
Abstract
Malignant Brenner tumor is a rare primary ovarian carcinoma subtype that may present diagnostic and therapeutic conundrums. Here, we characterize the genomics of 11 malignant Brenner tumors, which represented 0.1% of 14,153 clinically advanced ovarian carcinomas submitted for genomic profiling during the course of clinical care. At the time of molecular profiling, there was no evidence of a primary urothelial carcinoma of the urinary tract in any case. Cases with transitional-like morphologic features in the setting of variant ovarian serous or endometrioid carcinoma morphology were excluded from the final cohort. Malignant Brenner tumors exhibited CDKN2A/2B loss and oncogenic FGFR1/3 genomic alterations in 55% of cases, respectively; including recurrent FGFR3 S249C or FGFR3-TACC3 fusion in 45% of cases. FGFR3-mutated cases had an associated benign or borderline Brenner tumor pre-cursor components, further confirming the diagnosis and the ovarian site of origin. Malignant Brenner tumors were microsatellite stable, had low tumor mutational burden and exhibited no evidence of homologous recombination deficiency. PIK3CA mutations were enriched with FGFR3 alterations, while FGFR3 wild-type cases featured MDM2 amplification or TP53 mutations. The FGFR3 S249C short variant mutation was absent in 14,142 non-Brenner, ovarian carcinomas subtypes. In contrast to malignant Brenner tumors, FGFR1/2/3 alterations were present in ~5% of non-Brenner, ovarian serous, clear cell and endometrioid carcinoma subtypes, most often as FGFR1 amplification in serous carcinoma or FGFR2 short variant alterations in clear cell or endometrioid carcinomas, respectively. Finally, malignant Brenner tumors had overall distinct genomic signatures compared to FGFR-mutated ovarian serous, endometrioid, and clear cell carcinoma subtypes. This study provides insights into the molecular pathogenesis of malignant Brenner tumors, contrasts the extent of FGFR1/2/3 alterations in ovarian serous, clear cell and endometrioid carcinomas and emphasizes the potential value of novel and FDA-approved, anti-FGFR inhibitors, such as erdafitinib and pemigatinib, in refractory, FGFR3-mutated malignant Brenner tumors.
Similar content being viewed by others

Introduction
Malignant Brenner tumor is an exceedingly rare type of primary ovarian carcinoma, which morphologically resembles urothelial/transitional cell carcinoma from the bladder and urinary tract [1]. In the past, malignant Brenner tumor was distinguished from both primary transitional cell carcinoma of the ovary and metastatic urothelial carcinoma to the ovary by the presence of benign or borderline Brenner tumor pre-cursor components [2, 3]. However, in the most recent version of the “WHO Classification of Tumors of the Female Reproductive Organs”, primary ovarian transitional cell carcinoma is no longer recognized as a unique entity and is now considered to be a variant of serous or endometrioid carcinoma with transitional-like features. In contrast, malignant Brenner tumor still stands as a distinctively recognized entity, but it may be difficult to diagnose without the identification of the precursor benign or Borderline Brenner tumor components [1].
Due to its rarity, no consensus exists for the optimal treatment of malignant Brenner tumor. However, adjuvant platinum-taxane based chemotherapy after surgical excision according to current recommendations for other epithelial ovarian cancers may yield favorable results [4]. Recurrences of advanced-stage malignant Brenner tumors can be more difficult to treat as recurrences may become refractory to conventional chemotherapy [4]. Currently, opportunities for targeted therapies against malignant Brenner tumor are not known since the cause and molecular pathogenesis of this rare ovarian epithelial malignancy are not well defined.
Specific genomic alterations in the FGFR (fibroblast growth factor receptor) family are well-known to be characteristic driver events of certain cancer types, such as FGFR3 mutations in primary urothelial carcinoma of the bladder and FGFR2 fusions in primary intrahepatic cholangiocarcinoma [5, 6]. Specific gain-of-function FGFR alterations suggest that optimal therapeutic benefits of FGFR inhibitors may be specific to certain cancer subtypes. Recently, novel anti-FGFR targeted therapy was approved by the FDA for metastatic bladder urothelial carcinoma (i.e., erdafitinib) [7] and for advanced primary intrahepatic cholangiocarcinoma (i.e., pemigatinib) [8]. However, the extent of FGFR3 genomic alterations in specific ovarian carcinoma subtypes is unknown.
In the current study, we mined a large genomic dataset of 14,153 clinically advanced ovarian carcinoma patients that had undergone comprehensive genomic profiling via DNA-based targeted next-generation sequencing during the course of clinical care and retrospectively identified and analyzed the genomic landscape of malignant Brenner tumors of the ovary. We found that malignant Brenner tumor mutational signatures were characterized by CDKN2A/B loss and well as two alternative genetic pathways: (1) FGFR3-driven or (2) MDM2/TP53-dependent. We further define the FGFR1/2/3 mutational landscape across the most frequent ovarian carcinoma subtypes (i.e., serous, clear cell, and endometrioid), and we propose a model for the pathogenesis of malignant Brenner tumors of the ovary, which may have diagnostic and targeted therapeutic implications.
Methods
Malignant Brenner tumor of the ovary cohort
Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). A retrospective database search of Foundation Medicine, Inc., a CLIA-cerfified and CAP-accredited reference molecular pathology laboratory, was performed for cases whose submitting diagnosis was malignant Brenner tumors of the ovary in female patients. Clinicopathological data including patient age, tumor size and tumor stage, and sites of metastasis were extracted from the accompanying pathology report or associated clinical records when available. The pathologic diagnosis of malignant Brenner tumor and morphological features were re-evaluated on routine H&E-stained slides of tissue sections submitted for DNA sequencing. Cases with transitional-like features in the setting of variant serous or endometrioid carcinoma morphology, as previously described [9], were excluded from the final malignant Brenner tumor cohort. Degree of atypia was scored as low-grade if nuclei resembled low-grade bladder urothelial carcinoma (nuclei uniformly enlarged with mild differences in shape, contour, and chromatin distribution) and high-grade if tumor cells met criteria for bladder high-grade urothelial carcinoma (moderate to marked pleomorphism, enlarged nuclei at least 4X the size of lymphocytes with variation in size, irregular contour, clumped chromatin and prominent nucleoli).
Internal 14,142 non-Brenner ovarian carcinoma cohort, including 11,433 serous carcinomas, 903 clear cell carcinomas, and 570 endometrioid carcinomas
Similarly to the malignant Brenner tumor cohort, we retrospectively identified 14,142 non-Brenner ovarian carcinoma cases from the archives of Foundation Medicine, Inc. These cases were also previously submitted for comprehensive genomic profiling from different institutions during the course clinical care between 2010 and 2020. This cohort included 11,433 serous carcinomas of which 85.8% were tubo-ovarian high-grade serous carcinomas, as well as 903 ovarian clear cell carcinomas and 570 ovarian endometrioid carcinomas, and in which FGFR1/2/3 mutational analysis was performed.
Genomic profiling
Comprehensive NGS-based genomic profiling was performed on hybridization-captured, adapter ligation–based libraries using DNA extracted from formalin-fixed paraffin-embedded tumor in a CLIA-certified and CAP-accredited laboratory (Foundation Medicine, Inc.). All samples forwarded for DNA extraction contained a minimum of 20% tumor cells. The samples were assayed via next-generation sequencing for up to 324 cancer-related genes, plus select introns from genes frequently rearranged in cancer, for all classes of genomic alterations [10,11,12]. To help visualize the mutation sequencing data results, an oncoprint plot was generated with online tools of the cbio portal as described by Gao et al. [13] and Cerami et al. [14]. Tumor mutational burden (TMB) was determined on 0.79–1.14 Mb of sequenced DNA using a mutation burden estimation algorithm [15]. In this study, low TMB scores was defined as <10 mut/Mb, since score of at least 10mut/Mb is a currently FDA-approved companion diagnostic biomarker for immunotherapy [16]. Assessment of microsatellite instability was performed from DNA next-generation sequencing across 114 loci [15].
Genomic ancestry analysis
Predominant ancestry was assessed using a SNP-based approach [17]. Briefly, germline single nucleotide polymorphisms (SNPs) were characterized in the publicly available 1000 Genomes data and used to train and validate a classifier to bin individuals into one of five inferred population groups, estimated to be of predominantly African, European, Admixed American, South Asian, or East Asian ethnic origin.
Homologous recombination deficiency status
We used validated methods for detecting genome-wide loss of heterozygosity (gLOH) scores as surrogate marker for homologous recombination deficiency (HRD) and PARP inhibitor effectiveness in ovarian cancer [18, 19]. We applied a genome-wide LOH score of at least 16% as the cutoff for HRD; as this cutoff has been analytically validated, prospectively tested in the Rucaparib maintenance setting as previously published in the ARIEL3 clinical trial for ovarian cancer via the same assay used in this study (NCT01968213) [19].
Statistical analysis
Fisher’s exact test was used in the statistical analysis comparing the frequency of genomic alterations in the cohort of ovarian FGFR3-mutated malignant Brenner tumor versus FGFR3-rearranged ovarian non-Brenner carcinomas. Statistical significance was defined as p < 0.05.
Analysis of FGFR3 mutations in publicly available non-Brenner ovarian carcinoma genomic datasets
Publicly available molecular data from the ovarian carcinoma TCGA study (n = 606) [20] and from the ovarian cancer samples from the MSK IMPACT 2017 Nature Medicine study (n = 224) [21] was analyzed with respect to FGFR3 alterations via the cBio Cancer Genomics Portal (http://cbioportal.org; Memorial Sloan Kettering Cancer Center, New York, NY) [13, 14]. The cohorts were queried for FGFR3 alterations in the cbio portal by using the advanced onco query language with respect to FGFR3 [13, 14]. Online analysis was performed between January 1st, 2020 and July 31st, 2020.
Results
Clinical and histopathological characteristics of malignant brenner tumor cohort
We performed a retrospective analysis of 14,153 consecutive ovarian carcinomas submitted for comprehensive genomic profiling (CGP) during the course of routine clinical care (2010–2020) and identified 11 unique malignant Brenner tumors cases. The median patient age was 63 years, range 41–74 (Table 1). Primary ovarian tumor size (retrieved from pathology reports) ranged from 5.1 to 30 cm with a median size of 13 cm. In our malignant Brenner tumor cohort, most tumors were aggressive, with either reported recurrences or metastasis beyond pelvis, and therefore clinically advanced, predominantly stage II or higher (Table 1). At the time of genomic profiling, there was no evidence of a primary urothelial carcinoma of the urinary tract in any case.
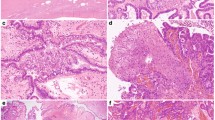
All malignant Brenner tumors displayed morphological features of urothelial carcinoma of the bladder and urinary tract, often with squamous differentiation (Fig. 1). Morphological features of the 11 malignant Brenner tumor cases are depicted in Table 2. Six of seven cases from primary tumor resections, exhibited either a benign or borderline Brenner tumor pre-cursor component (Table 1). Four cases were recurrences in which benign or borderline Brenner tumor components could not be assessed, but the patient had a reported history of malignant Brenner tumor (Table 1). Case #8 (Table 1) showed a pre-cursor component as well as a malignant Brenner tumor with areas transitioning to small-cell neuroendocrine carcinoma (Fig. 1). Cases exhibiting transitional-like features that were deemed to be variants of high-grade serous or endometrioid carcinomas were excluded from the final 11 malignant Brenner tumor cohort.

A Pre-cursor borderline Brenner tumor in a patient with FGFR3-mutated malignant Brenner tumor, exhibiting papillary architecture, pink cytoplasm, low-grade nuclear atypia and no invasion. B Different area of the tumor from (A) with stromal invasion and squamous differentiation consistent with malignant Brenner tumor. C Lymph node metastasis of a FGFR3-mutated malignant Brenner tumor resembling urothelial carcinoma. D MDM2-amplified malignant Brenner tumor. TP53 and RB1-mutated malignant Brenner tumor (E) and other areas with transformation to small-cell neuroendocrine carcinoma (F).
Genomic landscape of malignant Brenner tumor
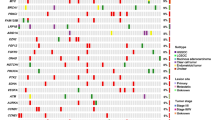
Genomic profiling revealed the most frequently altered genes in malignant Brenner tumors were CDKN2A/B, which were inactivated via homozygous deletion in 55% (6 of 11) of cases (Fig. 2), followed by activating, oncogenic FGFR3 alterations in 45.5% (5 of 11) of cases (Fig. 2). Specifically, 36% (4 of 11) of cases had a FGFR3 S249C missense mutation and 9% (1 of 11) had a FGFR3-TACC3 fusion (Fig. 2). The FGFR3 rearrangement identified in this tumor is predicted to result in a fusion including the N-terminal portion of FGFR3 (exons 1-17), which retains the full kinase domain (Fig. 3). When evaluable, all FGFR3-mutated malignant Brenner tumors had an associated benign or borderline Brenner tumor pre-cursor component (cases #1–4, Table 1, and Fig. 1), thus confirming the diagnosis and the ovarian site of origin. A benign or borderline precursor component could not be evaluated in one FGFR3-mutated case since it was a metastatic recurrence (Case #5, Table 1). One additional FGFR3 wild-type case (9%, 1 of 11) harbored an activating FGFR1 alteration, NM_023110:c.448 + 1 G > A_p.splice site 448 + 1 G > A (Case #6, Table 1). Therefore, overall genomic alterations leading to activation of the FGFR pathway occurred in 55% (6 of 11) of malignant Brenner tumor cases.

PIK3CA alterations co-occurred with FGFR3, while MDM2/TP53 alterations occurred in FGFR3 wild-type cases. Purple rectangle: gene rearrangement. Blue rectangle: homozygous deletion; red rectangle: amplification; black square: truncating mutation; Green square: oncogenic missense mutation.

A Schematic diagram showing the protein domains of FGFR3, and recurrent activating S249C short variant mutation identified in malignant Brenner tumors. B Schematic diagrams demonstrating activating DNA fusions involving FGFR3 identified in a malignant Brenner tumor.
Composite biomarker analysis revealed that malignant Brenner tumors were microsatellite stable (11 of 11) and exhibited low TMB (10 of 11), which are two well-established biomarkers for immunotherapy. Mean and median TMB were 3.6 and 2.5 mut/Mb, respectively. Case #1 (Table 1) exhibited 10.5 mut/Mb, while all other cases had scores of <10 mut/Mb. The FDA-approved CDx indication via same assay used in this study for recommending pembrolizumab for the treatment of solid tumor patients is TMB greater or equal to 10 mut/Mb [16].
Malignant Brenner tumors exhibited no evidence of homologous recombination deficiency (HRD), which was assessed by genome-wide loss of heterozygosity score (gLOH), a biomarker for HRD and PARP inhibitor therapy. gLOH scores >15% using same assay in this study are associated with improved progression-free survival (PFS) from Rubraca (rucaparib) maintenance therapy, based on the ARIEL3 clinical trial for ovarian cancer [19], previously leading to the FDA approval of high gLOH scores (>15%) in ovarian carcinomas for PARP inhibitor therapy. In 5 cases in which gLOH could be evaluated, median gLOH score was 1%, and no malignant Brenner tumor harbored gLOH >15%. In addition, genomic alterations of HRD genes were predominantly absent in our cohort, including in BRCA1, BRCA2, BARD1, ATM, ATR, BRIP1, CHEK1, CHEK2, CDK12, FANCA, FANCC, FANCG, FANCL, MRE11A, PALB2, RAD51B/C/D, RAD52, RAD54L. Two of five (40%) FGFR3-mutated malignant Brenner tumors contained homozygous inactivating BAP1 alterations (Fig. 2); however, gLOH scores for both cases were <15%, at 0% and 1%, respectively.
Activating PIK3CA mutations (E545K or C420R) co-occurred with FGFR3 alterations in 3 of 5 (60%) malignant Brenner tumors (Fig. 2), while FGFR3 wild-type cases frequently exhibited alterations in MDM2 via amplification or inactivating TP53 mutations (R213*, R110P, R273H) in 5 of 6 (83%) cases (Fig. 2). No case featured more than on PIK3CA mutation. Patients with FGFR3-mutated malignant Brenner tumors were older (mean age 67.4 years), compared with patients with FGFR3 wild-type cases (mean age 52.8 years, p = 0.0148 via student t test). FGFR3-mutated malignant Brenner tumors tended to have higher frequency of lower-grade malignant components often with squamous differentiation compared with FGFR3-wild-type tumors (Table 2). Limited outcome and survival data are available for the malignant Brenner tumor patient cohort (Table 2).
One FGFR3 wild-type malignant Brenner tumor (case #8, Table 1), which also showed a de-differentiated small-cell neuroendocrine carcinoma component, exhibited TP53 (R213*, R110P) and RB1 (132 fs*4) inactivating alterations consistent with the diagnosis, reminiscent of urothelial cancers with small-cell variant histology [22]. The presence of a benign Brenner tumor pre-cursor component in this case supports an ovarian origin. Overall distinct genomic profiles and clinical features indicate that FGFR3 and MDM2/TP53 are two different pathways for malignant Brenner tumor pathogenesis, and, together, they molecularly stratify 91% of cases, also reminiscent of bladder urothelial carcinoma pathogenesis [23].
Ancestral analysis of malignant Brenner tumor cohort
Genomic ancestry analysis revealed that 4 of 5 (80%) FGRF3-mutated malignant Brenner tumor patients were from European descent, while 1 of 5 (20%) FGFR3-mutated patient was of admixed American descent. In contrast, 2 of 5 (40%) patients with FGFR3 wild-type malignant Brenner tumors were of European descent, 2 of 5 (40%) were of admixed American descent, while 1 of 5 (20%) was of African descent.
Analysis of FGFR3 S249C and FGFR3-fusions in 14,142 non-Brenner ovarian carcinoma subtypes and publicly available non-Brenner ovarian carcinoma genomic datasets
Within our internal ovarian carcinoma genomic database of cases that were previously molecularly profiled during the course of clinical care at Foundation Medicine, FGFR3 S249C was not detected in 14,142 non-Brenner ovarian carcinomas. We validated the specificity of these internal results with publicly available genomic datasets of ovarian carcinomas. Activating FGFR3 short variant alterations, including FGFR3 S249C, was not identified in 606 samples from 594 patients with ovarian serous carcinomas from the ovarian carcinoma Cancer Genome Atlas (TCGA, Firehose Legacy, accessed via the cbio portal) [20]. In contrast, a FGFR3 fusion, FGFR3-MLLT10, was identified in 1 of 585 (0.02%) TGCA ovarian serous carcinoma samples (TCGA, PanCancer Atlas). In another independent, publicly available cohort of non-Brenner ovarian carcinomas, FGFR3 short variant alterations and fusions were not identified in 224 non-Brenner ovarian cancer samples from 216 patients from the MSKCC Nature Medicine 2017 study [21] (accessed via the cbio portal). The results from our cohort and external validation cohorts suggest that FGFR3 S249C may be specific to malignant Brenner tumors.
FGFR3 fusions in malignant Brenner tumor and other ovarian carcinoma subtypes
To determine whether FGFR3 fusions were specific to malignant Brenner tumors, we interrogated our 14,142 internal non-Brenner ovarian carcinomas for FGFR3 fusions and rearrangements. At a similar rate to the ovarian carcinoma TCGA cohort, we identified FGFR3 fusions or rearrangements in 0.01% (10 of 14,142) non-Brenner ovarian carcinoma subtypes (5 high-grade serous, 3 clear cell carcinoma, and 2 high-grade endometrioid adenocarcinomas). In this FGFR3-rearranged, non-Brenner ovarian carcinoma cohort, mean and median patient age were 62 and 61 years, respectively. We identified known activating FGFR3 fusions such as FGFR3-TACC3 (n = 5), internal FGFR3 rearrangements (n = 2) involving intron 17 or exon 18, as well as novel rearrangements (n = 3) FGFR3-ING1, FGFR3-LETM1, and FGFR3-LARGE. In contrast to FGFR3-mutated malignant Brenner tumor, FGFR3-rearranged non-Brenner ovarian carcinomas exhibited a high frequency of TP53 mutations (80%, p = 0.09), low frequency of CDKN2A/2B loss (10%, p = 0.08) and lacked PIK3CA mutations (0%, p = 0.02, Fisher exact test). Although FGFR3 fusions may occur in non-Brenner ovarian carcinoma subtypes, our results suggest that FGFR3-rearranged, non-Brenner ovarian carcinomas exhibit different genomic profiles compared to FGFR3-mutated ovarian malignant Brenner tumors.
FGFR1/2/3 landscape in non-Brenner ovarian carcinoma (serous, endometrioid, and clear cell) subtypes
Finally, we assessed the extent of activating FGFR1/2/3 genomic alterations in the most frequent non-Brenner ovarian carcinoma subtypes (i.e. serous, clear cell, and endometrioid types). There were 11,433 ovarian serous-type carcinomas in the internal Foundation Medicine dataset, of which 85.8% were tubo-ovarian high-grade serous carcinomas. 5% of ovarian serous carcinomas (593 of 11,433) harbored any activating FGFR1/2/3 alteration, most of which were FGFR1 amplification (2.7%) (Fig. 4). FGFR3 and FGFR2 amplification occurred less frequently, at 0.9% and 0.7%, respectively. FGFR1/2/3 short variant (0.8%) and fusions/rearrangements (0.3%) were also infrequent. Most recurrent short variant alterations included FGFR2 S252W (0.3%), FGFR2 N549K (0.01%), FGFR1 S546K (0.01%), FGFR1 T141R ( < 0.01%), FGFR3 A500T (<0.01%), FGFR3 D785fs*31 (<0.01%). Rare recurrent FGFR fusions/rearrangements occurring at <0.01% each included FGFR2 internal rearrangement, FGFR2-ATE1 fusion, FGFR1-TACC1, and FGFR3-TACC3. Notably, other gene alterations that co-occurred with activating FGFR1/2/3 alterations in ovarian serous-type carcinomas included inactivating TP53 mutations (94%), NSD3 amplification (48%), CCNE1 amplification (26%), MYC amplification (21%) and NOTCH3 amplification (9%) (Supplementary Fig. 1), consistent with profiles of aggressive tubo-ovarian high-grade serous carcinomas [20, 24, 25].

Frequency of FGFR1/2/3 alterations in non-Brenner ovarian carcinoma serous (A), clear cell (B), and endometrioid (C) subtypes with relative proportion of alterations of specific FGFR gene (right). Some cases had more than one alteration, thus the total of the right chart may be more than 100%.
Among 903 cases of ovarian clear cell carcinoma with comprehensive genomic profiling data available at Foundation Medicine, 4.5% (41 of 903) harbored activating FGFR1/2/3 alterations, most of which occurred in FGFR2 (2.4%) (Fig. 4) via FGFR2 short variant alterations S252W (1.3%) and P253 (0.5%). Other recurrent FGFR alterations in ovarian clear cell carcinoma included FGFR1 amplification (0.6%), FGFR3 amplification (0.5%), FGFR3-TACC3 fusion (0.2%). FGFR-mutated ovarian clear cell carcinoma was characterized by frequent co-alterations in PIK3CA (59%), ARID1A (51%), TERT (33%), TP53 (22%), PPP2RA1 (15%), and ARID1B (14%) (Supplementary Fig. 1), compatible with other known drivers of ovarian clear cell carcinoma tumorigenesis [26, 27].
In the internal Foundation Medicine dataset, there were 570 endometrioid-type ovarian carcinomas. Activating FGFR1/2/3 alterations occurred in 6.6% (38 of 570), and the majority were in FGFR2 (4.3%, 25 of 570) (Fig. 4), via recurrent short variant alterations in FGFR2 S252W (1.9%), FGFR2 N549K (1.2%), FGFR2 R664W (0.5%), FGFR2 amplification (0.3%) and internal FGFR2 rearrangement (0.3%). FGFR1 alterations occurred in 1.5% of ovarian endometrioid carcinomas with recurrent FGFR1 amplification in 1% of cases. FGFR3 alterations occurred in 0.7% of via recurrent FGFR3 amplification or FGFR3-TACC3 fusion. Frequently co-altered genes in FGFR-mutated ovarian endometrioid carcinomas were PTEN (58%), PIK3CA (58%), ARID1A (58%), TP53 (40%), PIK3R1 (18%), ATM (16%), CCND1 (16%), and CTNNB1 (16%) (Supplementary Fig. 1). Taken together, our results demonstrate an overall distinct FGFR1/2/3 mutational landscape and genomic signatures of malignant Brenner tumors compared to different FGFR-mutated ovarian serous, endometrioid, and clear cell carcinoma subtypes.
Discussion
We queried a unique database of 14,153 ovarian carcinomas that had undergone comprehensive genomic profiling during the course of routine clinical care to explore the genomic landscape of malignant Brenner tumors of the ovary. Malignant Brenner tumors were exceedingly rare and represented 0.1% of 14,153 clinically advanced primary ovarian carcinomas submitted for genomic profiling between 2010 and 2020. The histogenesis of Brenner tumors has classically been linked to Walthard cell rests [28], which are benign clusters of epithelial cells that resemble the urothelium of the urinary tract, and which can be found at the serosa of fallopian tubes, mesovarium, and ovarian hilum (Fig. 5). An alternative cell of origin for this tumor has been the ovarian surface epithelium and the underlying stroma through transitional/urothelial cell metaplasia [29]. Brenner tumors may be classified as benign, “atypical proliferative” of borderline malignancy or frankly malignant (Fig. 5). Based on morphological criteria and molecular data, benign and borderline Brenner tumors may be considered pre-cursors to their malignant counterpart in the stepwise progression of this ovarian carcinoma subtype.

H&E images were taken from our study cohort. Activating PIK3CA mutations and CDKN2A loss may be early events based on prior studies. FGFR3 and MDM2/TP53 alterations indicate two different pathways for malignant Brenner tumor pathogenesis and are likely late events. Together, alterations in either FGFR3 or MDM2/TP53 molecularly stratify 91% of cases, reminiscent of molecular pathways in bladder urothelial carcinoma development.
In this study, homozygous deletion of CDKN2A and activating FGFR3 alterations were the most frequent genomic alterations in malignant Brenner tumors. Prior literature has demonstrated that one malignant Brenner tumor case harboring CDKN2A loss and MDM2 amplification has been previously shown to overexpress MDM2 and lose p16 (encoded by CDKN2A) proteins by immunohistochemistry, thus supporting the validity of our genomic results [30]. In this prior case, p16 was also lost in the borderline Brenner tumor component [30]. In addition, another prior study has previously demonstrated CDKN2A homozygous deletion in 7 borderline Brenner tumors but not in 13 benign Brenner tumors [31]. These results suggest that homozygous deletion of CDKN2A occurs early in the pathogenesis of malignant Brenner tumors, likely in the transformation of benign Brenner tumors to tumors of borderline malignancy (Fig. 5).
Similarly, PIK3CA mutations may occur early in the progression of benign Brenner tumor to borderline Brenner tumors. Activating PIK3CA mutations have been detected in the epithelial component of 2 of 7 (29%) of borderline Brenner tumors but not in benign Brenner tumors [31]. These results have been corroborated by another study in which PIK3CA mutations were not identified in 3 benign Brenner tumors, but instead were present in 1 recurrent borderline Brenner tumor and 1 malignant Brenner tumor [32]. The results of these two prior studies suggest that activation of the PI3K/AKT pathway occurs early in the transformation of benign Brenner tumors (Fig. 5). In our study, activating PIK3CA alterations were present in 27% of malignant Brenner tumors and they co-occurred with FGFR3 mutations.
Our results suggest two alternative genetic pathways in the pathogenesis of malignant Brenner tumors of the ovary via alterations in either 1) FGFR3 or 2) MDM2/TP53 (Fig. 5). From results of prior studies, these alternative pathways may occur late in the progression of borderline Brenner tumors to frankly malignant Brenner tumors. For instance, absence of FGFR3 mutations has been previously reported in 21 Brenner tumors of borderline malignancy [33]. In a malignant Brenner tumor case report harboring MDM2 amplification, MDM2 protein was overexpressed by immunohistochemistry in the malignant component, but it was negative in the benign and borderline pre-cursor components as well as in 5 additional benign Brenner tumors [30]. In a different study, MDM2 amplification has also been reported in 3 of the 4 malignant Brenner tumor cases, but it was negative in 17 benign and 2 borderline Brenner tumors, respectively [34]. Similarly, TP53 mutations have not been previously identified in benign or borderline Brenner tumors [34, 35]. MDM2 and TP53 are within the same genomic and signaling axis, in which MDM2 targets p53 for ubiquitin-mediated proteasomal degradation, thereby resulting in p53 loss of function [36].
Our results together with previously published studies suggest a genetic model of malignant Brenner tumor pathogenesis in which CDKN2A loss and PIK3CA mutations may occur early in the transformation of benign Brenner tumors to atypical proliferative tumors of borderline malignancy (Fig. 5). In the stepwise progression from borderline to malignant Brenner tumors, we propose that, similarly to bladder urothelial carcinoma [23], FGFR3 or MDM2/TP53 may be two alternative genomic pathways in the pathogenesis of malignant Brenner tumors (Fig. 5). Overall, microsatellite instability, high TMB, or homologous recombination deficiency do not appear to play a role in the molecular pathogenesis of malignant Brenner tumors of the ovary (Fig. 5).
Our proposed molecular model is reminiscent of bladder urothelial carcinoma, in which FGFR3 and TP53 have been reported to be the most frequently mutated genes in bladder cancer, and urothelial carcinomas may develop through at least two molecular pathways, one related to FGFR3, typically in less invasive tumors, and one related to TP53, characterized by higher grade, invasive tumors [23, 37]. One point in contrast with malignant Brenner tumors is the well-known association of bladder urothelial carcinoma with cigarette smoking, which is linked to significantly higher TMB and corresponding responsiveness to immunotherapy and FDA-approved immune checkpoint inhibitors for advanced bladder cancer patients.
Our data shed insights into the potential value of targeted therapies in refractory, FGFR3-mutated malignant Brenner tumors, a tumor that currently presents a therapeutic conundrum due to its rarity. FGFR3 (fibroblast growth factor receptor 3) encodes a targetable receptor tyrosine kinase that typically promotes cell cycle progression and angiogenesis via activation of downstream signaling pathways, including RAS-MAPK and AKT [38,39,40]. Anti-FGFR inhibitors, such as erdafitinib, are currently approved by the FDA for the treatment of metastatic urothelial carcinoma with FGFR2 or FGFR3 alterations that have progressed after chemotherapy, based on the clinical trial BLC2001 (NCT02365597) [7]. FGFR3 mutation and amplification have been reported in 26–59% and 18% of bladder urothelial carcinoma cases, respectively [5, 41, 42]. FGFR3 S249C has been reported to be the most frequent mutation in urothelial tumors, with similar incidences of 62% and 58% in bladder tumors and upper urothelial tract tumors, respectively [41]. Based on morphological and now molecular resemblance of malignant Brenner tumors of the ovary to bladder urothelial carcinoma, our data suggests that FGFR3-mutated malignant Brenner tumors may also be sensitive to FGFR inhibitors. Patients with FGFR3-mutated malignant Brenner tumors may be eligible for clinical trials, such as the MATCH Screening Trial (NCT02465060), in which ovarian cancer patients with FGFR alterations may be eligible for erdafitinib. In addition, anti-FGFR targeted therapy with the drug pemigatinib has also been approved by the FDA for intrahepatic cholangiocarcinomas that are driven by FGFR2 fusions [8].
Other genomic alterations in malignant Brenner tumors for which FDA-approved therapies are available include CDKN2A and PIK3CA. Recently, a PIK3CA-specific inhibitor, alpelisib, has been approved by the FDA for PIK3CA-mutated, hormone receptor-positive, advanced breast cancers [43], suggesting that Alpelisib may be effective in PIK3CA/FGFR3 co-mutated malignant Brenner tumors. In this regard, our study suggests combination treatment with both PIK3CA and FGFR inhibitors should be further investigated in FGFR-mutated cases, as well as in FGFR-altered ovarian clear cell and endometrioid carcinomas. In addition, high frequency of homozygous deletion of CDKN2A, which encodes p16 (a CDK4/6 inhibitor), suggests that malignant Brenner tumors may be sensitive to CDK4/6 inhibition. The CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib are FDA-approved for the treatment of hormone receptor-positive and Her2-negative breast cancer [44, 45], and CDK4 inhibitors have previously shown to be effective in case reports of tumors with CDKN2A loss [46, 47]. Lastly, although in early phase, emerging MDM2 inhibitors, such as Idasanutlin, may also be investigated in MDM2-amplified malignant Brenner tumor patients [36].
In conclusion, here we reveal insights into the genomic landscape of malignant Brenner tumors of the ovary and propose a model of molecular pathogenesis, in which FGFR activation, in particular via FGFR3 S249C mutation, is a driver in the tumorigenesis of at least 50% of malignant Brenner tumors. A weakness of this study is lack of long-term follow-up to assess survival differences in FGFR3-mutated versus wild-type cases. In addition, we have not directly micro-dissected and sequenced pre-cursor components to definitively assess early genomic events. However, these potential weaknesses are also opportunities for future studies. Our proposed model is based on data from our study and analysis of previous literature in which a limited number of gene mutations were assessed in benign and borderline Brenner tumors. Given the overall morphological and molecular similarities to urothelial carcinoma, including recurrent FGFR3 S249C mutation and FGFR3-TACC3 fusion, our data emphasizes the potential value of FDA-approved, anti-FGFR inhibitors such as erdafitinib and pemigatinib, in refractory, FGFR3-mutated malignant Brenner tumors. Our data demonstrate the usefulness of comprehensive genomic profiling in characterizing this rare subtype of ovarian carcinomas, as correct identification of malignant Brenner tumors may have important therapeutic implications. Finally, we provide a key resource to guide future clinical investigations on the utility of anti-FGFR inhibitors as single agents or in combination strategies for the treatment of malignant Brenner tumors as well as of a subset (~5%) of FGFR-mutated ovarian serous, clear cell and endometrioid carcinomas.
References
Kurman RJ, Carcangiu ML, Herrington CS, Young RH. WHO classification of tumours of female reproductive organs. 4th edition; 2014;35–7.
Austin RM, Norris HJ. Malignant Brenner tumor and transitional cell carcinoma of the ovary: a comparison. Int J Gynecol Pathol. 1987;6:29–39.
Eichhorn JH, Young RH. Transitional cell carcinoma of the ovary: a morphologic study of 100 cases with emphasis on differential diagnosis. Am J Surg Pathol. 2004;28:453–63.
Zhang Y, Staley SA, Tucker K, Clark LH. Malignant Brenner tumor of the ovary: Case series and review of treatment strategies. Gynecol Oncol Rep. 2019;28:29–32.
Gust KM, McConkey DJ, Awrey S, Hegarty PK, Qing J, Bondaruk J, et al. Fibroblast growth factor receptor 3 is a rational therapeutic target in bladder cancer. Mol Cancer Ther. 2013;12:1245–54.
Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun. 2015;6. https://doi.org/10.1038/ncomms7087.
Loriot Y, Necchi A, Park SH, Garcias-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl J Med. 2019;381:338–48.
Hoy SM. Pemigatinib: first approval. Drugs. 2020;80:923–9.
Ritterhouse LL, Nowak JA, Strickland KC, Garcia EP, Jia J, Lindeman NI, et al. Morphologic correlates of molecular alterations in extrauterine Müllerian carcinomas. Mod Pathol. 2016;29:893–903.
Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–4.
He J, Abdel-Wahab O, Nahas MK, Wang K, Rampal R, Intlekofer AM, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–14.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31.
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Cerami E, Gao J, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2012;2:401–4.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
FDA approves pembrolizumab for adults and children with TMB-H solid tumors. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors.
Connelly CF, Carrot-Zhang J, Stephens PJ, Frampton GM Abstract 1227: Somatic genome alterations in cancer as compared to inferred patient ancestry. In: Cancer research. American Association for Cancer Research (AACR); 2018. pp. 1227–1227.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–61.
Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
Hoffman-Censits J, Choi W, Pal S, Trabulsi E, Kelly WK, Hahn NM, et al. Urothelial cancers with small cell variant histology have confirmed high tumor mutational burden, frequent TP53 and RB mutations, and a unique gene expression profile. Eur Urol Oncol. 2020;19:30168–3.
BWG VanRhijn, Van Der Kwast TH, Vis AN, Kirkels WJ, Boeve ER, Jobsis AC, et al. FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Res. 2004;64:1911–4.
Jones DH, Lin DI. Amplification of the NSD3‑BRD4‑CHD8 pathway in pelvic high‑grade serous carcinomas of tubo‑ovarian and endometrial origin. Mol Clin Oncol. 2017;7:301–7.
Goundiam O, Gestraud P, Popova T, De la Motte Rouge T, Fourchotte V, Gentien D, et al. Histo-genomic stratification reveals the frequent amplification/overexpression of CCNE1 and BRD4 genes in non-BRCAness high grade ovarian carcinoma. Int J Cancer. 2015;2015:1890–900.
Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol. 2012;25:615–24.
Wu RC, Ayhan A, Maeda D, Kim KR, Clarke BA, Shaw P, et al. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J Pathol. 2014;232:473–81.
Roma AA, Masand RP. Ovarian Brenner tumors and Walthard nests: a histologic and immunohistochemical study. Hum Pathol. 2014;45:2417–22.
Logani S, Oliva E, Amin MB, Folpe AL, Cohen C, Young RH. Immunoprofile of ovarian tumors with putative transitional cell (urothelial) differentiation using novel urothelial markers: histogenetic and diagnostic implications. Am J Surg Pathol. 2003;27:1434–41.
Wang L, Allison D, Shukla S. Amplification of MDM2 and Loss of p16 expression: do they have a role in malignant transformation of ovarian brenner tumor? A morphologic and immunohistochemical study. Am J Clin Pathol. 2020;154:133–41.
Kuhn E, Ayhan A, Shih IM, Seidman JD, Kurman RJ. The pathogenesis of atypical proliferative Brenner tumor: An immunohistochemical and molecular genetic analysis. Mod Pathol. 2014;27:231–7.
Cuatrecasas M, Catasus L, Palacios J, Prat J. Transitional cell tumors of the ovary. Am J Surg Pathol. 2009;33:556–67.
Van Rhijn BWG, Montironi R, Zwarthoff EC, Jobsis AC, van der Kwast TH. Frequent FGFR3 mutations in urothelial papilloma. J Pathol. 2002;198:245–51.
Pfarr N, Darb-Esfahani S, Leichsenring J, Taube E, Boxberg M, Braicu I, et al. Mutational profiles of Brenner tumors show distinctive features uncoupling urothelial carcinomas and ovarian carcinoma with transitional cell histology. Genes Chromosom Cancer. 2017;56:758–66.
Simons M, Simmer F, Bulten J, Ligtenberg MJ, Hollema H, van Vliet S, et al. Two types of primary mucinous ovarian tumors can be distinguished based on their origin. Mod Pathol. 2020;33:722–33.
Li W, Peng X, Lang J, Xu C. Targeting mouse double minute 2: current concepts in DNA damage repair and therapeutic approaches in cancer. Front Pharmacol. 2020;11:631.
Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nat Rev Cancer. 2005;5:713–25.
Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–97.
Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–49.
Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–213.
van Oers JMM, Zwarthoff EC, Rehman I, Azzouzi AR, Cussenot O, Meuth M, et al. FGFR3 mutations indicate better survival in invasive upper urinary tract and bladder tumours. Eur Urol. 2009;55:650–8.
Dodurga Y, Tataroglu C, Kesen Z, Satiroglu-Tufan NL. Incidence of fibroblast growth factor receptor 3 gene (FGFR3) A248C, S249C, G372C, and T375C mutations in bladder cancer. Genet Mol Res. 2011;10:86–95.
André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA -mutated, hormone receptor–positive advanced breast cancer. N. Engl J Med. 2019;380:1929–40.
Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N Engl J Med. 2015;373:209–19.
Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N. Engl J Med. 2018;379:1926–36.
Elvin JA, Gay LM, Ort R, Shuluk J, Long J, Shelley L, et al. Clinical benefit in response to palbociclib treatment in refractory uterine leiomyosarcomas with a common CDKN2A alteration. Oncologist. 2017;22:416–21.
Tramontana TF, Marshall MS, Helvie AE, Schmitt MR, Ivanovich J, Carter JL, et al. Sustained complete response to palbociclib in a refractory pediatric sarcoma with BCOR - CCNB3 fusion and germline CDKN2B variant. JCO Precis Oncol. 2020;4:466–71.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors of the manuscript are employees of Foundation Medicine, Inc., which is a whole subsidiary of Roche.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lin, D.I., Killian, J.K., Venstrom, J.M. et al. Recurrent urothelial carcinoma-like FGFR3 genomic alterations in malignant Brenner tumors of the ovary. Mod Pathol 34, 983–993 (2021). https://doi.org/10.1038/s41379-020-00699-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00699-1
This article is cited by
-
Ultrasonographic and clinicopathologic features of benign Brenner tumors of the ovary*
Oncology and Translational Medicine (2022)
-
Genomic alterations in gynecological malignancies: histotype-associated driver mutations, molecular subtyping schemes, and tumorigenic mechanisms
Journal of Human Genetics (2021)
