
Abstract
The publication of the “Pan-Cancer Atlas” by the Pan-Cancer Analysis of Whole Genomes Consortium, a partnership formed by The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC), provides a wonderful opportunity to reflect on where we stand in our understanding of the genetics of pancreatic cancer, as well as on the opportunities to translate this understanding to patient care. From germline variants that predispose to the development of pancreatic cancer, to somatic mutations that are therapeutically targetable, genetics is now providing hope, where there once was no hope, for those diagnosed with pancreatic cancer.
Similar content being viewed by others

Introduction
Little more than a decade has passed since the exomes of a series of pancreatic cancers, by which we mean ductal adenocarcinomas, were first sequenced [1]. This decade saw an unimaginable explosion in knowledge of the genetic drivers of all types of cancer, culminating in a tour de force series of publications describing integrated analyses of 2658 cancers across 38 cancer types by the Pan-Cancer Analysis of Whole Genomes Consortium of The Cancer Genome Atlas and the International Cancer Genome Consortium [2,3,4,5,6,7]. These publications provided unprecedented insight into the germline and somatic drivers of cancer—including intragenic mutations, mutational signatures, amplifications, larger structural variants, and distinct patterns of gene expression. The publications went beyond “the usual suspects,” and described the importance of non-coding changes, chromothripsis, and retrotransposons.
The Pan-Cancer Analysis of Whole Genomes Consortium reported that the average cancer has four or five driver gene mutations, that chromothripsis is a frequent and early event occurring in 20% of cancers, that deleterious germline variants are relatively common, and that these germline variants, in turn, affect the subsequent patterns of somatic mutations that occur in these cancers [2,3,4,5,6,7]. Addressing evolutionary processes that drive neoplasia, the consortium also bettered our understanding of the “clonal sweeps,” which occur when a driver gene mutation gives a neoplastic cell a growth advantage [4]. Driver gene mutations tend to occur early in disease progression, while whole-genome duplications are late events [4].
The genetic changes driving ductal adenocarcinoma of the pancreas mirror those described in the Pan-Cancer Atlas by the Pan-Cancer Analysis of Whole Genomes Consortium [8]. Among them, two findings have potential to improve early detection and affect treatment options. First, a number of germline genetic variants that predispose to pancreatic cancer have been discovered. These can now be used to quantify risk, and several of these changes also create potential targets for therapeutic intervention [9]. Second, some of the somatic genetic changes are also therapeutically targetable, and, progress has even been made targeting the famously “undruggable” KRAS pathway [10,11,12,13,14,15,16].
Germline variants
One of the most consistent and remarkable findings across the sequencing of all cancer types is that a significant fraction (~ 10%) of all cancers arise in individuals with a germline variant that is associated with an increased risk of cancer [5]. Pancreatic cancer is no exception, and heritability studies have indicated that >20% of pancreatic cancer is due to inherited sequence variation [8, 17,18,19]. Looking back, this should not have been surprising. It has been known for decades that pancreatic cancer runs in some families, and that having multiple family members with pancreatic cancer increases an individual’s risk of pancreatic cancer [20,21,22,23,24,25]. Prospective studies of families in which there had been a pancreatic cancer have allowed epidemiologists to develop risk prediction models, such as PancPRO, that can be used to estimate a person’s risk of developing pancreatic cancer based on their family’s cancer history [26, 27]. Prospective studies have also shown that relatives of patients with familial pancreatic cancer (defined as two first-degree relatives in a kindred with pancreatic cancer), also have an increased risk of death from breast, ovarian, and bile duct cancers [20]. Based on this strong epidemiologic background, “candidate gene approaches,” the targeted sequencing of known cancer genes, were then undertaken and revealed that germline variants in BRCA1, BRCA2, CDKN2A, PRSS1, STK11 and in the DNA mismatch repair genes can all increase the risk of pancreatic cancer [28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. Whole-exome and whole-genome sequencing followed and confirmed the importance of these genes, while also revealing that germline variants in PALB2, ATM, and other lower-prevalence genes increase the risk of developing pancreatic cancer (Table 1) [5, 8, 17, 60,61,62]. Importantly, as discussed below in the sections on specific genes, knowledge of each of these pancreatic cancer susceptibility genes now allows us to quantify cancer risk, and some of these variants create therapeutic targets [29,30,31, 41,42,43, 59, 63,64,65,66,67,68,69].
Fanconi anemia pathway genes
Fanconi anemia, first described in 1927 by the Swiss pediatrician Guido Fanconi, is an autosomal recessive disease characterized by congenital abnormalities, defective hematopoiesis, and an increased risk of developing leukemia and solid malignancies [70]. The Fanconi anemia pathway functions to repair DNA damage, especially DNA interstrand crosslinks [70]. Deleterious germline variants in genes coding for members of the Fanconi anemia pathway, including BRCA1, BRCA2, and PALB2, increase the risk of developing cancer, including cancers of the breast, ovary, and, in some studies, prostate [33, 34, 66]. These germline variants also increase the risk of pancreatic cancer. Indeed, genes coding for members of the Fanconi anemia pathway account for the largest fraction (10–20%) of the known pancreatic cancer susceptibility genes [35, 39, 71]. The demonstration of a second somatic hit, one inactivating the wild-type allele of the gene having the germline variant, further helped to establish a causative role for these genes in the development of pancreatic cancer [36, 59, 72, 73].
The identification of Fanconi anemia pathway genes as pancreatic cancer susceptibility genes allows one to now quantify risk. The risk of pancreatic cancer is elevated 2.4–6.2-fold in individuals who carry a deleterious germline BRCA2 variant [31, 35, 66], and these BRCA2 pathogenic variants occur in 2–7% of pancreatic cancer patients [31, 42, 43, 64, 65, 74,75,76]. The increased risk of pancreatic cancer is slightly lower (2.58-fold (95% CI, 1.54–4.05)) in individuals who carry a deleterious germline BRCA1 variant, and these pathogenic variants in BRCA1 occur in ~ 1% of patients with pancreatic cancer [31, 42, 43, 64, 75]. The risk of pancreatic cancer in carriers of pathogenic PALB2 variants is not well established, with estimates ranging from 2.37 (95% CI, 1.24–4.50) in families identified through a breast cancer patient to 14.82 (95% CI 8.12–26.2) in pancreatic cancer patients undergoing genetic testing [42, 60, 75, 76]. This uncertainty is due to the relative rarity of pathogenic PALB2 variants which occur in ~ 1% of all pancreatic cancer patients and up to 3% of familial pancreatic cancer patients [42, 60, 75, 76]. Finally, there have been isolated reports suggesting that other genes coding for members of this pathway, including FANCA, FANCC, FANCG, and FANCM, may contribute to the development of pancreatic cancer [43, 54, 77, 78].
Deleterious germline variants in BRCA1 and BRCA2 are more common in individuals of Ashkenazi Jewish heritage, helping to explain why pancreatic cancer is more common in this group [28, 36, 37, 59, 79]. Remarkably, deleterious germline variants in genes coding for members of the Fanconi anemia pathway have been reported in ~ 5% of pancreatic cancer patients without a family history of cancer [18, 29, 31, 44, 59, 67, 68, 75, 80, 81]. Because family history cannot be used to rule out a germline change, the National Comprehensive Cancer Network (NCCN) now recommends that all patients with pancreatic cancer undergo germline testing [82, 83].
Although the breast cancers arising in individuals with a deleterious BRCA germline variant are reported to often be triple-negative and to harbor high numbers of tumor-infiltrating lymphocytes, the histopathologic features of the pancreatic cancers that arise in individuals with a deleterious BRCA germline variant do not appear to be distinctive [84, 85]. Most are typical ductal adenocarcinomas.
As nicely described in the “Pan-Cancer Atlas,” deleterious germline variants in BRCA1, BRCA2, and PALB2 impact the subsequent somatic mutations that occur in the cancers [5, 86,87,88]. These deleterious germline variants are associated with increased numbers of small somatic deletions and somatic tandem duplications in the cancers [5]. In addition, these cancers harbor structural variants characteristic of “cycles of template insertions.” These structural variants occur when small fragments of DNA copied from across the genome are joined together and then inserted into a derivative chromosome [5]. Pancreatic cancers from patients carrying one of these deleterious germline variants and having somatic inactivation of the wild-type allele, typically have an “unstable” genetic phenotype, with multiple chromosomal rearrangements, and are remarkably sensitive to treatment with DNA cross-linking agents, such as platinum-based drugs, and poly (adenosine diphosphate-ribose) polymerase (PARP) inhibitors [12, 32, 88,89,90]. For example, when compared with patients lacking a germline variant, complete pathologic responses are more likely in BRCA2 carriers following neoadjuvant FOLFIRINOX therapy [91].
Unfortunately, resistant clones can emerge following these targeted therapies. Sequencing has identified some resistance mechanisms. It is likely that, among the billions of cancer cells in a tumor, there exist a few having a secondary mutation in the BRCA2 gene, one that restores the gene back into a normal reading frame and re-establishes coding to synthesize a full-length protein [32, 92]. This returns normal (or near normal) function to the protein product, and under selective pressure, this small preexisting clone emerges as a dominant clone resistant to therapy [32, 92].
ATM
The ATM gene encodes a phosphoinositide 3-kinase (PI3K) that functions in the PI3K kinase (PIKK) pathway [93, 94]. Through this pathway the protein product of the ATM gene coordinates the repair of double-stand DNA breaks [93, 94]. When a double-strand DNA break occurs, the ATM protein phosphorylates and thereby activates, a number of downstream proteins, which in turn result in DNA repair, cell cycle arrest, and in some cells apoptosis [93, 94]. Pathogenic germline ATM variants, including whole gene deletions, have been reported in ~ 3% of patients with familial pancreatic cancer [29, 62, 63, 68, 75, 76]. These pathogenic germline ATM variants are also present in ~ 2% of unselected patients with pancreatic cancer, indicating that, just as is true for patients with germline pathogenic BRCA gene variants, not all patients with a deleterious germline ATM variant have a family history of cancer [18, 29, 43, 62, 68, 76, 80, 94]. Germline pathogenic variants in ATM are estimated to increase the risk of pancreatic cancer ~ 5.71-fold (95% CI, 4.38–7.33) [31].
At the light microscope level, there is a hint that patients with a deleterious germline ATM variant are slightly more likely to develop cancers with a colloid morphology, but the majority of pancreatic cancers that arise in patients with a germline ATM mutation are standard infiltrating ductal adenocarcinomas [95]. Pathogenic germline ATM mutations have also been described in 1.6% of patients with intraductal papillary mucinous neoplasms (IPMNs) who underwent surgical resection, suggesting that the IPMN precursor pathway may be taken by some of these pancreatic cancers [96]. Although this fits with the observation of an increased prevalence of colloid carcinomas in patients with deleterious germline ATM variants, as almost all colloid carcinomas of the pancreas arise in association with intestinal-type IPMNs, the complete progression from germline ATM variant, to intestinal-type IPMN to invasive colloid carcinoma has not been established [97].
Just as was true for germline pathogenic BRCA variants, germline pathogenic ATM variants may confer specific therapeutic sensitivities. ATM inactivation is predicted to inhibit the repair of double-stand DNA breaks, thereby increasing genomic instability and increasing sensitivity to radiation therapy and to platinum-containing drugs, as these therapies induce double-strand breaks [93, 94, 98]. Although ATM mutated cells were reported to be sensitive to PARP inhibitors in vitro, studies to date have not demonstrated these drugs to be effective in genetic models or in patients with ATM mutations [99, 100]. In addition, a number of compounds are now available to target the ATR-checkpoint kinase 1 (Chk1) pathway, including compounds that target ATR and Chk1 (a serine/threonine kinase functioning downstream of ATR), and pancreatic cancers with loss of ATM-mediated DNA repair may show increased sensitivity to one of these [93, 94].
CDKN2A
Germline mutations in the CDKN2A gene cause the familial atypical multiple mole melanoma (FAMMM) syndrome [101]. FAMMM, as the name suggests, is characterized by multiple melanocytic nevi, some of which are dysplastic, and an increased risk of melanoma (Fig. 1). In addition, individuals who inherit a germline CDKN2A gene mutation have a 17% risk of developing pancreatic cancer by the age of 75 [43, 45, 46, 102,103,104,105]. Indeed, deleterious germline variants in the CDKN2A gene are found in about ~ 1% of pancreatic cancer patients; the risk of pancreatic cancer has been estimated to be 12–46-fold greater in carriers of one of these variants than in the general population [18, 29, 31, 42, 54, 68, 80, 103, 106]. The risk of pancreatic cancer may be even higher in carriers of deleterious CDKN2A variants who smoke cigarettes [49]. Multifocal pancreatic cancers have been reported in some patients with pathogenic germline CDKN2A variants [102]. Not everyone with a deleterious germline variant has a personal or family history of melanoma, adding support to the NCCN guidelines recommending universal germline testing in all patients with pancreatic cancer [45, 50, 75].

Note the numerous melanocytic nevi. (Courtesy of Elise Ng, MD).
The pathology of the pancreatic cancers that develop in these patients does not appear to be specific, however, there have been reports of cancers with prominent squamous features [107]. Since melanomas can be detected with a simple skin exam, the identification of a germline CDKN2A variant does allow one to go back to carriers in an affected family and detect early, curable melanomas [50].
Lynch syndrome
Lynch syndrome, also known as hereditary non-polyposis colorectal cancer, is characterized by an increased risk of developing numerous cancers especially colorectal, endometrial, brain, ovary, gastric, small intestine, urothelial, and pancreatic cancer [56, 108, 109]. Germline variants in genes coding for proteins that function in DNA mismatch repair, e.g., MLH1, MSH2, MSH6, and PMS2 cause Lynch syndrome [18, 29, 32, 54, 55, 58, 64, 68, 75, 76, 80, 110]. Pathogenic germline variants in the genes associated with Lynch syndrome occur in <1% of pancreatic cancer patients, and individuals with Lynch syndrome have a 6–8.6-fold increased risk of developing pancreatic cancer [31, 56, 58, 111]. As is true for the other germline changes associated with an increased risk of pancreatic cancer, germline variants in DNA mismatch repair genes have been reported in unselected individuals with pancreatic cancer [29, 68, 80]. The pancreatic cancers that arise in patients with the Lynch syndrome often have a distinct “medullary” histology (Fig. 2), although acinar cell and other carcinomas can also occur, and not all pancreatic cancers with a medullary appearance have an alteration in a DNA mismatch repair gene [112,113,114,115]. Of note, the vastly increased numbers of somatic mutations that accumulate in these cancers result in the expression of altered proteins that can serve as “neoantigens” to the immune system [86]. Therefore, these cancers are often accompanied by a brisk immune response, which includes activated CD8-positive T cells [86]. All of this, of course, has profound therapeutic implications, as microsatellite-unstable (MSI-high) cancers often respond well to treatment with immune checkpoint inhibitors [11, 12, 15, 116, 117].

These cancers are often microsatellite unstable. Note the pushing boarder and syncytial growth pattern (a), and the loss of expression of the mismatch repair protein MLH1 (b). (a hematoxylin and eosin stain; b immunolabeling for MLH1).
Peutz–Jeghers
The Peutz–Jeghers syndrome, described as early as 1895, was first characterized by Jan Peutz in 1921 [118]. The full syndrome was then described in 1949 by Harold Jeghers [118]. Peutz–Jeghers is characterized by melanocytic macules involving the lips and buccal mucosa (Fig. 3a), distinctive hamartomatous polyps of the gastrointestinal tract (Fig. 3b), and a dramatically increased risk of cancer [119]. Peutz–Jeghers syndrome is caused by germline pathogenic variants in the STK11 gene (also known as LKB1) on chromosome 19p. LKB1 codes for a serine threonine/kinase that plays a role in energy metabolism and cell polarity. Individuals having the Peutz–Jeghers syndrome have an increased risk of developing cancer of the lung, breast, ovary, and pancreas [52, 53, 120]. The risk of pancreatic cancer is 75–135 times greater than that of the general population [52, 53, 120]. Korsse et al. estimated that patients with Peutz–Jeghers have a 26% chance of developing pancreatic cancer by the age of 70 [53]. Although the risk in each individual is greatly increased, the syndrome itself is relatively rare, and it accounts for <1% of all pancreatic cancers [52, 53, 120]. The pathology of the Peutz–Jeghers syndrome is well-described. In addition to the characteristic hamartomatous polyps of the gastrointestinal tract, some patients develop IPMNs of the pancreas [121]. The invasive cancers of the pancreas are usually conventional ductal adenocarcinomas.

Note the melanocytic macules on the lips and buccal mucosa in a, and the classic hamartomatous polyp in b. (a courtesy of Sewon Kang, MD. b is a hematoxylin and eosin stain).
Hereditary pancreatitis
Hereditary pancreatitis is characterized by the early onset of recurrent bouts of severe pancreatitis. It is caused by germline pathogenic variants in PRSS1, SPINK1, and other genes [51]. Individuals with hereditary pancreatitis have an extraordinarily high risk of developing pancreatic cancer. Their risk is elevated 50–60-fold, and some series report that the risk approaches 40% by the age of 70 [122,123,124,125,126,127]. The risk of developing pancreatic cancer is especially high in individuals with hereditary pancreatitis who also smoke, and the cancers that arise in these smokers develop years earlier than those that arise in non-smokers [123, 124]. The pancreas pathology associated with hereditary pancreatitis has been described and, in addition to the changes of chronic pancreatitis, includes pancreatic intraepithelial neoplasia (PanIN) lesions [128, 129]. In the report by Rebours et al., these included high-grade PanIN lesions, while Singhi et al. reported only low-grade PanIN (PanIN-1A) [128, 129]. Unlike the other familial pancreatic cancer syndromes, the risk of cancer in individuals with hereditary pancreatitis is confined to the pancreas. Since increased risk of cancer is confined to pancreas, and the repeated bouts of pancreatitis badly damage the pancreas, some patients have elected to undergo prophylactic pancreatectomy. An excess of pathogenic variants in the pancreatitis susceptibility gene CPA1 and the related gene, CPB1, have been observed in patients with pancreatic cancer compared with controls, suggesting that these variants can predispose to pancreatic cancer without the clinical syndrome of pancreatitis [17, 130].
Other pancreatic cancer susceptibility genes
Germline variants in a number of other genes are associated with an increased risk of pancreatic cancer, but, judging from these reports, pathogenic variants in these genes are less prevalent as compared with those described above. These genes include APC, BARD1, BUB1, BUB3, BAP1, CHEK2, FAM175A, FAN1, NEK1, POLQ, RABL3, RAD50, RHNO1, SKIL, SMG1, TP53, TUBB5, and others [18, 29,30,31, 43, 59, 61, 68, 75, 80, 131,132,133,134]. Several of these genes are of note. Hu et al. estimated that germline TP53 mutations increase the risk of pancreatic cancer 6.7-fold [31]. Although APC is included on this list, one has to be careful interpreting the cancers that arise in individuals with germline APC mutations as these patients have an increased risk of duodenal and ampullary cancers, and both of these cancers can invade into the pancreas and mimic pancreatic cancer [135]. SKIL and TUBB5 are on this list because of the remarkable report of an infant with germline SKIL and TUBB5 variants who developed a large IPMN [61, 136]. Of note, TUBB5 is in the robo-slit pathway, and somatic mutations in genes coding for members of this pathway have been described in sporadic pancreatic cancers [136]. Certainly, based on the findings of whole-genome sequencing, there are other, yet to be discovered, pancreatic cancer susceptibility genes [17].
Moderate/low-risk pancreatic cancer loci
While we have focused on the rare deleterious germline variants having high penetrance, it should be noted that there are also many more common, lower-penetrance genetic variants that impact the risk of pancreatic cancer ever so slightly. These have been identified by genome-wide association studies (GWAS) in which the prevalence of these genetic variants in large populations of individuals with the phenotype (pancreatic cancer) were compared with the prevalence of these variants in individuals free of pancreatic cancer. A number of such GWAS have been performed, revealing genetic loci associated with a slightly increased pancreatic cancer risk [137]. In European populations, these loci include 1q32.1 (NR5A2), 1p36.33 (NOC2L), 2p13.3 (ETAA1), 3q29 (TP63), 5p15.33 (CLPTM1L, TERT), 7p14.1 (INHBA), 8q21.11 (HNF4G) 8q24.21 (MYC), 9q34.2 (ABO), 13q12.2 (PDX1), 13q22.1 (KLF5), 16q23.1 (BCAR1), 17q12 (HNF1B) 17q25.1 (LINC00673), 18q21.32 (GRP), and 22q12.1 (ZNRF) [137,138,139,140]. Additional GWAS studies have been conducted in both in Chinese and Japanese populations, with five loci 21q21.3, 5p13.1, 21q22.3, 22q13.32, and 10q26.11 reported to be associated with pancreatic cancer in the Chinese population, and 6p25.3, 12p11.21, and 7q36.2 in the Japanese [141, 142]. The ABO locus is particularly interesting as non-O blood type increases risk [137, 143]. The mechanisms driving this association are unclear.
Screening in familial syndromes
In addition to guiding precision therapy for patients known to have pancreatic cancer, an understanding of the genetic drivers of the familial aggregation of pancreatic cancer has implications for early detection. As genes are identified, the risk each variant carriers can be quantified, and used to prioritize people for screening with the goal of detecting early, curable pancreatic and extra-pancreatic neoplasms [104]. Individuals with both a family history and a pathogenic germline variant may benefit most, as they have been found to be at the highest risk of pancreatic cancer in screening studies, compared with individuals with a family history by no identifiable mutation [144]. Several screening approaches have been taken, but most rely on endoscopic ultrasound (EUS) combined with magnetic resonance imaging or computed tomography (CT) scanning [145,146,147]. Preliminary results from EUS-based screening trials have demonstrated that asymptomatic precursor lesions and, rarely, early curable cancers can be detected, and SEER data hint that more early stage cancers are now being detected nationally [145,146,147,148,149]. Consensus guidelines currently recommend pancreatic surveillance for individuals at the highest estimated risk, it has, however, yet to be demonstrated that screening for pancreatic cancer saves lives [150]. Because of the role of environment and chance in the development of cancer, accurately predicting an individual’s risk of developing cancer remains elusive [150, 151]. As discussed later, there is also a risk that screening will detect harmless lesions and, in so doing, will lead to patient harm through unnecessary interventions [152, 153].
Precancers
A number of morphologically distinct precancerous lesions have been identified and characterized in the pancreas. These range from microscopic PanIN lesions, to larger macroscopic lesions such as IPMNs and mucinous cystic neoplasms (MCNs) [135, 154, 155]. Rather than review each lesion, here we will focus on key recent advances. We will then expand on the clinical implications of these advances, with an emphasis on clinical cyst fluid testing.
The four most common cystic neoplasms of the pancreas
There are four main cystic neoplasms of the pancreas [135, 155]. In addition to IPMNs and MCNs, these include solid pseudopapillary neoplasms (SPNs) and serous cystic neoplasms (SCNs). Of these four, IPMNS and MCNs are precancerous lesions; some IPMNs and some MCNs progress over time to invasive carcinoma. By contrast, all SPNs are low-grade malignancies, and essentially all SCNs are benign [135, 155]. Distinguishing among these cystic neoplasms is therefore important.
IPMNs are of particular note, for they are detectable using currently available imaging and are now the most commonly surgically resected cystic neoplasm of the pancreas [156]. IPMNs therefore probably represent the best opportunity to detect and treat precancers before they progress to an incurable invasive cancer. The challenge is that IPMNs are remarkably common, and most do not progress to invasive cancer, and there therefore is a real risk of causing harm by over treating those lesions that would remain harmless [157].
With the goal of improving the preoperative classification of these neoplasms, the exomes of a series of well-characterized cystic neoplasms of the pancreas were sequenced a decade ago [158, 159]. IPMNs were found to have somatic mutations in GNAS and RNF43, as well as mutations in three of the genes frequently targeted in invasive ductal adenocarcinomas of the pancreas (KRAS, TP53, and CDKN2A) [158,159,160,161]. By contrast, it was recently shown that the morphologically distinct intraductal neoplasm, the intraductal oncocytic papillary neoplasm (IOPN), does not harbor these changes and, instead, IOPNs have distinctive translocations targeting the PRKACA or PRKACB genes (Fig. 4) [162, 163]. Importantly, as shown in Table 2 and illustrated in Fig. 5, these patterns of mutations differ significantly from those seen other cystic neoplasms in the pancreas [158,159,160,161]. SPNs have mutations in the gene coding for beta-catenin (CTNNB1), SCNs have somatic mutations in VHL, and the mutational spectrum seen in MCNs closely matches that of IPMNs, except GNAS mutations are rare in MCNs [158,159,160,161, 164]. Of note, mutations and allelic losses in IPMNS and in MCNs appear to accumulate with the degree of dysplasia, such that higher degrees of aneuploidy are seen in lesions with high-grade dysplasia than in lesions with low-grade dysplasia [158, 165, 166].

The fusion has resulted in loss of the 5′ probe (red) for PRKACA in this fluorescent in situ hybridization. (3′PRKACA[19p13.12](Green)/5′PRKACA[19p13.12](Red)) (Courtesy of Michael Torbenson).

VEGFA vascular endothelial growth factor-A.
Importantly, neoplastic cells shed mutant DNA into cyst fluid, suggesting that sequencing of cyst fluid could be used clinically to determine cyst type and potentially even the grade of dysplasia [160,161,162, 167, 168]. This indeed appears to be the case. Cysts can be aspirated at the time of EUS, and genetic analysis of cyst fluid can help define the cyst type [160, 161, 167, 168]. The sensitivity and specificity of these analyses can be improved when genetic findings are combined together with protein biomarkers and with clinical findings [167, 168]. For example, the cyst fluid of SPNs harbor a CTNNB1 gene mutation, and SPNs almost always occur in young women [164, 167, 168]. By contrast, high levels of vascular endothelial growth factor-A are present in the cyst fluid of SCNs, SCNs often harbor VHL gene mutations, and SCNs have a characteristic sponge-like appearance on CT scanning [167,168,169]. These findings provide great hope for improved preoperative diagnoses. As discussed in the next section, however, the pathology of cystic neoplasms presents real challenges to clinical cyst testing.
Challenges in the genetic analysis of cyst fluid
The first challenge is that clinical management of cystic neoplasms is not simply driven by cyst type. For example, IPMNs are common, and while some will progress to invasive cancer, most do not [149, 170, 171]. The goal is to resect those IPMNs that are likely to progress, and to observe those that are unlikely to progress [172]. Because the grade of dysplasia is an indicator of the risk of progression, preoperative tools to determine the degree of dysplasia are needed. This turns out to be much harder than determining the cyst type. Although some somatic mutations, such as inactivating mutations in the TP53 gene, are more common in IPMNs with high-grade dysplasia, no single specific genetic change correlates perfectly with dysplasia [160, 161, 167, 168]. Instead, the accumulation of multiple genetic changes, including the development of significant aneuploidy, appears to be most useful [165, 173, 174]. Ultimately, it is likely that a combination of clinical features (imaging, symptoms) and cyst fluid biomarkers (intragenic mutations, aneuploidy, and protein) will be required to assess risk [172].
A second significant hurdle in clinical cyst fluid analysis is that most cystic neoplasms do not form a single locule [135, 155]. Instead, they form multiple locules, and the degree of dysplasia in one locule often does not match the degree of dysplasia in the other locules (Fig. 6) [175,176,177]. The implications of this are profound. It means that, even with a perfect test, the findings in the fluid aspirated from one locule of a multi-locular cystic neoplasm may not reflect the pathology in the other locules.

Note the dramatically different degrees of dysplasia in the three locules. (Hematoxylin and eosin stain).
Third, in addition to heterogeneity among the different locules, there appears to also be significant genetic heterogeneity within individual locules [174, 175, 177]. This has been demonstrated with sequencing and has been visualized with mutation-specific in situ hybridization probes. For example, single-cell sequencing of ten IPMNs revealed that different mutations in the same driver gene frequently occur in the same IPMN [177]. Different neoplastic clones within a single IPMN will grow at different rates, and new clones can emerge and out compete existing clones [174, 177]. While this heterogeneity can complicate cyst fluid analysis, it can also provide insight into the progression of a neoplasm as there is often a loss of heterogeneity as a single-dominant high-grade clone emerges and outgrows the clones with lower grade dysplasia [174, 175, 177].
Further complicating cyst fluid analysis, there can be heterogeneity within the broader pancreas. Perhaps the best example of this is the observation that invasive cancers that arise adjacent to a well-defined IPMN may be genetically unrelated to that IPMN [174, 176]. Clinically important neoplasms can arise from areas of the pancreas unrelated to the cyst itself, and the likelihood that two lesions are genetically distinct grows with greater anatomic separation. Thus, even if we characterize an IPMN perfectly, we are not characterizing the entire pancreas in its totality.
We have saved the pièce de résistance for the last part of this discussion, as it is now clear that neoplastic cells can move within the duct system [178, 179]. This had been observed earlier with invasive carcinoma. In a process called cancerization of the ducts, invasive carcinoma within the stroma can grow into and along the internal surface of a preexisting pancreatic duct [178]. Similarly, the spread of neoplastic cells of non-invasive IPMNs within the duct system has been demonstrated through the genetic analyses of IPMNs that recurred after surgery [179, 180]. Surgically resected non-invasive IPMNs with negative surgical margins can recur in the remnant pancreas, and in some, but not all, instances the recurrent IPMN in the remnant pancreas harbors identical mutations as those present in the originally resected IPMN [179, 180]. The movement of neoplastic cells within the duct system means that the neoplastic cells, even of a well-delineated IPMN, are not always limited to the grossly visible lesion. The remainder of the grossly uninvolved pancreas remains at risk owing to “seeding” from the original IPMN. Indeed, the movement of neoplastic cells within the duct system of the pancreas partially explains why the entire pancreas appears to be at risk when a patient has an IPMN [181,182,183]. This is especially true for main-duct IPMNs [182]. In one study of over a thousand patients who were followed after the surgical resection of an IPMN, 14.4% developed a recurrence [183]. Clinically, this suggests that patients with an IPMN need to be followed carefully, even if that IPMN is resected.
Thus, although great progress has been made, there are still significant unanswered questions that fundamentally impact the management of precancerous lesions. Perhaps the big unanswered question is—why is the entire pancreas at risk in patients with an IPMN [102, 179]?
Invasive cancers
Recent advances
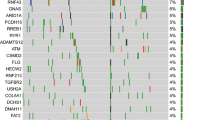
The genes targeted with somatic mutations in pancreatic cancer have been well-characterized (Table 3). These include the now famous “four mountains” (present in >50% of the cancers), KRAS, TP53, CDKN2A, and SMAD4, as well as a long list of genes targeted less frequently [1, 8, 88]. Alterations in some of these genes, as illustrated in Fig. 7, can be used to aid diagnoses.

The patient’s primary pancreatic cancer showed loss of SMAD4 as did the adenocarcinoma involving both ovaries. This pattern supports the diagnosis that the adenocarcinoma in the ovaries was a metastasis from the patient’s pancreatic primary. (Immunolabeling for SMAD4).
The whole-exome and whole-genome sequencing efforts of the last decade have created a more complete list of targeted genes, and a more complex picture of pancreatic cancer is now emerging [1, 8, 88]. This picture includes intragenic mutations, homozygous deletions, amplifications, dramatic shattering of chromosomes (chromothripsis), the activation of transposons, and intragenic mutations coupled with copy number changes of the same gene [3, 184].
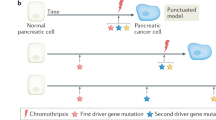
Chromothripsis is the dramatic sudden shattering of a chromosome, or of part of a chromosome, followed by massive genomic rearrangements as the bits of the shattered pieces reassemble [3, 184]. Chromothripsis can result in the amplification of oncogenes, and the inactivation of tumor suppressor genes. Chromothripsis is pervasive across many cancer types, and occurs in almost two-thirds of pancreatic cancers [3, 184]. The suddenness of chromothripsis makes it tempting to speculate that chromothriptic events cause the apparent sudden aggressiveness that can be seen clinically in some patients with pancreatic cancer [184]. However, the magnitude of the overall contribution of chromothripsis to the pathogenesis of pancreatic cancer is yet to be determined, as many chromothriptic events in pancreatic cancer do not target well-defined oncogenes or tumor suppressor genes [3, 184, 185]. New technologies could help define the impact of chromothripsis. Currently, the detection of chromothripsis requires deep sequencing. The development of novel methods to detect chromothripsis in situ would allow investigators to determine when chromothripsis occurs in neoplastic progression, and then perhaps, correlate the timing with the patient’s clinical status. For example, if chromothripsis occurs as a primary cancer explosively metastasizes, this temporal association would suggest that the event is clinically important.
The last decade was also marked by advances in our understanding of the role played by mobile DNA elements in the development of pancreatic cancer [7, 186, 187]. The open reading frame 1 protein, ORF1p, is encoded by a long interspersed element-1 (LINE-1) retrotransposon, and novel LINE-1 insertions and ORF1p overexpression have been documented in pancreatic cancer [186, 187]. These somatically acquired LINE-1 insertions could, plausibly, influence gene expression, and Ardeljan et al. recently suggested that these insertions may create a unique vulnerability that could be exploited therapeutically [188]. The complexity of mobile DNA elements makes them hard to study; emerging technologies, however, offer promise to clarify the mechanisms and roles that these elements have in disease [189, 190].
For decades, activating point mutations in the KRAS oncogene were known to be important drivers of pancreatic cancer [191]. More recently, it has been shown that copy number changes in the KRAS gene are also important [14, 191, 192]. The intragenic mutations that activate the KRAS protein have a number of downstream effects, including increasing glucose uptake, a shift to aerobic glycolysis, and ultimately the promotion of proliferation, invasion, and cell survival [14]. Recently, single-cell analyses supported suggestions that somatic copy number changes in the KRAS gene also promote tumorigenesis [192]. For example, Chan-Seng-Yue et al. described amplifications of mutant KRAS, relative to the wild-type allele, which create allelic imbalances favoring the mutant allele [192]. Genomic duplication appears to drive these imbalances in mutant KRAS copy number, and it has been reported that primary pancreatic cancers with KRAS copy number events are more likely to be of the more aggressive basal subtype, often with squamous differentiation (see below) [192]. Studies of recurrent PDAC find evidence of convergent evolution on the KRAS pathway with mechanisms including multiple routes to copy number gain of mutant KRAS, multiple different KRAS mutations in different metastatic clones and mutations in other components of the KRAS/MAPK/ERK pathway [193].
Copy number changes, such as those seen with KRAS, are a component of the larger and nearly ubiquitous (except for the cancers with MSI) phenomenon of aneuploidy in pancreatic cancer. The FAM190A gene occupies a hotspot of genomic homozygous deletions, producing deletions of internal exons, and, in turn, creating in-frame deletions of the protein-coding sequence [194, 195]. In addition, more than a third of pancreatic cancers have similar in-frame deletions within FAM190A transcripts, which are manifest without an identifiable genomic mutation. Functional studies of genetically engineered cells and protein localization techniques show that Fam190a normally functions in mitosis to ensure the creation of mononuclear daughter cells after the abscission (separation) phase of mitotic division, thus avoiding a simple cause of polyploidy [196]. The common Fam190a abnormalities thus might spur chromosomal instability during tumorigenesis and, specifically, could help explain the common finding of tetraploidy in pancreatic cancers [192].
A great deal of effort in the last decade has gone into defining “molecular” (gene expression) subtypes of pancreatic cancer with mixed results [197,198,199,200]. It is now clear that contaminating non-neoplastic cells often confound these studies, and subtypes with “exocrine differentiation” almost certainly simply reflect the contamination of cancers with non-neoplastic acinar cells [8]. When non-neoplastic contamination is taken into account, two broad subtypes do consistently emerge [8, 199]. Cancers of the “classical” subtype often have GATA6 gene amplification, and a classic ductal differentiation. The “basal-like” subtype, as noted above, are more likely to have KRAS gene copy number changes, and a worse prognosis [8, 192, 199, 201]. While the subtyping of pancreatic cancer based on patterns of RNA expression has proven prognostically valuable, it should be noted that expression patterns in a cancer are not fixed [192]. They can change over time, and can differ in different areas of a tumor [192]. We believe that the subtyping of pancreatic cancer will be most impactful when RNA and protein expression patterns are correlated with tumor morphology.
The clinical application of molecular pathology to pancreatic cancer is becoming increasingly challenging. Low tumor cellularity (<10%) puts detection of mutations just around the limit of detection of most next generation sequencing assays (commonly ~ 5%). This is becoming even more difficult in the era of neoadjuvant therapy, which if successful, can result in extremely low neoplastic cellularity in the resected tumor.
Early detection
Most patients with pancreatic cancer are not diagnosed until their disease is advanced [202]. Not surprisingly, it has been predicted that pancreatic cancer will soon become the second leading cause of cancer death in the United States and in other countries [203, 204]. As a result, there is a great deal of interest in harnessing the power of genetics for the earlier detection of pancreatic cancer [205,206,207]. Given the specificity of somatic mutations for the presence of a clone of cells, the detection of circulating tumor DNA (ctDNA) provides a promising foundation for the early detection of pancreatic cancer [207]. Extremely sensitive and specific tests for ctDNA have been developed, and when applied clinically these have been very specific, but only moderately sensitive in detecting ctDNA in patients known to have a cancer [205,206,207,208]. Other approaches, including those based on detecting fragmented DNA of specific lengths and the detection of aneuploidy, have been developed that should improve the accuracy and usefulness of blood tests [205, 209, 210]. In addition, a number of genes are abnormally methylated in pancreatic cancer, and several approaches have been developed to detect aberrant methylation patterns in blood, pancreatic juice, and pancreatic cyst fluid [211,212,213,214,215,216].
As exciting as these advances are, significant hurdles remain to developing an effective screening test for pancreatic cancer. One major challenge confronting the screening for pancreatic cancer is its rarity in the general population at any point in time [152, 153]. As a result, even a highly specific screening test will generate many false positive results relative to the true positives [152, 153]. Indeed, the costs, including financial, psychological, and patient harm from unnecessary procedures, currently outweigh the benefits [152, 153].
Even if a perfect test were developed, it might never be possible to detect curable pancreatic cancers, as the window of opportunity for the detection of curable pancreatic cancer is likely extremely short in some patients. For example, several studies have shown that even early pancreatic cancers invade veins within the pancreas [202, 217, 218]. These veins drain directly into the liver, suggesting that early liver metastases may be almost universal in pancreatic cancer [202, 217, 218]. As a result, today, the clinical use of ctDNA testing is limited to following the response of patients with a known cancer to therapy [219]. Given the increase in therapeutic options and given the speed at which a given patient develops resistance to therapy, it seems likely that the use of ctDNA to guide therapy will increase in the coming decade. Given that ctDNA values commonly increase in advance of radiographic progression, intuitively a change in therapy should be implemented in patients with rising values, however there is some reluctance to change therapy based on ctDNA measurement alone.
Personalized therapy
Some of the genetic alterations described above produce cellular changes in the neoplastic cells that are potentially targetable therapeutically. For example, somatic BRAF mutations occur in 1–3% of pancreatic cancers, and BRAF mutations have proven targetable in a number of tumor types [220, 221]. In fact, the Food and Drug Administration recently approved the combination of Encorafenib and Cetuximab for metastatic colon cancer with BRAF mutations [220]. Furthermore, although rare, some pancreatic cancers harbor potentially targetable gene fusions [15, 222,223,224,225,226]. For example, pancreatic cancers with NTRK gene fusions have been treated with tropomyosin receptor kinase inhibitors [222, 223]. Similarly, some KRAS wild-type pancreatic cancers harbor somatic NRG1 gene fusions, and some of these cancers have been reported to respond to treatment with a HER-family kinase inhibitor [225, 226].
Overall, however, although theoretically targetable genetic alterations are present in 30–40% of pancreatic cancers, the results of targeted therapies have mostly been disappointing, with, at best, mixed results [11,12,13, 15]. The poor track record in treating potentially targetable somatic mutations is, in part, due to the short life expectancy for many patients with pancreatic cancer. There just is not the time to sequence their cancers and develop a treatment plan based on the somatic mutations in most cases. The exceptions to this have been the targetable germline alterations described above in the sections on familial pancreatic cancer [11,12,13, 15]. On a positive note, the “Know Your Tumor” trial of over 1500 patients with pancreatic cancer reported an improved outcome in the patients who received targeted therapies based on the molecular profiles of their cancers [227]. Cancer centers are currently building the infrastructure needed to test cancers quickly and to efficiently institute personalized care.
The elephant in the room for targeted therapy remains KRAS [10]. After decades of investments, KRAS was deemed “undruggable” by many [10, 228, 229]. The protein lacks an efficient small-molecule binding pocket and has a high affinity for guanosine triphosphate, which is at high concentration in the cytoplasm [229]. Recently, however, several significant advances were made in targeting KRAS, especially the KRAS G12C alterations, and several inhibitors specifically inhibiting KRAS G12C are now in clinical trials [229]. Unfortunately, KRAS G12C mutations are only a small fraction of KRAS mutations in pancreatic cancers. Broader KRAS inhibitors could have real impact, as 95% of pancreatic cancers harbor KRAS gene mutations [191].
Future directions
The last decade has witnessed dramatic progress, but the death rate from pancreatic cancer is still unacceptably high, and much needs to be done. Looking forward, we envision major advances in five broad areas.
First, as outlined above, the discovery of deleterious germline variants that predispose to pancreatic cancer has had enormous clinical impact. However, many individuals carry a variant of uncertain significance in a pancreatic cancer susceptibility gene, and it is unclear how these individuals should be managed [17]. In the next decade, we believe that most germline variants will be categorized into benign or deleterious, improving the clinical management of all patients. Furthermore, as the functional pathways perturbed by deleterious germline variants are defined, we envision the development of novel therapies that specifically target these pathways.
Second, we envision the development of new combinations of targeted therapies. The emergence of drug resistance when a single targeted agent is given should not be surprising. It was, in fact, entirely predictable based on previous experience treating patients with tuberculosis and treating patients infected with the human immunodeficiency virus [230]. As understanding of pancreatic cancer grows, combinations of therapies, each exploiting a unique weakness, will be developed. These combination therapies will greatly reduce the emergence of drug resistance.
Third, we predict that the next decade will witness understandings that will bring insight into why pancreatic cancer almost universally invades veins in the pancreas [202]. In so doing, it is our hope that new therapies will be developed to impede this process, thereby reducing metastases.
Fourth, we predict that early detection will reduce pancreatic cancer deaths. Early detection approaches will include more sensitive and more specific biomarkers to distinguish low-grade precursor lesions from high-grade precursor lesions, as well as novel strategies that address sampling/multifocality issues in precursor lesions [231].
Fifth, while this review has focused on recent advances in genetics, other fields, particularly imaging, have advanced in parallel. The next decade will see better integration of diverse clinical data, creating a more integrated understanding of a patient’s disease. For example, CT data will be integrated with laboratory values such as blood glucose levels, and germline and somatic sequencing data (including structural variants, chromothripsis, genome doubling, etc.). As these data sets get bigger, artificial intelligence will allow for the discovery of new patterns, patterns that otherwise would not be appreciated by the human brain [232]. This is already happening in radiology, and the integration of genetics and digital pathology into deep learning algorithms is just around the corner. Parenthetically, while we view artificial intelligence as intellectually exciting, it does tear at our hearts to think that humans will become more separated from patient care.
In sum, and in short, we have come a long way, but, paraphrasing Robert Frost, we have miles to go before we sleep.
References
Jones S, Zhang X, Parsons DW, Lin JCH, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6.
Li Y, Roberts ND, Wala JA, Sapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature. 2020;578:112–21.
Cortes-Ciriano I, Lee JJ, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet. 2020;52:331–41.
Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrok D, et al. The evolutionary history of 2,658 cancers. Nature. 2020;578:122–8.
Consortium ITP-CAoWG, Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93.
PCAWG Transcriptome Core Group, Calabrese C, Davidson NR, Demircioğlu D, Fonseca NA, He Y, Kahles A, et al. Genomic basis for RNA alterations in cancer. Nature. 2020;578:129–36.
Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, Zamora J, Supek F, Demeulemeester J, et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat Genet. 2020;52:306–19.
Cancer Genome Atlas Research Network, Raphael BJ, Hruban RH, Aguiree AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32:185–203.e13.
Rainone M, Singh I, Salo-Mullen EE, Stadler ZK, O’Reilly EM. An emerging paradigm for germline testing in pancreatic ductal adenocarcinoma and immediate implications for clinical practice: a review. JAMA Oncol. 2020. Online ahead of print.
Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Geurin TM, et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. 2020;80:1630–43.
Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, et al. Genomics-driven precision medicine for advanced pancreatic cancer: early results from the COMPASS trial. Clin Cancer Res. 2018;24:1344–54.
Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Kimpson S, et al. Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res. 2015;21:2029–37.
Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: potential actionability and correlation with clinical phenotype. Clin Cancer Res. 2017;23:6094–100.
Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153–68.
Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology. 2019;156:2242–53.e4.
McCormick F. Sticking it to KRAS: covalent inhibitors enter the clinic. Cancer Cell. 2020;37:3–4.
Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6:166–75.
Mandelker D, Zhang L, Kemel Y, Stadler ZK, Joseph V, Zehir A, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–35.
Chen F, Childs EJ, Mocci E, Bracci P, Gallinger S, Li D, et al. Analysis of heritability and genetic architecture of pancreatic cancer: a PanC4 study. Cancer Epidemiol Biomarkers Prev. 2019;28:1238–45.
Wang L, Brune KA, Visvanathan K, Laheru D, Herman J, Wolfgang C, et al. Elevated cancer mortality in the relatives of patients with pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:2829–34.
Jacobs EJ, Rodriguez C, Newton CC, Bain EB, Patel AV, Feigelson S, et al. Family history of various cancers and pancreatic cancer mortality in a large cohort. Cancer Causes Control. 2009;20:1261–9.
Jacobs EJ, Chanock SJ, Fuchs CS, Lacroix A, McWilliams RR, Seplowsi E, et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer. 2010;127:1421–8.
Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus JA, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64:2634–8.
Schulte A, Pandeya N, Fawcett J, Fritschi L, Klein K, Risch HA, et al. Association between family cancer history and risk of pancreatic cancer. Cancer Epidemiol. 2016;45:145–50.
Kohli DR, Smith KR, Wong J, Yu Z, Boucher K, Faigel DO, et al. Familial pancreatic cancer risk: a population-based study in Utah. J Gastroenterol. 2019;54:1106–12.
Wang W, Chen S, Brune KA, Hruban RH, Parmigiani G, Klein AP. PancPRO: risk assessment for individuals with a family history of pancreatic cancer. J Clin Oncol. 2007;25:1417–22.
Leonardi G, Marchi S, Falconi M, Zerbo A, Ussia V, de Bortoli N, et al. “PancPro” as a tool for selecting families eligible for pancreatic cancer screening: an Italian study of incident cases. Dig Liver Dis. 2012;44:585–8.
Stadler ZK, Salo-Mullen E, Patil SM, Pietanza C, Vijai J, Saloustros E, et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer. 2012;118:493–9.
Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2015;148:556–64.
Hu C, Hart SN, Bamlet WR, Moore RM, Nandakumar K, Eckloff BW, et al. Prevalence of pathogenic mutations in cancer predisposition genes among pancreatic cancer patients. Cancer Epidemiol Biomarkers Prev. 2016;25:207–11.
Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319:2401–9.
Lowery MA, Kelsen DP, Stadler ZK, Yu KH, Janjigian YY, Ludwig E, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist. 2011;16:1397–402.
Moran A, O’Hara C, Khan S, Shack L, Woodward E, Maher ER, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer. 2012;11:235–42.
Noh JM, Choi DH, Baek H, Nam SJ, Lee JE, Kim JW, et al. Associations between BRCA mutations in high-risk breast cancer patients and familial cancers other than breast or ovary. J Breast Cancer. 2012;15:283–7.
Iqbal J, Ragone A, Lubinski J, Lynch HT, Moller P, Ghadirian P, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2012;107:2005–9.
Lucas AL, Shakya R, Lipsyc MD, Mitchel EB, Kumar S, Hwang C, et al. High prevalence of BRCA1 and BRCA2 germline mutations with loss of heterozygosity in a series of resected pancreatic adenocarcinoma and other neoplastic lesions. Clin Cancer Res. 2013;19:3396–403.
Lucas AL, Frado LE, Hwang C, Kumar S, Khanna LG, Levinson EJ, et al. BRCA1 and BRCA2 germline mutations are frequently demonstrated in both high-risk pancreatic cancer screening and pancreatic cancer cohorts. Cancer. 2014;120:1960–7.
Slater EP, Langer P, Fendrich V, Habbe N, Chaloupka B, Matthäi E, et al. Prevalence of BRCA2 and CDKN2a mutations in German familial pancreatic cancer families. Fam Cancer. 2010;9:335–43.
Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10:10–27.
Cho JH, Bang S, Park SW, Chung JB, Song SY. BRCA2 mutations as a universal risk factor for pancreatic cancer has a limited role in Korean ethnic group. Pancreas. 2008;36:337–40.
Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–9.
Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17:569–77.
Schwartz M, Korenbaum C, Benfoda M, Mary M, Colas C, Coulet F, et al. Familial pancreatic adenocarcinoma: a retrospective analysis of germline genetic testing in a French multicentre cohort. Clin Genet. 2019;96:579–84.
Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol. 2015;33:3124–9.
Ghiorzo P, Fornarini G, Sciallero S, Battistuzzi L, Belli F, Bernard L, et al. CDKN2A is the main susceptibility gene in Italian pancreatic cancer families. J Med Genet. 2012;49:164–70.
Mukherjee B, Delancey JO, Raskin L, Everett J, Jeter J, Begg CB, et al. Risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers. J Natl Cancer Inst. 2012;104:953–6.
Potjer TP, van der Stoep N, Houwing-Duistermaat JJ, Konings ICAW, Aalfs CM, van den Akker PC, et al. Pancreatic cancer-associated gene polymorphisms in a nation-wide cohort of p16-Leiden germline mutation carriers; a case-control study. BMC Res Notes. 2015;8:264.
Middlebrooks CD, Stacey ML, Li Q, Snyder C, Shaw TG, Richardson-Nelson T, et al. Analysis of the CDKN2A gene in FAMMM syndrome families reveals early age of onset for additional syndromic cancers. Cancer Res. 2019;79:2992–3000.
Potjer TP, Kranenburg HE, Bergman W, de Vos tot Nederveen Cappel WH, von Monsjou HS, et al. Prospective risk of cancer and the influence of tobacco use in carriers of the p16-Leiden germline variant. Eur J Hum Genet. 2015;23:711–4.
Harinck F, Kluijt I, van der Stoep N, Oldenburg RA, Wagner A, Aalfs CM, et al. Indication for CDKN2A-mutation analysis in familial pancreatic cancer families without melanomas. J Med Genet. 2012;49:362–5.
Midha S, Sreenivas V, Kabra M, Chattopadhyay KC, Joshi YK, Garg PK, et al. Genetically determined chronic pancreatitis but not alcoholic pancreatitis is a strong risk factor for pancreatic cancer. Pancreas. 2016;45:1478–84.
Su GH, Hruban RH, Bansal RK, Bova GS, Tang DJ, Shekher MC, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol. 1999;154:1835–40.
Korsse SE, Harinck F, van Lier MG, Biermann K, Offerhaus GJA, Krak N, et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance. J Med Genet. 2013;50:59–64.
Earl J, Galindo-Pumarino C, Encinas J, Barreto E, Castillo ME, Pachón V, et al. A comprehensive analysis of candidate genes in familial pancreatic cancer families reveals a high frequency of potentially pathogenic germline variants. EBioMedicine. 2020;53:102675.
Borelli I, Casalis Cavalchini GC, Del Peschio S, Micheletti M, Venesio T, Sarotta I, et al. A founder MLH1 mutation in Lynch syndrome families from Piedmont, Italy, is associated with an increased risk of pancreatic tumours and diverse immunohistochemical patterns. Fam Cancer. 2014;13:401–13.
Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30:958–64.
Dong X, Li Y, Chang P, Hess KR, Abbruzzese JL, Li D. DNA mismatch repair network gene polymorphism as a susceptibility factor for pancreatic cancer. Mol Carcinog. 2012;51:491–9.
Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–5.
Lowery MA, Wong W, Jordan EJ, Lee JW, Kemel Y, Vijai J, et al. Prospective evaluation of germline alterations in patients with exocrine pancreatic neoplasms. J Natl Cancer Inst. 2018;110:1067–74.
Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Williams Parsons D, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217.
Jiao Y, Lumpkins K, Terhune J, Hruban RH, Klein A, Kinszler KW, et al. Intraductal papillary mucinous neoplasm in a neonate with congenital hyperinsulinism and a de novo germline SKIL gene mutation. Pancreatology. 2015;15:194–6.
Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy M, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–6.
Arts HH, Lynch L, Grafodatskaya D, Eng B, Malloy L, Duck J, et al. ATM whole gene deletion in an Italian family with hereditary pancreatic cancer: challenges to cancer risk prediction associated with an 11q22.3 microdeletion. Cancer Genet. 2020;240:1–4.
Catts ZA, Baig MK, Milewski B, Keywan C, Guarino M, Petrelli N. Statewide retrospective review of familial pancreatic cancer in delaware, and frequency of genetic mutations in pancreatic cancer kindreds. Ann Surg Oncol. 2016;23:1729–35.
Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78:490–4.
Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J Clin Oncol. 2020;38:674–85.
Borecka M, Zemankova P, Vocka M, Soukupova J, Kleiblova P, Sevcik J, et al. Mutation analysis of the PALB2 gene in unselected pancreatic cancer patients in the Czech Republic. Cancer Genet. 2016;209:199–204.
Hu C, LaDuca H, Shimelis H, Polley EC, Lilyquist J, Hart SN, et al. Multigene hereditary cancer panels reveal high-risk pancreatic cancer susceptibility genes. JCO Precis Oncol. 2018;2:10.
Goldstein JB, Zhao L, Wang X, Ghelman Y, Oerman MJ, Javle MM, et al. Germline DNA sequencing reveals novel mutations predictive of overall survival in a cohort of patients with pancreatic cancer. Clin Cancer Res. 2020;26:1385–94.
Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet. 2003;40:1–10.
Kim DH, Crawford B, Ziegler J, Beattie MS. Prevalence and characteristics of pancreatic cancer in families with BRCA1 and BRCA2 mutations. Fam Cancer. 2009;8:153–8.
Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol. 2000;156:1767–71.
Al-Sukhni W, Rothenmund H, Borgida AE, Zogopoulos G, O’shea AM, Pllett A, et al. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Hum Genet. 2008;124:271–8.
Murphy KM, Brune KA, Griffin C, Sollenberger JE, Petersen GM, Bansal R, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res. 2002;62:3789–93.
Chaffee KG, Oberg AL, McWilliams RR, Majithia N, Allen BA, Kidd J, et al. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med. 2018;20:119–27.
Takai E, Yachida S, Shimizu K, Furuse J, Kubo E, Ohmoto A, et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget. 2016;7:74227–35.
Rogers CD, van der Heijden MS, Brune K, Yeo CJ, Hruban RH, Kern SE, et al. The genetics of FANCC and FANCG in familial pancreatic cancer. Cancer Biol Ther. 2004;3:167–9.
van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63:2585–8.
Hall MJ, Dignam JJ, Olopade OI. Family history of pancreatic cancer in a high-risk cancer clinic: implications for risk assessment. J Genet Couns. 2008;17:365–72.
Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;35:3382–90.
Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–4.
Tempero MA. NCCN guidelines updates: pancreatic cancer. J Natl Compr Canc Netw. 2019;17:603–5.
Daly MB, Pilarski R, Yurgelun MB, Berry MP, Buys SS, Dickson P, et al. NCCN guidelines insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 1.2020. J Natl Compr Canc Netw. 2020;18:380–91.
de Boo L, Cimino-Mathews A, Lubeck Y, Daletzakis A, Opdam M, Sanders J, et al. Tumour-infiltrating lymphocytes (TILs) and BRCA-like status in stage III breast cancer patients randomised to adjuvant intensified platinum-based chemotherapy versus conventional chemotherapy. Eur J Cancer. 2020;127:240–50.
Singhi AD, Ishida H, Ali SZ, Goggins M, Canto M, Wolfgang C, et al. A histomorphologic comparison of familial and sporadic pancreatic cancers. Pancreatology. 2015;15:387–91.
Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol. 2017;3:774–83.
Humphris JL, Patch AM, Nones K, Bailey PJ, Johns AL, McKay K, et al. Hypermutation in pancreatic cancer. Gastroenterology. 2017;152:68–74.e2.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501.
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–27.
Yarchoan M, Myzak MC, Johnson BA 3rd, De Jesus-Acosta A, Le DT, Jaffee EM, et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget. 2017;8:44073–81.
Golan T, Barenboim A, Lahat G, Nachmany I, Goykhman Y, Shacham-Shmueli E, et al. Increased rate of complete pathologic response after neoadjuvant FOLFIRINOX for BRCA mutation carriers with borderline resectable pancreatic cancer. Ann Surg Oncol. 2020. Online ahead of print.
Pishvaian MJ, Biankin AV, Bailey P, Chang DK, Laheru D, Wolfgang CL, et al. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br J Cancer. 2017;116:1021–6.
Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic Implications. Mol Cancer Ther. 2016;15:1781–91.
Nanda N, Roberts NJ. ATM serine/threonine kinase and its role in pancreatic risk. Genes (Basel). 2020;11:108.
Hutchings D, Jiang Z, Skaro M, Weiss MJ, Wolfgang CL, Makary MA, et al. Histomorphology of pancreatic cancer in patients with inherited ATM serine/threonine kinase pathogenic variants. Mod Pathol. 2019;32:1806–13.
Skaro M, Nanda N, Gauthier C, Felsenstein M, Jiang Z, Qiu M, et al. Prevalence of germline mutations associated with cancer risk in patients with intraductal papillary mucinous neoplasms. Gastroenterology. 2019;156:1905–13.
Seidel G, Zahurak M, Iacobuzio-Donahue C, Sohn TA, Adsay NV, Yeo CJ, et al. Almost all infiltrating colloid carcinomas of the pancreas and periampullary region arise from in situ papillary neoplasms: a study of 39 cases. Am J Surg Pathol. 2002;26:56–63.
Ayars M, Eshleman J, Goggins M. Susceptibility of ATM-deficient pancreatic cancer cells to radiation. Cell Cycle. 2017;16:991–8.
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res. 2020;26:2487–96.
Rafiei S, Fitzpatrick K, Liu D, Cai MY, Elmarakeby HA, Park J, et al. ATM loss confers greater sensitivity to ATR inhibition than parp inhibition in prostate cancer. Cancer Res. 2020;80:2094–2100.
Lynch HT, Shaw TG. Familial atypical multiple mole melanoma (FAMMM) syndrome: history, genetics, and heterogeneity. Fam Cancer. 2016;15:487–91.
Ibrahim I, Sibinga Mulder BG, Bonsing B, Morreau H, Sarasqueta AF, Inderson A, et al. Risk of multiple pancreatic cancers in CDKN2A-p16-Leiden mutation carriers. Eur J Hum Genet. 2018;26:1227–9.
de Snoo FA, Bishop DT, Bergman W, van Leeuwen I, vand der Drift C, van Nieuwpoort FA, et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res. 2008;14:7151–7.
Kluijt I, Cats A, Fockens P, Nio Y, Gouma DJ, Bruno MJ. Atypical familial presentation of FAMMM syndrome with a high incidence of pancreatic cancer: case finding of asymptomatic individuals by EUS surveillance. J Clin Gastroenterol. 2009;43:853–7.
Lynch HT, Brand RE, Hogg D, Deters CA, Fusaro RM, Lynch JF, et al. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families: the familial atypical mole melanoma-pancreatic carcinoma syndrome. Cancer. 2002;94:84–96.
Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer. 2000;87:809–11.
Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med. 1995;333:970–4.
Geary J, Sasieni P, Houlston R, Izatt L, Eeles R, Payne SJ, et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam Cancer. 2008;7:163–72.
Barrow E, Robinson L, Alduaij W, Shenton A, Clancy T, Lalloo F, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet. 2009;75:141–9.
Gargiulo S, Torrini M, Ollila S, Nasti S, Pastorino L, Cusano R, et al. Germline MLH1 and MSH2 mutations in Italian pancreatic cancer patients with suspected Lynch syndrome. Fam Cancer. 2009;8:547–53.
Ahmad-Nielsen SA, Bruun Nielsen MF, Mortensen MB, Detlefsen S. Frequency of mismatch repair deficiency in pancreatic ductal adenocarcinoma. Pathol Res Pract. 2020;216:152985.
Wilentz RE, Goggins M, Redston M, Marcus VA, Adsay NV, Sohn TA, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000;156:1641–51.
Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, et al. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol. 1998;152:1501–7.
Banville N, Geraghty R, Fox E, Leay DT, Green A, Keegan D, et al. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum Pathol. 2006;37:1498–502.
Liu W, Shia J, Gönen M, Lowery MA, O’Reilly EM, Klimstra DS. DNA mismatch repair abnormalities in acinar cell carcinoma of the pancreas: frequency and clinical significance. Pancreas. 2014;43:1264–70.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Luchini C, Brosens LAA, Wood LD, Chatterjee D, Sin JI, Sciammarella C, et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: histology, molecular pathology and clinical implications. Gut. 2020. Online ahead of print.
Keller JJ, Offerhaus GJ, Giardiello FM, Menko FH. Jan Peutz, Harold Jeghers and a remarkable combination of polyposis and pigmentation of the skin and mucous membranes. Fam Cancer. 2001;1:181–5.
Ma H, Brosens LAA, Offerhaus GJA, Giardiello FM, Leng WWJ, Montgomery EA. Pathology and genetics of hereditary colorectal cancer. Pathology. 2018;50:49–59.
Resta N, Pierannunzio D, Lenato GM, Stella A, Capacoaccia R, Bagnulo R, et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis. 2013;45:606–11.
Sahin F, Maitra A, Argani P, Sato N, Maehara N, Montgomery E, et al. Loss of Stk11/Lkb1 expression in pancreatic and biliary neoplasms. Mod Pathol. 2003;16:686–91.
Duell EJ, Lucenteforte E, Olson SH, Bracci PM, Risch HA, Silverman DT, et al. Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol. 2012;23:2964–70.
Lowenfels AB, Maisonneuve P, Whitcomb DC, Lerch MM, DiMagno EP. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. JAMA. 2001;286:169–70.
Lowenfels AB, Maisonneuve P, Whitcomb DC. Risk factors for cancer in hereditary pancreatitis. International Hereditary Pancreatitis Study Group. Med Clin North Am. 2000;84:565–73.
Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK Jr, Perrault J, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442–6.
Shelton CA, Grubs RE, Umapathy C, Yadav D, Whitcomb DC. Impact of hereditary pancreatitis on patients and their families. J Genet Couns. 2020. Online ahead of print.
Shelton CA, Umapathy C, Stello K, Yadav D, Whitcomb DC. Hereditary pancreatitis in the United States: survival and rates of pancreatic cancer. Am J Gastroenterol. 2018;113:1376.
Rebours V, Lévy P, Mosnier JF, Scoazec JY, Soubeyrand MS, Fléjou JF, et al. Pathology analysis reveals that dysplastic pancreatic ductal lesions are frequent in patients with hereditary pancreatitis. Clin Gastroenterol Hepatol. 2010;8:206–12.
Singhi AD, Pai RK, Kant JA, Bartholow TL, Zeh HJ, Lee KK, et al. The histopathology of PRSS1 hereditary pancreatitis. Am J Surg Pathol. 2014;38:346–53.
Tamura K, Yu J, Hata T, Suenaga M, Shindo K, Abe T, et al. Mutations in the pancreatic secretory enzymes CPA1 and CPB1 are associated with pancreatic cancer. Proc Natl Acad Sci USA. 2018;115:4767–72.
Nissim S, Leshchiner I, Mancias JD, Greenblatt MB, Maertens O, Cassa CA, et al. Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. Nat Genet. 2019;51:1308–14.
Smith AL, Alirezaie N, Connor A, Chan-Seng-Yue M, Grant R, Selander I, et al. Candidate DNA repair susceptibility genes identified by exome sequencing in high-risk pancreatic cancer. Cancer Lett. 2016;370:302–12.
Wang X, Szabo C, Qian C, Amadio PG, Thibodeau SN, Cerhan JR, et al. Mutational analysis of thirty-two double-strand DNA break repair genes in breast and pancreatic cancers. Cancer Res. 2008;68:971–5.
Wong C, Chen F, Alirezaie N, Wang Y, Cuggia A, Borgida A, et al. A region-based gene association study combined with a leave-one-out sensitivity analysis identifies SMG1 as a pancreatic cancer susceptibility gene. PLoS Genet. 2019;15:e1008344.
Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. Washington, DC: American Registry of Pathology; 2007. p. 422.
Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405.
Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47:911–6.
Zhong J, Jermusyk A, Wu L, Hoskins JW, Collins I, Mocci E, et al. A Transcriptome-Wide Association Study (TWAS) identifies novel candidate susceptibility genes for pancreatic cancer. J Natl Cancer Inst. 2020. Online ahead of print.
Zhang M, Wang Z, Obazee O, Jia J, Childs EJ, Hoskins J, et al. Three new pancreatic cancer susceptibility signals identified on chromosomes 1q32.1, 5p15.33 and 8q24.21. Oncotarget. 2016;7:66328–43.
Klein AP, Wolpin BM, Risch HA, Stolzenberg-Solomon RZ, Mocci E, Zhang M, et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nat Commun. 2018;9:556.
Wu C, Miao X, Huang L, Che X, Jiang G, Yu D, et al. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat Genet. 2011;44:62–6.
Low SK, Kuchiba A, Zembutsu H, Saito A, Takahashi A, Kubo M, et al. Genome-wide association study of pancreatic cancer in Japanese population. PLoS ONE. 2010;5:e11824.
Wolpin BM, Chan AT, Hartge P, Chanock SJ, Kraft P, Hunter DJ, et al. ABO blood group and the risk of pancreatic cancer. J Natl Cancer Inst. 2009;101:424–31.
Abe T, Blackford AL, Tamura K, Ford M, McCormick P, Chuidian M, et al. Deleterious germline mutations are a risk factor for neoplastic progression among high-risk individuals undergoing pancreatic surveillance. J Clin Oncol. 2019;37:1070–80.
Overbeek KA, Cahen DL, Canto MI, Bruno MJ. Surveillance for neoplasia in the pancreas. Best Pract Res Clin Gastroenterol. 2016;30:971–86.
Vasen H, Ibrahim I, Ponce CG, Slater EP, Matthäi E, Carrato A, et al. Benefit of surveillance for pancreatic cancer in high-risk individuals: outcome of long-term prospective follow-up studies from three European expert centers. J Clin Oncol. 2016;34:2010–9.
Konings I, Canto MI, Almario JA, Harinck F, Saxena P, Lucas Al, et al. Surveillance for pancreatic cancer in high-risk individuals. BJS Open. 2019;3:656–65.
Blackford AL, Canto MI, Klein AP, Hruban RH, Goggins M. Recent trends in the incidence and survival of stage 1A pancreatic cancer: a surveillance, epidemiology, and end results analysis. J Natl Cancer Inst. 2020. Online ahead of print.
Canto MI, Almario JA, Schulick RD, Yeo CJ, Klein A, Blackford A, et al. Risk of neoplastic progression in individuals at high risk for pancreatic cancer undergoing long-term surveillance. Gastroenterology. 2018;155:740–51.e2.
Goggins M, Overbeek KA, Brand R, Syngal S, Del Chiaro M, Bartsch DK, et al. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut. 2020;69:7–17.
Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355:1330–4.
Hruban RH, Lillemoe KD. Screening for pancreatic cancer gets a D, but the student is improving. JAMA Surg. 2019;154:795–7.
Lennon AM, Hruban RH, Klein AP. Screening for pancreatic cancer—is there hope? JAMA Intern Med. 2019. Online ahead of print.
Basturk O, Hong SM, Wood LD, Adsay NV, Albores-Saavedra J, Biankin AV, et al. A revised classification system and recommendations from the Baltimore Consensus Meeting for neoplastic precursor lesions in the pancreas. Am J Surg Pathol. 2015;39:1730–41.
Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of tumours of the digestive system. 4th ed. Lyon: International Agency for Research on Cancer; 2010.
Valsangkar NP, Morales-Oyarvide V, Thayer SP, Ferrone CR, Wargo JA, Warshaw AL, et al. 851 resected cystic tumors of the pancreas: a 33-year experience at the Massachusetts General Hospital. Surgery. 2012;152:S4–12.
Laffan TA, Horton KM, Klein AP, Berlanstein B, Siegelman SS, Kawamoto S, et al. Prevalence of unsuspected pancreatic cysts on MDCT. Am J Roentgenol. 2008;191:802–7.
Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci USA. 2011;108:21188–93.
Wu J, Matthaei H, Maitra A, de Wilde RF, Wood LD, Eshleman JR, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66.
Molin MD, Matthaei H, Wu J, Blackford A, Debeljak M, Rezaee N, et al. Clinicopathological correlates of activating GNAS mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg Oncol. 2013;20:3802–8.
Amato E, Molin MD, Mafficini A, Yu J, Malleo G, Rusev B, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol. 2014;233:217–27.
Singhi AD, Wood LD, Parks E, Torbenson MS, Felsenstein M, Hruban RH, et al. Recurrent rearrangements in PRKACA and PRKACB in intraductal oncocytic papillary neoplasms of the pancreas and bile duct. Gastroenterology. 2020;158:573–82.e2.
Vyas M, Hechtman JF, Zhang Y, Benayed R, Yavas A, Askan G, et al. DNAJB1-PRKACA fusions occur in oncocytic pancreatic and biliary neoplasms and are not specific for fibrolamellar hepatocellular carcinoma. Mod Pathol. 2020;33:648–56.
Abraham SC, Klimstra DS, Wilentz RE, Yeo CJ, Conlon K, Brennan M, et al. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am J Pathol. 2002;160:1361–9.
Springer S, Wang Y, Dal Molin M, Masica DL, Jiao Y, Kinde I, et al. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology. 2015;149:1501–10.
Miyabe K, Hori Y, Nakazawa T, Hayashi K, Naitoh I, Shimizu S, et al. Locus/chromosome aberrations in intraductal papillary mucinous neoplasms analyzed by fluorescence in situ hybridization. Am J Surg Pathol. 2015;39:512–20.
Springer S, Masica DL, Dal Molin M, Douville C, Thoburn CJ, Afsari B, et al. A multimodality test to guide the management of patients with a pancreatic cyst. Sci Transl Med. 2019;11:eaav4772.
Singhi AD, McGrath K, Brand RE, Khalid A, Zeh HJ, Chennat JS, et al. Preoperative next-generation sequencing of pancreatic cyst fluid is highly accurate in cyst classification and detection of advanced neoplasia. Gut. 2018;67:2131–41.
Yip-Schneider MT, Wu H, Dumas RP, Hancock BA, Agaram N, Radovich M, et al. Vascular endothelial growth factor, a novel and highly accurate pancreatic fluid biomarker for serous pancreatic cysts. J Am Coll Surg. 2014;218:608–17.
Choi SH, Park SH, Kim KW, Lee JY, Lee SS. Progression of unresected intraductal papillary mucinous neoplasms of the pancreas to cancer: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2017;15:1509–20.e4.
Oyama H, Tada M, Takagi K, Tateishi K, Hamada T, Nakai Y, et al. Long-term risk of malignancy in branch-duct intraductal papillary mucinous neoplasms. Gastroenterology. 2020;158:226–37.e5.
Tanaka M, Fernandez-Del Castillo C, Kamisawa T, Jang JY, Levy P, Ohtsuka T, et al. Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology. 2017;17:738–53.
Simpson RE, Cockerill NJ, Yip-Schneider MT, Ceppa EP, House MG, Zyromski NJ, et al. DNA profile components predict malignant outcomes in select cases of intraductal papillary mucinous neoplasm with negative cytology. Surgery. 2018;164:712–8.
Omori Y, Ono Y, Tanino M, Karasaki H, Yamaguchi H, Furukawa T, et al. Pathways of progression from intraductal papillary mucinous neoplasm to pancreatic ductal adenocarcinoma based on molecular features. Gastroenterology. 2019;156:647–61.e2.
Fischer CG, Beleva Guthrie V, Braxton AM, Zhen L, Wang P, Song Q, et al. Intraductal papillary mucinous neoplasms arise from multiple independent clones, each with distinct mutations. Gastroenterology. 2019;157:1123–37.e22.
Felsenstein M, Noe M, Masica DL, Hosoda W, Chianchiano P, Fischer CG, et al. IPMNs with co-occurring invasive cancers: neighbours but not always relatives. Gut. 2018;67:1652–62.
Kuboki Y, Fischer CG, Beleva Guthrie V, Huang W, Yu J, Chianchiano P, et al. Single-cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. J Pathol. 2019;247:347–56.
Hutchings D, Waters KM, Weiss MJ, Wolfgang CL, Makary MA, He J, et al. Cancerization of the pancreatic ducts: demonstration of a common and under-recognized process using immunolabeling of paired duct lesions and invasive pancreatic ductal adenocarcinoma for p53 and Smad4 expression. Am J Surg Pathol. 2018;42:1556–61.
Pea A, Yu J, Rezaee N, Luchini C, He J, Dal Molin M, et al. Targeted DNA sequencing reveals patterns of local progression in the pancreatic remnant following resection of intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg. 2017;266:133–41.
Nagai K, Mizukami Y, Omori Y, Kin T, Yane K, Takahashi K, et al. Metachronous intraductal papillary mucinous neoplasms disseminate via the pancreatic duct following resection. Mod Pathol. 2020;33:971–80.
Kang MJ, Jang JY, Lee KB, Chang YR, Kwon W, Kim SW. Long-term prospective cohort study of patients undergoing pancreatectomy for intraductal papillary mucinous neoplasm of the pancreas: implications for postoperative surveillance. Ann Surg. 2014;260:356–63.
Majumder S, Philip NA, Singh Nagpal SJ, Takahashi N, Mara KC, Kendrick ML, et al. High-grade dysplasia in resected main-duct intraductal papillary mucinous neoplasm (MD-IPMN) is associated with an increased risk of subsequent pancreatic cancer. Am J Gastroenterol. 2019;114:524–9.
Hirono S, Shimizu Y, Ohtsuka T, Kin T, Hara K, Kanno A, et al. Recurrence patterns after surgical resection of intraductal papillary mucinous neoplasm (IPMN) of the pancreas; a multicenter, retrospective study of 1074 IPMN patients by the Japan Pancreas Society. J Gastroenterol. 2020;55:86–99.
Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 2016;538:378–82.
Reiter JG, Iacobuzio-Donahue CA. Pancreatic cancer: pancreatic carcinogenesis—several small steps or one giant leap? Nat Rev Gastroenterol Hepatol. 2016;14:7–8.
Rodic N, Steranka JP, Makohon-Moore A, Moyer A, Shen P, Sharma R, et al. Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1060–4.
Rodic N, Sharma R, Sharma R, Zampella J, Dai L, Taylor MS, et al. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am J Pathol. 2014;184:1280–6.
Ardeljan D, Steranka JP, Liu C, Li Z, Taylor MS, Payer LM, et al. Cell fitness screens reveal a conflict between LINE-1 retrotransposition and DNA replication. Nat Struct Mol Biol. 2020;27:168–78.
Loh JW, Ha H, Lin T, Sun N, Burns KH, Xing J. Integrated mobile element scanning (ME-Scan) method for identifying multiple types of polymorphic mobile element insertions. Mob DNA. 2020;11:12.
Steranka JP, Tang Z, Grivainis M, Huang CRL, Payer LM, Rego FOR, et al. Transposon insertion profiling by sequencing (TIPseq) for mapping LINE-1 insertions in the human genome. Mob DNA. 2019;10:8.
Hruban RH, van Mansfeld AD, Offerhaus GJ, van Weering DH, Allison DC, Goodman SN, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol. 1993;143:545–54.
Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Flores E, Flores Figueroa E, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet. 2020;52:231–40.
Sakamoto H, Attiyeh MA, Gerold JM, Makohon-Moore AP, Hayashi A, Hong J, et al. The evolutionary origins of recurrent pancreatic cancer. Cancer Discov. 2020;10:792–805.
Calhoun ES, Hucl T, Gallmeier E, West KM, Arking DE, Maitra A, et al. Identifying allelic loss and homozygous deletions in pancreatic cancer without matched normals using high-density single-nucleotide polymorphism arrays. Cancer Res. 2006;66:7920–8.
Scrimieri F, Calhoun ES, Patel K, Gupta R, Huso DL, Hruban RH, et al. FAM190A rearrangements provide a multitude of individualized tumor signatures and neo-antigens in cancer. Oncotarget. 2011;2:69–75.
Patel K, Scrimieri F, Ghosh S, Zhong J, Kim MS, Ren YR, et al. FAM190A deficiency creates a cell division defect. Am J Pathol. 2013;183:296–303.
Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2019;16:207–20.
Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–3.
Moffitt RA, Marayati R, Flate EL, Volmar KE, Herrera Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168–78.
Rashid NU, Peng XL, Jin C, Moffitt RA, Volmar KE, Belt BA, et al. Purity independent subtyping of tumors (PurIST), a clinically robust, single-sample classifier for tumor subtyping in pancreatic cancer. Clin Cancer Res. 2020;26:82–92.
Brunton H, Caligiuri G, Cunningham R, Upstill-Goddard R, Bailey UM, et al. HNF4A and GATA6 loss reveals therapeutically actionable subtypes in pancreatic cancer. Cell Rep. 2020;31:107625.
Hruban RH, Gaida MM, Thompson E, Hong SM, Noë M, Brosens LA, et al. Why is pancreatic cancer so deadly? The pathologist’s view. J Pathol. 2019;248:131–41.
Ferlay J, Partensky C, Bray F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016;55:1158–60.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21.
Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–9.
Lennon AM, Buchanan AH, Kinde I, Warren A, Honushefsty A, Cohain AT, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science. 2020. Online ahead of print.
Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci USA. 2017;114:10202–7.
Macgregor-Das A, Yu J, Tamura K, Abe T, Suenaga M, Shindo K, et al. Detection of circulating tumor DNA in patients with pancreatic cancer using digital next-generation sequencing. J Mol Diagn. 2020;22:748–56.
Douville C, Cohen JD, Ptak J, Popoli M, Schaefer J, Silliman N, et al. Assessing aneuploidy with repetitive element sequencing. Proc Natl Acad Sci USA. 2020;117:4858–63.
Douville C, Springer S, Kinde I, Cohen JD, Hruban RH, Lennon AM, et al. Detection of aneuploidy in patients with cancer through amplification of long interspersed nucleotide elements (LINEs). Proc Natl Acad Sci USA. 2018;115:1871–6.
Bailleux C, Lacroix L, Barranger E, Delaloge S. Using methylation signatures on cell-free DNA for early cancer detection: a new era in liquid biopsy? Ann Oncol. 2020;31:665–7.
Majumder S, Raimondo M, Taylor WR, Yab TC, Bergere CK, Dukek BA, et al. Methylated DNA in pancreatic juice distinguishes patients with pancreatic cancer from controls. Clin Gastroenterol Hepatol. 2020;18:676–83.e3.
Eissa MAL, Lerner L, Abdelfatah E, Shankar N, Canner JK, Hasan NM, et al. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin Epigenetics. 2019;11:59.
Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–83.
Matsubayashi H, Canto M, Sato N, Klein A, Abe T, Yamashita K, et al. DNA methylation alterations in the pancreatic juice of patients with suspected pancreatic disease. Cancer Res. 2006;66:1208–17.
Hata T, Dal Molin M, Hong SM, Tamura K, Suenaga M, Yu J, et al. Predicting the grade of dysplasia of pancreatic cystic neoplasms using cyst fluid DNA methylation markers. Clin Cancer Res. 2017;23:3935–44.
Noe M, Rezaee N, Asrani K, Skaro M, Groot VP, Wu PH, et al. Immunolabeling of cleared human pancreata provides insights into three-dimensional pancreatic anatomy and pathology. Am J Pathol. 2018;188:1530–5.
Hong SM, Jung D, Kiemen A, Gaida MM, Yoshizawa T, Braxton AM, et al. Three-dimensional visualization of cleared human pancreas cancer reveals that sustained epithelial-to-mesenchymal transition is not required for venous invasion. Mod Pathol. 2020;33:639–47.
Groot VP, Mosier S, Javed AA, Teinor JA, Gemenetzis G, Ding D, et al. Circulating tumor DNA as a clinical test in resected pancreatic cancer. Clin Cancer Res. 2019;25:4973–84.
Bernabe-Ramirez C, Patel R, Chahal J, Saif MW. Treatment options in BRAF-mutant metastatic colorectal cancer. Anticancer Drugs. 2020;31:545–57.
Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov. 2018;8:1096–111.
O’Reilly EM, Hechtman JF. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann Oncol. 2019;30:viii36–40.
Solomon JP, Linkov I, Rosado A, Mullaney K, Rosen EY, Frosina D, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33:38–46.
Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MB, Shaw AT, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21:261–70.
Aguirre AJ. Oncogenic NRG1 fusions: a new hope for targeted therapy in pancreatic cancer. Clin Cancer Res. 2019;25:4589–91.
Jones MR, Williamson LM, Topham JT, Lee MKC, Goytain A, Ho J, et al. NRG1 gene fusions are recurrent, clinically actionable gene rearrangements in KRAS wild-type pancreatic ductal adenocarcinoma. Clin Cancer Res. 2019;25:4674–81.
Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020;21:508–18.
Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS mutant cancers with a covalent G12C-Specific Inhibitor. Cell. 2018;172:578–89.e17.
Nagasaka M, Li Y, Sukari A, Ou SHI, Najeeb Al-Hallak M, Azmi AS. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat Rev. 2020;84:101974.
Ryan F. The forgotten plague: how the battle against tuberculosis was won–and lost. 1st American ed. Boston: Little, Brown; 1993. xix, 460 p., 8p. of plates pp.
Yu J, Sadakari Y, Shindo K, Suenaga M, Brant A, Alejandro J, et al. Digital next-generation sequencing identifies low-abundance mutations in pancreatic juice samples collected from the duodenum of patients with pancreatic cancer and intraductal papillary mucinous neoplasms. Gut. 2017;66:1677–87.
Park S, Chu LC, Fishman EK, Yuille AL, Vogelstein B, Kinzler KW, et al. Annotated normal CT data of the abdomen for deep learning: challenges and strategies for implementation. Diagn Interv Imaging. 2020;101:35–44.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
RHH has the potential to receive royalty payments from Thrive Earlier Detection for the GNAS invention.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Thompson, E.D., Roberts, N.J., Wood, L.D. et al. The genetics of ductal adenocarcinoma of the pancreas in the year 2020: dramatic progress, but far to go. Mod Pathol 33, 2544–2563 (2020). https://doi.org/10.1038/s41379-020-0629-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0629-6
This article is cited by
-
Immunosuppression, immune escape, and immunotherapy in pancreatic cancer: focused on the tumor microenvironment
Cellular Oncology (2023)
-
Pancreatic cancer cells spectral library by DIA-MS and the phenotype analysis of gemcitabine sensitivity
Scientific Data (2022)
-
Neoplastic cell enrichment of tumor tissues using coring and laser microdissection for proteomic and genomic analyses of pancreatic ductal adenocarcinoma
Clinical Proteomics (2022)
-
Pancreatic cancer pathology viewed in the light of evolution
Cancer and Metastasis Reviews (2021)
-
Die Mikroarchitektur des Pankreaskarzinoms aus Sicht des Pathologen und des Radiologen
Der Pathologe (2021)
