
Abstract
DICER1 syndrome is a hereditary cancer predisposition syndrome caused by deleterious germline DICER1 mutations. Characteristic “hotspot” somatic mutations of DICER1 have been identified in DICER1-associated tumors. With the exception of genitourinary embryonal rhabdomyosarcoma and anaplastic sarcoma of the kidney, sarcomas are rarely reported in DICER1 syndrome. Herein, we report the clinical, histopathologic, and molecular findings of a germline DICER1-associated ovarian sarcoma in a 5-year-old female, a somatic DICER1-associated metastatic peritoneal sarcoma in a 16-year-old female, and a somatic DICER1-associated primary intracranial sarcoma in a 4-year-old male. A comprehensive review of the literature, including 83 DICER1-associated sarcomas, illustrates an unequivocal histologic pattern mimicking pleuropulmonary blastoma, regardless of the site of origin. The features include undifferentiated small round blue cells, poorly differentiated spindle cells, and large bizarre pleomorphic cells (anaplasia), often with rhabdomyoblastic and/or chondroid differentiation, and rare bone/osteoid formation. This unique heterogeneous histologic pattern should raise suspicion for pathogenic DICER1 mutation(s) warranting a detailed review of the family history and DICER1 mutation analysis. In addition to expanding the phenotypic spectrum of DICER1-associated conditions, identification of pathogenic DICER1 variants facilitates optimized genetic counseling, caregiver education and judicious imaging-based surveillance.
Similar content being viewed by others


Introduction
The DICER1 gene encodes an endoribonuclease in the ribonuclease (RNase) III family required for processing microRNA (miRNA). The miRNA regulates translation and degradation of messenger RNA, which plays an important role in gene expression. Dysregulation of miRNA by DICER1 mutations causes activation of oncogenes [1]. Characteristic “hotspot” somatic mutations (E1705, E1813, D1709, D1810, or G1809) at the four metal-binding sites of the RNase IIIb domain that affect the catalytic activity of DICER1 have been identified in DICER1-associated tumors [2]. In addition, DICER1 mosaicism may occur as a result of postzygotic acquisition of a de novo mutation and can result in two genetically distinct populations of cells. DICER1 syndrome is a hereditary cancer predisposition syndrome caused by deleterious germline mutations of the DICER1 gene (Online Mendelian Inheritance in Man number 601200). DICER1 syndrome is associated with various benign and malignant conditions, including lung cysts, pleuropulmonary blastoma, cystic nephroma, Wilms tumor, multinodular goiter, thyroid adenoma, differentiated thyroid cancer, juvenile-type intestinal polyps, ciliary body medulloepithelioma, nasal chondromesenchymal hamartoma, macrocephaly, pituitary blastoma, pineoblastoma, and ovarian tumors [2,3,4,5,6,7,8,9,10,11]. With the exception of genitourinary embryonal rhabdomyosarcoma of the bladder or uterine cervix and anaplastic sarcoma of the kidney, sarcomas are rarely reported in DICER1 syndrome [12,13,14,15,16,17,18,19,20,21,22,23,24].
Ovarian tumors associated with DICER1 syndrome are predominantly nonepithelial, sex cord-stromal tumors, and the majority of these are Sertoli–Leydig cell tumors [5, 25, 26]. The International Pleuropulmonary Blastoma Registry reported six patients or close relatives with an ovarian sex cord-stromal tumor (three Sertoli–Leydig cell tumors, one Sertoli cell tumor, one juvenile granulosa cell tumor, and one gynandroblastoma) among 325 children from 296 families with pleuropulmonary blastoma [5]. Heravi-Moussavi et al. demonstrated hotspot DICER1 somatic mutations in 30/102 (29%) nonepithelial ovarian tumors, including Sertoli–Leydig cell tumor (n = 26/43; 60%), yolk sac tumor (n = 2/15), juvenile granulosa cell tumor (n = 1/14), and mature teratoma (n = 1/8) [26]. Witkowski et al. performed a similar study and identified DICER1 somatic mutations in the RNase IIIb domain in 14/197 nonepithelial tumors, including 9/28 sex cord-stromal tumors, 5/118 gonadal germ cell tumors, and 0/43 extragonadal germ cell tumors [25].
While pleuropulmonary blastoma, rhabdomyosarcoma and nonrhabdomyosarcoma soft tissue sarcomas with central nervous system metastases have been previously reported, primary central nervous system sarcomas are exceedingly rare in the pediatric population. Central nervous system manifestations currently considered as components of DICER1 syndrome include pituitary blastoma, pineoblastoma, ciliary body medulloepithelioma, and macrocephaly [2]. Most recently, primary DICER1-associated central nervous system sarcoma and embryonal tumor with multilayered rosettes like infantile cerebellar tumor have also been recognized as central nervous system manifestations of DICER1 syndrome [27]. Other primary central nervous system tumors reported in pleuropulmonary blastoma kindreds include glioblastoma, meningeal sarcoma, primitive neuroectodermal tumor, and optic glioma [2].
In this report, we describe a germline DICER1-associated ovarian sarcoma in a 5-year-old female, a somatic DICER1-associated metastatic peritoneal sarcoma in a 16-year-old female, and a somatic DICER1-associated primary intracranial sarcoma in a 4-year-old male. Our aims in the present study are to expand the phenotypic spectrum of DICER1-associated sarcomas and to highlight the importance of detailed molecular profiling in determining the true association of a condition with DICER1 mutation.
Materials and methods
Clinical description
Following institutional review board approval, we compiled a list of patients who were diagnosed with a DICER1-associated sarcoma and treated at our institution. We then performed a retrospective analysis of their demographics, clinical presentation at diagnosis, relevant family history, laboratory, radiographic, histopathologic and molecular findings, treatment modality, and outcomes.
Histopathology and immunohistochemistry
Slide preparation was performed in our CLIA-certified laboratory in accordance with the institutional protocol. Immunohistochemistry for alpha fetoprotein, BCOR, BRG1, calretinin, CD10, CD34, CD99, carcinoembryonic antigen, CK (AE1/AE3), CK7, desmin, EMA, GFAP, inhibin A, INI-1, Ki67, myogenin, MyoD1, NKX2.2, Oligo2, SALL4, SMA, synaptophysin, S-100, TLE-1, and WT1 was performed according to the manufacturer’s instructions. The antibodies, clone numbers, vendors, and catalog numbers are summarized in Supplementary Table 1. Selected stains including SOX10 (Patient 1), and myogenin, desmin, and MyoD1 (Patient 2) were performed at other CLIA-certified laboratories. All positive and negative controls were appropriately stained. Anaplasia was defined according to the Wilms tumor criteria of marked nuclear enlargement (>3× of normal nuclear size), hyperchromatism, and atypical mitotic figures [28].
DNA and RNA extraction
For frozen tissue, DNA was extracted using the Qiagen Gentra Puregene Tissue Kit, and RNA was extracted with the Qiagen RNeasy Mini RNA Extraction Kit (Qiagen Inc., Valencia, CA). For formalin-fixed paraffin-embedded tissue, DNA was extracted using the Qiagen QIAamp® DNA formalin-fixed paraffin-embedded Tissue Kit, and RNA was extracted from the patient’s formalin-fixed paraffin-embedded tissue specimen using the FormaPure RNA kit (Beckman Coulter, Brea, CA). If available, DNA was extracted from the patient’s peripheral blood using the Qiagen Gentra Puregene Blood kit.
Sequencing and copy number analysis
Next-generation sequencing of DNA and RNA was performed with the OncoKids® panel, a comprehensive DNA- and RNA-based next-generation sequencing panel for pediatric malignancies, as previously described [29]. Chromosomal microarray analysis of tumor DNA was performed according to the manufacturer’s protocols (Affymetrix OncoScan formalin-fixed paraffin-embedded Assay) [30]. For Patient 1, targeted Sanger sequencing of DNA extracted from peripheral blood was performed using primers for exons 15 and 26 of the DICER1 gene. Sanger sequencing was performed using the BigDye Terminator v1.1 (Thermo Fisher Scientific, Waltham, Massachusetts) on an automated fluorescent sequencer (ABI 3730 Genetic Analyzer, Thermo Fisher Scientific).
Literature review
A PubMed literature search was performed using keywords “DICER1” and “sarcoma” through February 2019. Key articles were reviewed, and relevant information was abstracted to identify individuals with DICER1-associated sarcomas. All reviewed papers had their bibliographies searched manually to recognize further citations of interest and to ensure that no relevant articles were omitted. Data from these papers were aggregated and reviewed to broadly assess age at sarcoma diagnosis, gender, site and type of sarcoma, other co-existing conditions, key gross pathology, microscopic features, immunohistochemistry, and molecular pathology for each DICER1-associated sarcoma.
Results
Herein, we present three patients with DICER1-associated sarcomas treated at our institution.
Clinical description
Patient 1
A 5-year-old Caucasian female presented with fatigue, nausea, decreased appetite, diffuse abdominal pain, and decreased urine output. Review of systems was otherwise negative. Family history was significant for benign thyroid nodules and thyroidectomy in her mother, thyroid cancer in a paternal grandmother, and brain tumor at 9 years of age in a paternal great uncle who died of disease. Imaging revealed a 6 × 4 × 4 cm right adnexal mass complicated by right ovarian torsion with pre-operative rupture. Pre-operative tumor marker evaluation revealed a mildly elevated cancer antigen-125 (36 U/mL) and normal serum alpha fetoprotein, beta-human chorionic gonadotropin, and lactate dehydrogenase. She underwent an exploratory laparotomy with right salpingo-oophorectomy. Pathologic examination, prior to molecular testing, revealed a high-grade pleomorphic sarcoma. Peritoneal cytology showed no overt malignant cells. Intra- and post-operative staging work-up demonstrated no metastatic disease. The International Federation of Gynecology and Obstetrics staging was stage IC2. The patient received adjuvant chemotherapy and is alive without any evidence of disease 13-months off therapy.
Patient 2
A 16-year-old African American female presented with a 2-week history of nausea, vomiting, and worsening abdominal pain. Review of systems was otherwise negative. Imaging revealed an 8.8 × 6.6 × 9.3 cm pelvic mass, omental and peritoneal metastases, complex ascites, small pericardial and bilateral pleural effusions. Tumor marker evaluation revealed an elevated lactate dehydrogenase (1046 U/L) and cancer antigen-125 (464 U/mL), with normal serum alpha fetoprotein, beta-human chorionic gonadotropin, and carcinoembryonic antigen. Approximately 6 months prior to this presentation, the patient had undergone a right salpingo-oophorectomy and peritoneal mass resection at another institution. Pathology of both specimens was reportedly a mature cystic teratoma. Re-review of the original pathology material at our institution favored the diagnosis of an ovarian corpus luteum cyst for the oophorectomy specimen. Pathology of the peritoneal mass, prior to molecular testing, was consistent with an adenosarcoma with rhabdomyosarcomatous overgrowth. At the time of recurrence, her hospital course was complicated by abdominal compartment syndrome requiring intra-abdominal drainage; respiratory failure requiring intubation and mechanical ventilation; hypotension requiring pressor support; and oliguria requiring continuous veno-venous hemofiltration. Rapid clinical deterioration necessitated emergent adjuvant chemotherapy. On day 12 of hospitalization, she developed new-onset seizure activity. Due to her clinical instability, neuroimaging could not be performed. An electroencephalogram demonstrated minimal evidence of brain activity. A brain death examination demonstrated cessation of all functions of the brain, including the brainstem. Life support was withdrawn. The family consented to an autopsy in which a papillary thyroid microcarcinoma (0.6 cm) was incidentally noted.
Patient 3
A 4-year-old Hispanic male presented with a 5-day history of headache, vomiting, and refusal to bear weight. Review of systems was otherwise negative. Family history was notable for a maternal great aunt with a history of unknown cancer. At the time of his initial presentation, he was noted to have hypertension with bradycardia, bilaterally fixed, and dilated pupils, extensor posturing, and altered mental status. Imaging revealed a large left frontal mass with midline shift. Due to concern for herniation, he was intubated for mechanical ventilation and airway protection. An external ventricular drain was placed to relieve increased intracranial pressure, and an emergent left decompressive hemicraniectomy with gross total resection of the left frontal mass was performed. Pathologic examination, prior to molecular testing, revealed a primitive embryonal tumor with embryonal rhabdomyosarcomatous elements. Postoperatively, imaging demonstrated extensive intraparenchymal infarction. Life support was withdrawn on hospital day 5. Parents declined an autopsy.
Gross pathology
The gross features (Patients 1–3) are summarized in Table 1.
Microscopic features and immunohistochemistry
The microscopic features and immunohistochemistry (Patient 1–3) are summarized in Table 1 and Supplementary Table 2.
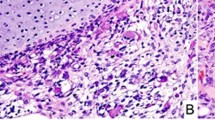
The hematoxylin and eosin sections from the ovarian sarcoma (Patient 1) revealed an ill-defined tumor arising in a hemorrhagic edematous ovarian tissue consistent with torsion. The tumor comprised undifferentiated small round blue cells, spindle cells, large bizarre pleomorphic cells (anaplasia), and islands of malignant cartilage. The undifferentiated small round blue cells (Fig. 1A1) and spindle cells had relatively monotonous, hyperchromatic nuclei (Fig. 1A2). The anaplasia was characterized by markedly enlarged (at least >3× larger than small round blue cells), hyperchromatic bizarre nuclei with irregular nuclear membranes (Fig. 1A2), and occaional prominent nucleoli. Multinucleation and mitotic figures, including atypical mitoses (Fig. 1A3 box), were often identified. The malignant cartilage (chondroid/chondrosarcomatous differentiation) contained pleomorphic, often multinucleated tumor cells within the lacunae (Fig. 1A4). There was also focal bone formation (Fig. 1A5).

Microphotographs of the DICER1-associated ovarian sarcoma (a Patient 1), peritoneal sarcoma (b Patient 2), and primary intracranial sarcoma (c Patient 3). Hematoxylin and eosin sections, ×40: A5; ×200: A1, A4, B1, B4, B5, B6, and B7; ×400: A2, A3, B2, B3, C1, C2, and C3; ×400, Immunohistochemistry, ×400: A6, A7, C4, C5, and C6. Patient 1 (A1–A7): The tumor was ill-defined and comprised a heterogeneous population of loosely cohesive tumor cells demonstrating areas with undifferentiated small round blue cells (A1, box: high power), spindle cells (A2), large, bizarre pleomorphic cells with frequent mitoses (anaplasia; A3, box: mitoses) and islands of malignant cartilage scattered throughout the tumor (A4). Focal bone formation was present (A5). Immunohistochemistry showed that the undifferentiated small round blue cells and spindle cells were partially, strongly positive for desmin (A6), and a smaller number of cells were positive for myogenin (A7). Patient 2 (B1–B7): Peritoneal mass. The sections from the peritoneal mass contained cystic walls in variable thicknesses (Fig. 1B1–B5), lined by bland ciliated columnar cells (B1). The underlying stroma was expanded (overgrown) and demonstrated a heterogeneous population including undifferentiated small round blue cells (B2), spindle cells (B3), and scattered pleomorphic cells (anaplasia, B2 box). The small round blue cells formed a hypercellular zone, “cambium layer” (B1), immediately beneath the epithelium. Loose myxoid stroma with occasional islands of immature-appearing cartilage (B4) and focal osteoid formation (B5) were identified in the background. Autopsy (post chemotherapy, B6 and B7). The tumor was largely necrotic, but single and clusters of viable tumor cells were present throughout. The tumor cells demonstrated prominent rhabdomyoblastic differentiation with abundant eosinophilic cytoplasm and “strap cells” (B6). Pleomorphic cells were occasionally seen. The tumor also contained multiple broad areas of chondrosarcomatous differentiation with abundant chondroid matrix and occasional pleomorphic cells within the lacunae, which were occasionally multinucleated (B7). Patient 3 (C1–C6): The hematoxylin and eosin sections revealed a cellular, heterogenous population of tumor cells with occasional necrosis demonstrating areas with undifferentiated small round blue cells (C1) with scattered large pleomorphic cells (C1 box) and spindle cells (C2). Some areas show abundant myxoid to chondroid matrix in the background (C3). Immunohistochemistry staining showed that tumor cells were partially strongly positive for desmin (C4), MyoD1 (C5) and myogenin (C6). The immunoprofile is supportive of rhabdomyoblastic differentiation
The peritoneal mass (Patient 2) contained cystic walls in variable thicknesses, lined by ciliated columnar cells (Fig. 1B1). The underlying stroma was overgrown and demonstrated a heterogeneous population of small round blue cells (Fig. 1B2), spindle cells (Fig. 1B3), and scattered pleomorphic cells (anaplasia, Fig. 1B2 box). The small round blue cells formed a hypercellular “cambium layer” (Fig. 1B1) beneath the epithelium. Occasional islands of immature-appearing cartilage (Fig. 1B4) and focal osteoid formation (Fig. 1B5) were also seen. The post-chemotherapy sarcoma (autopsy) was largely necrotic. The scattered viable tumor cells demonstrated prominent rhabdomyoblastic differentiation with abundant eosinophilic cytoplasm and “strap cells” (Fig. 1B6) and occasional pleomorphism. The tumor also showed extensive chondrosarcomatous differentiation similar to that seen in the sarcoma of Patient 1 (Fig. 1B7).
The intracranial sarcoma (Patient 3) revealed a hypercellular heterogeneous tumor with small round blue cells with scattered large pleomorphic cells (Fig. 1B1) and spindle cells (Fig. 1B2) with occasional necrosis. Areas with abundant chondromyxoid matrix were also seen (Fig. 1C3).
Immunohistochemistry results are summarized in Table 1. Desmin (Fig. 1A6, 1C4) and myogenin (Fig. 1A7, 1C6) positivity supported rhabdomyoblastic differentiation in all three patients. MyoD1 was positive in Patients 2 and 3 (Fig. 1C5). For Patient 1, Inhibin A and calretinin staining were performed on multiple sections and the consistently negative results ruled out Sertoli–Leydig cell tumor with heterologous sarcomatous growth from the differential.
Molecular pathology
Patient 1
Two DICER1 variants of strong clinical significance were identified in this patient’s tumor specimen; a DICER1 c.2223_2230del (p.Ser742Thrfs*16) frameshift inactivating mutation and a DICER1 c.5425G>A (p.Gly1809Arg) missense mutation; confirming the diagnosis of a DICER1-associated sarcoma. Sanger sequencing for these two DICER1 mutations performed on the peripheral blood revealed that the patient was heterozygous for the DICER1 c.2223_2230del (p.Ser742Thrfs*16) frameshift mutation. Chromosomal microarray was not performed.
Patient 2
Molecular testing was performed on the tumor specimen from autopsy. Two DICER1 variants of strong clinical significance were detected in the tumor at similar variant allele frequencies—a DICER1 c.3682C>T (p.Gln1228Ter) nonsense mutation and a hotspot DICER1 c.5428G>T (p.Asp1810Tyr) missense mutation; confirming the diagnosis of a DICER1-associated sarcoma. In addition, an activating missense mutation was identified in the NRAS proto-oncogene—NRAS c.181C>A (p.Gln61Lys). Chromosomal microarray and germline DICER1 targeted mutation testing were not performed.
Patient 3
The tumor specimen contained a DICER1 c.5439G>C (p.Glu1813Asp) frameshift inactivating mutation of strong clinical significance, with 97% variant allele frequency. In addition, variants of unknown clinical significance were observed in GLI2, IGF1R, and TSC2. Chromosomal microarray from this tumor specimen demonstrated a variety of copy number alterations and regional loss of heterozygosity. Notably, there was a region of loss of heterozygosity involving the distal short arm of chromosome 11, which is commonly observed in embryonal rhabdomyosarcoma. A key finding was a deletion of the distal long arm of chromosome 14 that includes the DICER1 gene. There were also five copies of part of chromosome 8, which has also been reported in DICER1-associated tumors. The molecular and cytogenetic findings affecting DICER1 may be the result of a heterozygous germline DICER1 c.5439 (p.Glu1813Asp) variant followed by somatic deletion affecting the wild-type DICER1 allele; highly suggestive of the diagnosis of a DICER1-associated sarcoma. A matched normal tissue or peripheral blood sample was not available for germline testing.
In summary, all three DICER1-associated sarcomas demonstrated a unique heterogeneous histologic pattern, including undifferentiated small round blue cells, poorly differentiated spindle cells, pleomorphic (anaplastic) cells, rhabdomyoblastic differentiation, and myxoid/chondroid or chondrosarcomatous differentiation, regardless of the site. Except for the presence of bone/osteoid formation identified in Patients 1 and 2, the tumor histology bears a striking resemblance to pleuropulmonary blastoma (Type II/III) of the lung. There was no histological or immunohistochemistry evidence of sex cord-stromal tumor or ectodermal/endodermal components suggestive of teratoma/germ cell tumor in Patients 1 and 2. Due to its variegated appearance, Patient 2 was initially misdiagnosed as mature cystic teratoma. These three cases highlight the diversity of extrapulmonary sites of DICER1-associated sarcomas and the importance of awareness of their common morphologic denominator to facilitate the diagnosis.
Discussion
Pathogenic germline DICER1 variants or characteristic somatic “hotspot” mutations are key mutational events in DICER1-associated tumorigenesis [1, 31]. Patients with germline or mosaic DICER1 gene mutations are at an increased risk of developing a range of childhood-, adolescent, and young adult-onset tumors, infrequently including sarcomas like genitourinary embryonal rhabdomyosarcoma of the bladder or uterine cervix and anaplastic sarcoma of the kidney [2, 31]. Our report includes a germline DICER1-associated ovarian sarcoma, a somatic DICER1-associated metastatic peritoneal sarcoma, and a somatic DICER1-associated primary intracranial sarcoma, expanding the phenotypic spectrum of DICER1-associated sarcomas. Although germline testing was not performed in two patients, the coexistence of a papillary thyroid microcarcinoma with a DICER1-associated metastatic peritoneal sarcoma in Patient 2, and the molecular and cytogenetic findings in Patient 3 were both highly suggestive of an underlying DICER1 syndrome.
Sarcomas are a rare group of histologically and genetically heterogeneous tumors of mesenchymal origin [32]. Since DICER1 screening began in 2009, at least 83 DICER1-associated sarcomas have been reported in the literature to date [3, 8, 12,13,14,15,16,17, 19,20,21,22,23,24, 26, 31, 33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49]. A comprehensive review of 86 DICER1-associated sarcomas, including our three patients, is summarized in Table 2 and Supplementary Table 2. Clinical and histopathologic characteristics of patients with DICER1-associated sarcomas are summarized in Table 2. The median age at sarcoma diagnosis was 12 years (range 0–92 years). The gender distribution of DICER1-associated sarcomas in the literature included 21 males and 62 females. Age at diagnosis and gender were not reported in 11 and 3 patients, respectively. Gross descriptions were available for 57/86 cases (Supplementary Table 2). The sizes of the sarcomas were reported in 25 cases and ranged from 2.3 to 20 cm in the greatest dimension. Many of the cervical embryonal rhabdomyosarcomas were described as “vaginal polyp.” Terms like “heterogeneous,” “cystic,” “multicystic/multiloculated,” “cystic degeneration (necrosis),” “solid,” and mixed “solid and cystic” were used to describe the masses. Microscopic features were available for 63/86 cases (Supplementary Table 2). Some cases had a simplified description, such as “high-grade.” Other series had a combined summary describing multiple cases together. Anaplasia (pleomorphism), rhabdomyoblastic differentiation, chondroid, chondromyxoid, myxoid or chondrosarcomatous differentiation was reported in many cases (Table 2). Bone/osteoid formation was identified in Patients 1 and 2 from current series, case 64 (anaplastic sarcoma of the kidney), and cases 56-62 (anaplastic sarcoma of the kidney). There were no confirmed associations reported between DICER1 mutation and alveolar rhabdomyosarcoma in the literature. Regardless of the sites, the sarcomas demonstrated a recurring, heterogeneous histologic appearance including undifferentiated small round blue cells, spindle cells and/or large anaplastic (pleomorphic) cells with/without rhabdomyoblastic, chondroid, myxoid, or chondrosarcomatous differentiation, and bone/osteoid formation.
The histologic patterns of DICER1-associated sarcomas in this report strongly resembles that of pleuropulmonary blastoma, more than 70% of which are associated with an underlying DICER1 mutation [2, 3, 50, 51]. Type I pleuropulmonary blastoma is composed of cystic lesions containing malignant cells. Type II pleuropulmonary blastoma contains cystic and solid areas and Type III pleuropulmonary blastoma is purely solid. Pleuropulmonary blastoma demonstrates a heterogeneous histologic pattern (Supplementary Fig. 1A–F), including areas with small round blue cells (blastemal; Supplementary Fig. 1A, B) and spindle cells (Supplementary Fig. 1C), rhabdomyoblastic differentiation (Supplementary Fig. 1D) with or without “cambium layer” formation (Supplementary Fig. 1A), and cartilaginous nodules with primitive to sarcomatous features (Supplementary Fig. 1E). Pleomorphic/anaplastic cells (Supplementary Fig. 1F) can be seen in any of the above components [2]. DICER1 sarcoma differs from pleuropulmonary blastoma, in that osteoid formation has yet to be described. The otherwise similar histologic pattern shared by pleuropulmonary blastoma and DICER1-associated sarcoma suggests analogous tumorigenesis affecting mesenchymal cell.
In our literature review (Supplementary Table 2), all patients had either somatic (n = 51), germline (n = 15), or both (n = 20) DICER1 mutations reported. Alterations in other genes (testing performed/reported = 49, testing not performed/not reported = 37) are summarized in Supplementary Table 2. In our review, frequently reported additional genomic alterations include TP53 (n = 22), KRAS (n = 11), NRAS (n = 5), and NF1 (n = 9) mutations (Supplementary Table 2). The reported histologic features were similar regardless of the type and number of additional genetic alterations. Exome sequencing of pleuropulmonary blastoma has previously demonstrated frequent biallelic loss of TP53 and some cases with mutations of NRAS or BRAF in addition to the mutation(s) in the DICER1 gene [52]. Copy number changes in critical genomic regions including PTEN, ATM, and WT1 are also common [52].
Identification of pathogenic somatic, germline, and mosaic mutations in the DICER1 gene is imperative to discern the true causality of DICER1-associated conditions. Recently Liu et al. reported a metachronous Type I pleuropulmonary blastoma and atypical choroid plexus papilloma in a 17-month-old male; raising a strong suspicion for an underlying germline DICER1 mutation or DICER1 mosaicism [53]. Subsequently, extensive molecular profiling demonstrated a somatic hotspot mutation in exon 25 (c.5438A>G, p.E1813G) and a frameshift mutation in exon 15 (c.2392_2393insA, p.T798Nfs*33) DICER1 mutation in the pleuropulmonary blastoma tumor tissue and none in the atypical choroid plexus papilloma tumor tissue or in normal lung tissue [54]. Whole exome sequencing demonstrated a pathogenic variant in c.263dupT, p.(I88*fs) in SBDS, which is unlikely to be a plausible cause for the atypical choroid plexus papilloma. Therefore, the authors concluded that in this patient atypical choroid plexus papilloma is not a phenotype of DICER1 syndrome.
In summary, this report of three DICER1-associated sarcomas and comprehensive literature review that included 83 other DICER1-associated sarcomas, revealed a recurring histologic pattern, regardless of the site of origin. The features include undifferentiated small round blue cells, poorly differentiated spindle cells and large, bizarre pleomorphic cells (anaplasia), often with rhabdomyoblastic, and/or chondroid/chondrosarcomatous differentiation that resembles pleuropulmonary blastoma. Bone/osteoid formation appears to be a novel finding in DICER1-associated sarcomas, not shared by pleuropulmonary blastoma. Given that DICER1 mutations underlie the genetic basis of only a small fraction of sarcomas, identification of this unique histologic pattern should raise the suspicion for the presence of a pathogenic DICER1 mutation(s), warranting a detailed review of the family history and DICER1 mutation analysis. Identification of pathogenic DICER1 variants would facilitate optimized genetic counseling and germline testing, individual and caregiver education, and judicious imaging-based surveillance. This will result in further elucidating the evolving features of DICER1 syndrome and expanding the spectrum of associated tumors.
References
Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–72.
Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, et al. DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res. 2018;24:2251–61.
Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965.
Bahubeshi A, Bal N, Frio TR, Hamel N, Pouchet C, Yilmaz A, et al. Germline DICER1 mutations and familial cystic nephroma. J Med Genet. 2010;47:863–6.
Schultz KAP, Pacheco MC, Yang J, Williams GM, Messinger Y, Hill DA, et al. Ovarian sex cord -stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol. 2011;122:246–50.
de Kock L, Sabbaghian N, Druker H, Weber E, Hamel N, Miller S, et al. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol. 2014;128:583–95.
de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014;128:111–22.
Frio TR, Bahubeshi A, Kanellopoulou C, Hamel N, Niedziela M, Sabbaghian N, et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli–Leydig cell tumors. JAMA. 2011;305:68–77.
Priest JR, Williams GM, Manera R, Jenkinson H, Bründler M-A, Davis S, et al. Ciliary body medulloepithelioma: four cases associated with pleuropulmonary blastoma—a report from the International Pleuropulmonary Blastoma Registry. Br J Ophthalmol. 2011;95:1001–5.
Stewart DR, Messinger Y, Williams GM, Yang J, Field A, Schultz KAP, et al. Nasal chondromesenchymal hamartomas arise secondary to germline and somatic mutations of DICER1 in the pleuropulmonary blastoma tumor predisposition disorder. Hum Genet. 2014;133:1443–50.
Wu MK, Sabbaghian N, Xu B, Addidou-Kalucki S, Bernard C, Zou D, et al. Biallelic DICER1 mutations occur in Wilms tumours. J Pathol. 2013;230:154–64.
Fremerey J, Balzer S, Brozou T, Schaper J, Borkhardt A, Kuhlen M. Embryonal rhabdomyosarcoma in a patient with a heterozygous frameshift variant in the DICER1 gene and additional manifestations of the DICER1 syndrome. Fam Cancer. 2017;16:401–5.
Chen J, Wang Y, McMonechy MK, Anglesio MS, Yang W, Senz J, et al. Recurrent DICER1 hotspot mutations in endometrial tumours and their impact on microRNA biogenesis. J Pathol. 2015;237:215–25.
Foulkes WD, Bahubeshi A, Hamel N, Pasini B, Asioli S, Baynam G, et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat. 2011;32:1381–4.
Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol. 2012;25:602–14.
Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. 2012;59:558–60.
Tomiak E, de Kock L, Grynspan D, Ramphal R, Foulkes WD. DICER1 mutations in an adolescent with cervical embryonal rhabdomyosarcoma (cERMS): DICER1 in cervical embryonal rhabdomyosarcoma. Pediatr Blood Cancer. 2014;61:568–9.
de Kock L, Boshari T, Martinelli F, Wojcik E, Niedziela M, Foulkes WD. Adult-onset cervical embryonal rhabdomyosarcoma and DICER1 mutations. J Low Genit Tract Dis. 2016;20:e8–e10. https://doi.org/10.1097/LGT.0000000000000149. Accessed 13 Mar 2018.
Cowan M, Suntum T, Olivas AD, Perpich M, Applebaum MA, Lastra RR, et al. Second primary rhabdomyosarcoma of the uterine cervix presenting with synchronous ovarian Sertoli–Leydig cell tumor: an illustrative case of DICER1 syndrome. Gynecol Oncol Rep. 2018;25:94–7.
Wu MK, Goudie C, Druker H, Thorner P, Traubici J, Grant R, et al. Evolution of renal cysts to anaplastic sarcoma of kidney in a child with DICER1 syndrome. Pediatr Blood Cancer. 2016;63:1272–5.
Wu MK, Cotter MB, Pears J, McDermott MB, Fabian MR, Foulkes WD, et al. Tumor progression in DICER1-mutated cystic nephroma—witnessing the genesis of anaplastic sarcoma of the kidney. Hum Pathol. 2016;53:114–20.
Wu MK, Vujanic GM, Fahiminiya S, Watanabe N, Thorner PS, O’Sullivan MJ, et al. Anaplastic sarcomas of the kidney are characterized by DICER1 mutations. Mod Pathol. 2018;31:169–78.
Yoshida M, Hamanoue S, Seki M, Tanaka M, Yoshida K, Goto H, et al. Metachronous anaplastic sarcoma of the kidney and thyroid follicular carcinoma as manifestations of DICER1 abnormalities. Hum Pathol. 2017;61:205–9.
Gomi K, Hamanoue S, Tanaka M, Matsumoto M, Kitagawa N, Niwa T, et al. Anaplastic sarcoma of the kidney with chromosomal abnormality: first report on cytogenetic findings. Hum Pathol. 2010;41:1495–9.
Witkowski L, Mattina J, Schönberger S, Murray MJ, Choong CS, Huntsman DG, et al. DICER1 hotspot mutations in non-epithelial gonadal tumours. Br J Cancer. 2013;109:2744–50.
Heravi-Moussavi A, Anglesio MS, Cheng S-WG, Senz J, Yang W, Prentice L, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2012;366:234–42.
de Kock L, Priest JR, Foulkes WD, Alexandrescu S. An update on the central nervous system manifestations of DICER1 syndrome. Acta Neuropathol. 2019. https://doi.org/10.1007/s00401-019-01997-y. Accessed 29 Apr 2019.
Moch H, Humphrey PA, Ulbright TM, Reuter VE. Tumours of the kidney. In: Humphrey PA, Reuter VE, editors. WHO classification of tumours of the urinary system and male genital organs. 4th ed. Lyon, France: International Agency for Research on Cancer; 2016. p. 49–53.
Hiemenz MC, Ostrow DG, Busse TM, Buckley J, Maglinte DT, Bootwalla M, et al. OncoKids: a comprehensive next-generation sequencing panel for pediatric malignancies. J Mol Diagn. 2018;20:765–76.
Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, et al. Cross-laboratory validation of the OncoScan® FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med Genom. 2015;8:5.
de Kock L, Foulkes WD. Sarcoma and germline DICER1 mutations. Lancet Oncol. 2016;17:e470.
Fletcher CDM. The evolving classification of soft tissue tumours - an update based on the new 2013 WHO classification. Histopathology. 2014;64:2–11.
de Kock L, Druker H, Weber E, Hamel N, Traubici J, Malkin D, et al. Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum Pathol. 2015;46:917–22.
Schultz KAP, Harris A, Messinger Y, Sencer S, Baldinger S, Dehner LP, et al. Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Fam Cancer. 2016;15:105–10.
Melendez-Zajgla J, Mercado-Celis GE, Gaytan-Cervantes J, Torres A, Gabiño NB, Zapata-Tarres M, et al. Genomics of a pediatric ovarian fibrosarcoma. Association with the DICER1 syndrome. Sci Rep. 2018;8:3252.
Moke DJ, Thomas SM, Hiemenz MC, Nael A, Wang K, Shillingford N, et al. Three synchronous malignancies in a patient with DICER1 syndrome. Eur J Cancer. 2018;93:140–3.
Hodgson A, Amemiya Y, Seth A, Djordjevic B, Parra-Herran C. High-grade Müllerian adenosarcoma. Am J Surg Pathol. 2017;41:1513–22.
Mullen MM, Divine LM, Hagemann IS, Babb S, Powell MA. Endometrial adenosarcoma in the setting of a germline DICER1 mutation: a case report. Gynecol Oncol Rep. 2017;20:121–4.
Piscuoglio S, Burke KA, Ng CK, Papanastasiou AD, Geyer FC, Macedo GS, et al. Uterine adenosarcomas are mesenchymal neoplasms. J Pathol. 2016;238:381–8.
Koelsche C, Mynarek M, Schrimpf D, Bertero L, Serrano J, Sahm F, et al. Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathol. 2018;136:327–37.
Bean GR, Anderson J, Sangoi AR, Krings G, Garg K. DICER1 mutations are frequent in müllerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol. 2018;32:280–9.
Chen KS, Stuart SH, Stroup EK, Shukla AS, Wang J, Rajaram V, et al. Distinct DICER1 hotspot mutations identify bilateral tumors as separate events. JCO Precis Oncol. 2018;2:1–9.
Yang J, Sarita-Reyes C, Kindelberger D, Zhao Q. A rare malignant thyroid carcinosarcoma with aggressive behavior and DICER1 gene mutation: a case report with literature review. Thyroid Res. 2018;11:11.
de Kock L, Terzic T, McCluggage WG, Stewart CJR, Shaw P, Foulkes WD, et al. DICER1 mutations are consistently present in moderately and poorly differentiated Sertoli–Leydig cell tumors. Am J Surg Pathol. 2017;41:1178–87.
Schrader KA, Cheng DT, Joseph V, Prasad M, Walsh M, Zehir A, et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016;2:104–11.
de Kock L, Geoffrion D, Rivera B, Wagener R, Sabbaghian N, Bens S, et al. Multiple DICER1-related tumors in a child with a large interstitial 14q32 deletion. Genes Chromosomes Cancer. 2018;57:223–30.
Lee JC, Villanueva-Meyer JE, Ferris SP, Sloan EA, Hofmann JW, Hattab EM, et al. Primary intracranial sarcomas with DICER1 mutation often contain prominent eosinophilic cytoplasmic globules and can occur in the setting of neurofibromatosis type 1. Acta Neuropathol. 2019. https://doi.org/10.1007/s00401-019-01960-x. Accessed 24 Jan 2019.
Fernández-Martínez L, Villegas JA, Santamaría Í, Pitiot AS, Alvarado MG, Fernández S, et al. Identification of somatic and germ-line DICER1 mutations in pleuropulmonary blastoma, cystic nephroma and rhabdomyosarcoma tumors within a DICER1 syndrome pedigree. BMC Cancer. 2017;17:146.
Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–46.
Priest JR, McDermott MB, Bhatia S, Watterson J, Manivel JC, Dehner LP. Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer. 1997;80:147–61.
Messinger YH, Stewart DR, Priest JR, Williams GM, Harris AK, Schultz KAP, et al. Pleuropulmonary blastoma: a report on 350 central pathology–confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer. 2015;121:276–85.
Pugh TJ, Yu W, Yang J, Field AL, Ambrogio L, Carter SL, et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene. 2014;33:5295–302.
Liu DJ, Perrier R, Wei X-C, Joseph JT, Strother D. Metachronous type I pleuropulmonary blastoma and atypical choroid plexus papilloma in a young child. Pediatr Blood Cancer. 2016;63:2240–2.
Chong A-S, Fahiminiya S, Strother D, Priest J, Albrecht S, Rivera B, et al. Revisiting pleuropulmonary blastoma and atypical choroid plexus papilloma in a young child: DICER1 syndrome or not? Pediatr Blood Cancer. 2018;65:e27294.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Warren, M., Hiemenz, M.C., Schmidt, R. et al. Expanding the spectrum of dicer1-associated sarcomas. Mod Pathol 33, 164–174 (2020). https://doi.org/10.1038/s41379-019-0366-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0366-x
This article is cited by
-
Reclassification of two germline DICER1 splicing variants leads to DICER1 syndrome diagnosis
Familial Cancer (2023)
-
Genomic characterization of DICER1-associated neoplasms uncovers molecular classes
Nature Communications (2023)
-
DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma
Modern Pathology (2022)
-
DICER1-associated sarcomas: towards a unified nomenclature
Modern Pathology (2021)
-
Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup
Modern Pathology (2021)
