (Memorial Sloan Kettering Cancer Center) for ATC.
For the analysis of microarray data derived from Landa et al. (GEO Series accession number GSE76039; [6]), GEO2R was used.
Deep-sequencing and differential gene expression
Total RNA was isolated from fresh frozen tissues by using the miRNeasy Kit (Qiagen), according to the manufacturer’s instructions. Total RNA-sequencing library preparation and sequencing was performed at the IKFZ (Leipzig, Germany). For total RNA-seq low-quality read ends as well as remaining parts of sequencing adapters were clipped using Cutadapt (v 1.4.2 or 1.6). Subsequently, reads were aligned to the human genome (UCSC GRCh19) using TopHat (v 2.0.12; [18]) or Bowtie2 (V 2.2.4; [19]), respectively. FeatureCounts (v 1.4.6; [20]) was used for summarizing gene-mapped reads. Ensembl (GRCh37.75; [21]) was used for annotations. Differential gene expression (DE) was determined by the R package edgeR (v 3.12.1; [22]) using TMM normalization, essentially as described previously [23].
Presented data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE126729. RNA-seq data are also available via the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl; datasets: “Tumor Thyroid Carcinoma – Huettelmaier”) for interactive use.
GSEA analysis
Gene set enrichment analyses (GSEA) were performed as described previously [24]. The gene set hallmarks collection (H) was used for a list of all protein-coding genes ranked according to fold changes. The respective data were visualized by using the R package clusterProfiler [25].
Statistics
Statistical analysis was performed using GraphPad Prism software (V7.0). Statistical significance was determined by using nonparametric Mann–Whitney test. Positive/negative predictive values (PPV, NPV) and diagnostic odds ratios (DOR) were determined by using MedCalc (V19.1.3). A principal component analysis was performed by using the R package pcaExplorer [26].
Results
IGF2BP1 is de novo expressed in ATC
The ATC is the most fatal thyroid malignancy (Supplementary Fig. S1a), but with the exception of few analyzed samples, comprehensive transcriptome analyses aiming to identify selective biomarkers are still rare [6, 9, 27, 28]. To identify novel markers of ATC, thyroid carcinoma gene expression was analyzed by RNA-seq in a test cohort (Table 1; clinical characteristics of the studied tumor cohorts). The protein-coding transcriptome of ten ATCs was compared with gene expression of six PTCs, six FTCs, and six NTs. A principle component analysis of transcriptome data illustrated that primary ATC samples cluster well together and are strikingly distinct from PTC, FTC and NT (Fig. 1a). In detail, a comparison of numbers on differentially expressed genes to NT revealed approx. 8000 differentially expressed genes (FDR ≤ 0.01) for ATC, but only approx. 500 and 100 for PTC and FTC, respectively (Supplementary Fig. S1b).

a Principle component (PC) analysis on RNA-seq data derived from the test cohort, including 10 ATC, 6 PTC, 6 FTC, and 6 NT samples. b Dot plot presentation of cancer hallmark gene sets upon a GSEA for differentially expressed genes for 10 ATCs vs 18 noATCs (6 PTC, 6 FTC, and 6 NT samples), investigated in (a). NES = normalized enrichment score. c Volcano plot of log2 mRNA fold changes plotted against the −log10 FDR (false discovery rate) for 10 ATCs versus 18 noATCs, investigated in (a). Horizontal dashed line indicates threshold (false discovery rate, FDR ≤ 0.01). Indicated in red are the top 20 ATC-exclusive genes, identified as described in the text.
Cancer hallmark gene set enrichment analysis (GSEA) comparing ATC to PTC, FTC and NT, collectively referred to as “noATC,” dissected the molecular pathology aside frequently described mutations (Fig. 1b). These gene sets classify differentially expressed genes into well-defined hallmark pathways of cancer. Thus, essentially deregulated genes represented the invasive/pro-metastatic potential (epithelial-to-mesenchymal transition (EMT)), supported by dissociation of the apical junctional complexes, but also represented high rates of proliferation by ensuring fast cell cycle progression (G2M checkpoint; E2F targets). In support, genes directly activated by the MYC oncogene are upregulated, which also drive EMT and proliferation. ATCs are further distinguished by an increased expression of markers for inflammatory response and a severe alteration of metabolic processes, most prominently elevated glycolysis [29]. Pro-mesenchymal dedifferentiation of ATC was further supported by the reduced expression of thyroid markers like TSHR, epithelial markers like E-cadherin (CDH1) and the upregulation of stemness and EMT-associated markers MYC, SNAI2, TWIST1, OCT3/4 (POU5F1), LIN28B and NANOG (Supplementary Fig. S1c) [30,31,32]. Collectively, this indicated severe deregulation of the protein-coding transcriptome in ATC and suggested protein markers distinguishing this malignancy.
To identify exclusive protein markers of ATC, we assessed the de novo expression (mean FPM in noATC samples < 1; fold change in ATC against noATC samples > 50) of transcripts and evaluated the consistency of mRNA expression in ATC by ranking genes by increasing relative standard deviation (RSD) of expression in ATC (Fig. 1c; Table 2). The top 20-ranked de novo expressed protein-coding genes with low RSD of expression in ATC demonstrated the most consistent de novo expression for IGF2BP1 (Fig. 1c — top 20 genes in red). Interestingly, 12 of the 20 protein-coding genes, including IGF2BP1 and MAGEA (melanoma-associated antigen) proteins, are reported testis antigens [33]. These genes are of advanced interest in the focus of immunotherapy.
As our test cohort did not include PDTC samples, mRNA expression was analyzed in an independent microarray dataset, comparing the transcriptomes of ATC and PDTC [6]. Reinvestigation of this study revealed that IGF2BP1 mRNA was reliably observed in 40% (8/20) of ATC samples. In sharp contrast, IGF2BP1 mRNA remained at background levels in all 17 PDTC samples included in the study (Supplementary Fig. S1d). Similar findings could be drawn for the MAGEA representative MAGEA3, with 40% (8/20) positive ATC and 5.9% (1/17) PDTC samples (Supplementary Fig. S1e). These findings provided independent support of the gene expression analysis in the here presented test cohort suggesting that IGF2BP1, but also MAGEA3, are selective markers of ATC to distinguish this malignancy even from PDTC.
Exclusive expression of IGF2BP1 and MAGEA3 in ATC was initially validated by Western blotting in two ATC-derived cell lines (C643 and 8305C) and the individual samples of each thyroid cancer subtype comprised in the test cohort (Fig. 2a, b). The sharp and exclusive upregulation of both proteins in ATC samples was associated with enhanced expression of MYC, as well as the loss of CDH1 expression, confirming transcriptome studies. This suggested that IGF2BP1 provides a robust, positive marker for ATC at the mRNA as well as protein level. To evaluate this in further detail, representative tumor samples of the test cohort were analyzed by immunohistochemistry, confirming the selective de novo expression of IGF2BP1 protein in paraffinized ATC tissue (Fig. 2c; Supplementary Fig. S2). Importantly, in none of the other samples of the test cohort, IGF2BP1 protein expression was observed. This suggested IGF2BP1 as the first positive marker of ATC.

a Representative Western blot analysis of indicated proteins in two ATC-derived cell lines (C643 and 8305C) and protein lysates of individual samples analyzed by RNA-seq in (a). VCL served as loading control. b Scatter dot plot presentation of quantified log2 protein expression, determined for test cohort samples, investigated in (a). c IGF2BP1 expression analyzed by immunohistochemistry in representative samples investigated in (a). HE, hematoxylin eosin staining. Scale bars, 100 µm. Statistical significance was determined by Mann–Whitney test in (d) (***p ≤ 0.001; *p ≤ 0.05; n.s. not significant).
IGF2BP1 de novo expression is unlikely a consequence of chromosomal aberrations
Recently, the IGF2BP1 gene locus, located on the long arm of chromosome 17 (17q21.32), was found to be commonly gained and associated with poor survival probability in breast cancer and neuroblastoma [16, 34]. To further elucidate, whether alterations in copy numbers could be associated with de novo expression of IGF2BP1, shallow whole genome sequencing (sWGS) of the ATC samples from the initial test cohort was performed. However, copy numbers of IGF2BP1 gene locus remained unchanged in 90% (9/10) of all ATC samples from the test cohort (Supplementary Fig. S3). Remarkably, in one tumor we detected a breakpoint at the IGF2BP1 locus (sample #5).
IGF2BP1 distinguishes ATC from other thyroid carcinoma of follicular origin
To investigate the potential use of IGF2BP1 as a diagnostic marker of ATC, two independent thyroid carcinoma validation cohorts, one previously assembled in-house tissue microarray (TMA I: 20 ATC, 147 tumor samples total and 108 paired NT samples) and a commercial tissue microarray (TMA II: 6 ATC, 70 tumor samples total, and 10 unpaired NT samples) were analyzed for IGF2BP1 protein expression by immunohistochemistry (Table 3). In addition, the analysis of MAGEA3, as a second promising marker, and MYC, as a well-known upregulated gene in high-grade thyroid carcinoma [35], was considered to be included into the study. IGF2BP1 protein expression, determined by Histoscores, was observed in 70% (14/20) of analyzed ATC in TMA I and 50% (3/6) in the TMA II (Fig. 2a, b; Supplementary Fig. S4a–c). Less stringent and consistent upregulation was also observed for MAGEA3 in 35% (7/20) ATC in TMA I, whereas expression in TMA II could only be detected in 16.7% (1/6) of ATC samples. Further, MYC expression was detectable in the majority of ATC samples (TMA I: 75%, 15/20; TMA I: 33.3%, 2/6). However, MYC expression was, to a lower extend, also observed in all other types of thyroid carcinoma, excluding this oncogene as a selective marker.
Importantly, IGF2BP1 protein expression could not be observed in any other tested thyroid tissue/tumor sample, except for 5.6% (1/18) of PDTC samples with a Histoscore < 100. MAGEA3 protein expression was detectable in 0% (0/18) and MYC in 11.1% (2/18) of all PDTC samples.
To investigate IGF2BP1, but also MAGEA3 and MYC detection toward usability for diagnostic applications, we determined PPV and NPV, as well as DOR by testing with binary classification for the combination of the test cohort, TMA I and II. In sum, 75% (27/36) ATC samples were positive for IGF2BP1, whereas 5.6% (1/18) PDTC, 0% (0/132) of PTC and 0% (0/55) FTC samples revealed detectable IGF2BP1 protein expression. Diagnostic tests revealed overall PPV and NPV of ~100% and an exceptional overall DOR of 612 (95% CI: 74.6–5021) to identify ATC by IGF2BP1 detection (Fig. 3b, c). For MAGEA3 comparable PPV and NPV, but also a DOR of 411 (95% CI: 23.8–7098.7) were determined. Some detectable MYC protein expression in PDTC, PTC, and FTC led to a lower PPV of 61.9% (95% CI: 49.3–73.1) and a DOR of 30.7 (95% CI: 12.6–74.8). In conclusion, this indicated the potential use of IGF2BP1, but also MAGEA3, expression for discriminating ATC from other thyroid malignancies, including PDTC.

a IGF2BP1 expression analyzed by immunohistochemistry in representative samples derived from tissue microarray (TMA) I. HE, hematoxylin eosin staining. Scale bars, 100 µm. b Percentage view of IGF2BP1, MAGEA3 and MYC-histoscores for TMA I. Sample numbers are indicated.
This was strikingly supported by investigating IGF2BP1 protein expression in a patient-derived ATC sample with PTC content (Fig. 4c). In support of the ATC-selective expression of IGF2BP1, de novo expression was exclusively observed in the ATC area.

Plots of positive/negative predictive values (PPV/NPV) (a) and diagnostic odds ratio (DOR) (b) values for ATC diagnosis determined for indicated proteins by including all patient samples from the test cohort, tissue microarray I and II. Error bars indicate 95% confidence intervals (95% CI). c Detection of IGF2BP1 protein expression analyzed by immunohistochemistry on a patient-derived sample with ATC and PTC content.
Discussion
ATC is the most lethal malignancy of the thyroid, still lacking robust positive markers. In contrast to WDTC, the ATC is characterized by a rapid invasive growth, early metastasis and severe therapy resistance. Therefore, surgery in a limited stage is often the only potentially curative option [1, 2]. Aiming at a specific marker-driven approach to improve early ATC diagnosis, we combined a comparative RNA-seq analysis of distinct thyroid carcinomas of follicular origin and immunohistochemistry within a single methodological pipeline. This revealed robust and exclusive de novo expression of IGF2BP1, providing the first positive marker of this malignancy suitable for diagnosis on the mRNA and protein level.
Comparative transcriptome analyses and a subsequent cancer hallmark GSEA clearly dissected the molecular pathology of this distinct thyroid carcinoma from other subtypes, besides its frequently described mutations [7]. The GSEA outlines the molecular causes of histologic features for high rates of proliferation, but also an invasive behavior and in sum its high grade of dedifferentiation from its thyroid origin.
Further, the RNA-seq indicated de novo expression of testis antigens including MAGEA3 and IGF2BP1 as outstanding markers of ATC.
By sWGS we demonstrated that the de novo expression of IGF2BP1 in ATC can not be explained by gene gain, as previously observed in other tumors [16, 34]. Copy numbers of the IGF2BP1 gene locus remained unchanged in 90% (9/10) amongst ATC samples. But the here presented observation fits previous reports on copy number alterations in ATC, showing that the chromosomal region 17q21 is not frequently gained or lost [7, 9, 36]. This suggested that the de novo expression of IGF2BP1 in ATC largely results from epigenetic and/or transcriptional deregualtion.
In two independent TMAs immunohistochemistry analyses demonstrated that the majority of ATC samples were IGF2BP1 positive. This rate likely will be improved by optimizing the sensitivity of immunostaining, since IGF2BP1 mRNA expression was observed in all analyzed ATC from the initial test cohort. IGF2BP1 also performed well against MAGEA3 and MYC, for which less pronounced intensities were observed in fewer ATC and also some WDTC samples. Supporting a differential ATC diagnosis from PDTC, only 5.6% (1/18) of PDTC samples identified with a low IGF2BP1 histoscore by immunohistochemistry. Its mRNA expression was completely absent in an independent microarray analysis. Comparable findings were made in terms of MAGEA3 detection.
Accordingly, the protein detection of IGF2BP1 and MAGEA3 appeared to be suitable for the diagnosis of ATC with exceptional results from diagnostic tests, including PPV, NPV, and DOR. Finally MYC immunoreactivity could be excluded to be useful for differential diagnosis of ATC, although it was recently reported to correlate with dedifferentiation in thyroid neoplasias [35]. Still, IGF2BP1 revealed the highest consistency.
Further, IGF2BP1-positive samples identified in TMA I showed distinct WDTC/PDTC or just ATC content (Supplementary Table S1). Thus, we can largely exclude that IGF2BP1 expression in ATC is dependent on the disease origin. Nevertheless, our study stresses that IGF2BP1 immunohistochemistry has the potential to help not only defining a diagnosis but also to identify early dedifferentiation in areas of solid histoarchitecture in case of WDTC or even PDTC to prevent underestimation of tumor severity.
Notably, the histological diagnosis of ATC can be challenging due to heterogeneous histological appearance and similarity to cancers like the undifferentiated lung adenocarcinoma, lymphoma, or thyroid angiosarcoma [1, 37]. One should consider that IGF2BP1 was reported to be de novo expressed in several high-grade malignancies [14, 38]. Thus, IGF2BP1 detection could potentially lead to false-positive diagnosis in the case of a rarely observed lung cancer-derived metastasis to the thyroid [1, 39]. Nonetheless, established markers for immunostaining of ATC samples are cytokeratins and the only retained thyroid-specific transcription factor PAX8. These markers can often present with weak and focal immunoreactivity [1, 40]. Thus, positive markers will, again, help defining a diagnosis.
In view of the probably mutation-independent and sharp upregulation of IGF2BP1 in ATC, as well as its frequently reported expression in other aggressive cancers [14, 38], our study strongly suggests expediting the clinical evaluation and also improvement of IGF2BP1-directed inhibitors in cancer therapy. In this respect, the IGF2BP1-specific inhibitor BTYNB has recently been developed, which could prove promising in preclinical investigations [41].
Interestingly, we identify MAGEA proteins, including MAGEA3, which is induced in a variety of metastatic cancers and has been targeted most recently in a phase-II clinical trial [42]. However, it failed in an extensive phase-III clinical trial in immunotherapy, but remains as a promising candidate for novel targeted treatment opportunities of ATC [43].
Conclusively, our study provides new insights into the whole transcriptomic landscape of this highly aggressive neoplasia, which is distinct from other thyroid carcinomas, besides its frequently reported characteristic mutational burden, including TP53 or TERT-promoter mutations [1, 6, 7]. In consequence IGF2BP1, but also MAGEA3 are promising new candidates for fast clarification of disease severity, other than the established markers, although the here presented data requires additional confirmation by staining of whole sections and the conduction of extended studies in future.
References
Molinaro E, Romei C, Biagini A, Sabini E, Agate L, Mazzeo S, et al. Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies. Nat Rev Endocrinol. 2017;13:644–60.
Viola D, Valerio L, Molinaro E, Agate L, Bottici V, Biagini A, et al. Treatment of advanced thyroid cancer with targeted therapies: ten years of experience. Endocr Relat Cancer. 2016;23:R185–205.
Smallridge RC, Ain KB, Asa SL, Bible KC, Brierley JD, Burman KD, et al. American thyroid association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104–39.
Tiedje V, Stuschke M, Weber F, Dralle H, Moss L, Fuhrer D. Anaplastic thyroid carcinoma: review of treatment protocols. Endocr Relat Cancer. 2018;25:R153–R61.
Cabanillas ME, Zafereo M, Gunn GB, Ferrarotto R. Anaplastic thyroid carcinoma: treatment in the age of molecular targeted therapy. J Oncol Pract. 2016;12:511–8.
Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Investig. 2016;126:1052–66.
Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res. 2018;24:3059–68.
Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-Mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36:7–13.
Yoo SK, Song YS, Lee EK, Hwang J, Kim HH, Jung G, et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat Commun. 2019;10:2764.
Muller S, Glass M, Singh AK, Haase J, Bley N, Fuchs T, et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47:375–90.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Busch B, Bley N, Muller S, Glass M, Misiak D, Lederer M, et al. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016;44:3845–64.
Gutschner T, Hammerle M, Pazaitis N, Bley N, Fiskin E, Uckelmann H, et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology. 2014;59:1900–11.
Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci. 2013;70:2657–75.
Kobel M, Weidensdorfer D, Reinke C, Lederer M, Schmitt WD, Zeng K, et al. Expression of the RNA-binding protein IMP1 correlates with poor prognosis in ovarian carcinoma. Oncogene. 2007;26:7584–9.
Bell JL, Turlapati R, Liu T, Schulte JH, Huttelmaier S. IGF2BP1 harbors prognostic significance by gene gain and diverse expression in neuroblastoma. J Clin Oncol. 2015;33:1285–93.
Hirsch FR, Varella-Garcia M, Bunn PA Jr., Di Maria MV, Veve R, Bremmes RM, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21:3798–807.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30.
Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–6.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25.
Muller S, Bley N, Glass M, Busch B, Rousseau V, Misiak D, et al. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018;46:6285–303.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Marini F, Binder H. pcaExplorer: an R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinforma. 2019;20:331.
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20.
Yoo SK, Lee S, Kim SJ, Jee HG, Kim BA, Cho H, et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. 2016;12:e1006239.
Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25.
Carina V, Zito G, Pizzolanti G, Richiusa P, Criscimanna A, Rodolico V, et al. Multiple pluripotent stem cell markers in human anaplastic thyroid cancer: the putative upstream role of SOX2. Thyroid. 2013;23:829–37.
Guo Z, Hardin H, Lloyd RV. Cancer stem-like cells and thyroid cancer. Endocr Relat Cancer. 2014;21:T285–300.
Buehler D, Hardin H, Shan W, Montemayor-Garcia C, Rush PS, Asioli S, et al. Expression of epithelial-mesenchymal transition regulators SNAI2 and TWIST1 in thyroid carcinomas. Mod Pathol. 2013;26:54–61.
da Silva VL, Fonseca AF, Fonseca M, da Silva TE, Coelho AC, Kroll JE, et al. Genome-wide identification of cancer/testis genes and their association with prognosis in a pan-cancer analysis. Oncotarget. 2017;8:92966–77.
Doyle GA, Bourdeau-Heller JM, Coulthard S, Meisner LF, Ross J. Amplification in human breast cancer of a gene encoding a c-myc mRNA-binding protein. Cancer Res. 2000;60:2756–9.
Sakr HI, Chute DJ, Nasr C, Sturgis CD. cMYC expression in thyroid follicular cell-derived carcinomas: a role in thyroid tumorigenesis. Diagn Pathol. 2017;12:71.
Woodward EL, Biloglav A, Ravi N, Yang M, Ekblad L, Wennerberg J, et al. Genomic complexity and targeted genes in anaplastic thyroid cancer cell lines. Endocr Relat Cancer. 2017;24:209–20.
Kuhn E, Ragazzi M, Ciarrocchi A, Torricelli F, de Biase D, Zanetti E, et al. Angiosarcoma and anaplastic carcinoma of the thyroid are two distinct entities: a morphologic, immunohistochemical, and genetic study. Mod Pathol. 2019;32:787–98.
Degrauwe N, Suva ML, Janiszewska M, Riggi N, Stamenkovic I. IMPs: an RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016;30:2459–74.
Cao J, Yu YE, Li NN, Wu YX, Shi JN, Fang MY. Thyroid metastasis from non-small cell lung cancer. Int J Clin Exp Pathol. 2019;12:3013–21.
Bishop JA, Sharma R, Westra WH. PAX8 immunostaining of anaplastic thyroid carcinoma: a reliable means of discerning thyroid origin for undifferentiated tumors of the head and neck. Hum Pathol. 2011;42:1873–7.
Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10:818–27.
Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-Restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol. 2017;35:3322–9.
Dreno B, Thompson JF, Smithers BM, Santinami M, Jouary T, Gutzmer R, et al. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19:916–29.
Acknowledgements
This work was funded by the German Cancer Aid (DKH 70112631; to KL, CW, and SH) and the Deutsche Forschungsgemeinschaft (DFG RTG1591; to SH). Open access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Haase, J., Misiak, D., Bauer, M. et al. IGF2BP1 is the first positive marker for anaplastic thyroid carcinoma diagnosis. Mod Pathol 34, 32–41 (2021). https://doi.org/10.1038/s41379-020-0630-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0630-0
This article is cited by
-
PFKFB3 facilitates cell proliferation and migration in anaplastic thyroid carcinoma via the WNT/β‐catenin signaling pathway
Endocrine (2024)
-
Co-inhibition of glutaminolysis and one-carbon metabolism promotes ROS accumulation leading to enhancement of chemotherapeutic efficacy in anaplastic thyroid cancer
Cell Death & Disease (2023)
-
High-Grade Non-Anaplastic Thyroid Carcinomas of Follicular Cell Origin: A Review of Poorly Differentiated and High-Grade Differentiated Carcinomas
Endocrine Pathology (2023)
-
ACLY inhibitors induce apoptosis and potentiate cytotoxic effects of sorafenib in thyroid cancer cells
Endocrine (2022)
-
The biological function of IGF2BPs and their role in tumorigenesis
Investigational New Drugs (2021)
