
Abstract
Renal medullary carcinoma is a rare but highly aggressive type of renal cancer occurring in patients with sickle cell trait or rarely with other hemoglobinopathies. Loss of SMARCB1 protein expression, a core subunit of the switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex, has emerged as a key diagnostic feature of these tumors. However, the molecular mechanism underlying this loss remains unclear. We retrospectively identified 20 patients diagnosed with renal medullary carcinoma at two institutions from 1996 to 2017. All patients were confirmed to have sickle cell trait, and all tumors exhibited a loss of SMARCB1 protein expression by immunohistochemistry. The status of SMARCB1 locus was examined by fluorescence in situ hybridization (FISH) using 3-color probes, and somatic alterations were detected by targeted next-generation sequencing platforms. FISH analysis of all 20 cases revealed 11 (55%) with concurrent hemizygous loss and translocation of SMARCB1, 6 (30%) with homozygous loss of SMARCB1, and 3 (15%) without structural or copy number alterations of SMARCB1 despite protein loss. Targeted sequencing revealed a pathogenic somatic mutation of SMARCB1 in one of these 3 cases that were negative by FISH. Tumors in the 3 subsets with different FISH findings largely exhibited similar clinicopathologic features, however, homozygous SMARCB1 deletion was found to show a significant association with the solid growth pattern, whereas tumors dominated by reticular/cribriform growth were enriched for SMARCB1 translocation. Taken together, we demonstrate that different molecular mechanisms underlie the loss of SMARCB1 expression in renal medullary carcinoma. Biallelic inactivation of SMARCB1 occurs in a large majority of cases either via concurrent hemizygous loss and translocation disrupting SMARCB1 or by homozygous loss.
Similar content being viewed by others

Introduction
Renal medullary carcinoma is a rare but highly aggressive form of renal cancer primarily occurring in adolescents and young adults with sickle cell trait. First described as a distinct entity by Davis et al. [1], it was termed as the 7th sickle cell nephropathy given its almost exclusive association with sickle cell trait. However, rare cases have also been reported in patients with other hemoglobinopathies (e.g., SC or SS sickle cell disease) [2]. Patients with renal medullary carcinoma typically present at a young age and are often symptomatic and with advanced disease; despite surgery and chemotherapy, their disease progresses quickly with a dismal prognosis [2,3,4,5,6,7]. Beyond the strong clinical association with sickle cell trait, the underlying biology of this aggressive tumor is poorly understood. Consequently, the definition and pathologic diagnostic criteria for renal medullary carcinoma have remained somewhat controversial. As a medulla-centered tumor, it often exhibits significant clinicopathologic overlap with other subtypes of high-grade renal cell carcinoma involving renal medulla such as collecting duct carcinoma, hereditary leiomyomatosis renal cell carcinoma or fumarate hydratase-deficient renal cell carcinoma, unclassified renal cell carcinoma, and urothelial carcinoma of the renal pelvis [8,9,10,11,12,13].
In recent years, loss of SMARCB1 (also known as INI1 or BAF47) expression, has emerged as a key diagnostic feature of renal medullary carcinoma [14,15,16]. SMARCB1 protein is a core subunit of the switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complexs, which are essential modulators of a variety of cellular processes such as DNA damage repair, DNA replication, proliferation, and differentiation [17]. Loss of SMARCB1 expression has been described in a variety of malignant neoplasms with a propensity for rhabdoid cytomorphology, including pediatric renal and extrarenal malignant rhabdoid tumors, atypical teratoid/rhabdoid tumors of the central nervous system, epithelioid sarcomas, renal medullary carcinoma, and an increasing number of other malignant tumors [18, 19]. However, compared to tumors such as malignant rhabdoid tumors in which biallelic SMARCB1 alterations have been demonstrated [20], the mechanism underlying SMARCB1 protein loss in renal medullary carcinoma has remained relatively elusive: no SMARCB1 mutations were found in a small number of cases that were sequenced, while copy number or loss of heterozygosity analysis revealed only hemizygous loss [15, 16]. Calderaro et al. recently reported novel balanced translocations disrupting SMARCB1 as the second-hit in 4 of the 5 studied cases [21]. In that study, all 4 cases with SMARCB1 translocations occurred in patients with sickle cell trait or disease and involved various fusion partners, whereas the remaining case harbored a homozygous deletion of SMARCB1 and presented in a patient with normal hemoglobin. These results led the authors to suggest that translocations disrupting SMARCB1 is the hallmark recurrent genetic alterations in renal medullary carcinoma.
On the other hand, with the increasing utilization of next-generation sequencing in research and clinical samples, somatic SMARCB1 mutations are occasionally detected in other subtypes of renal cell carcinoma. For example, somatic mutations of SMARCB1 are found in about 1–2% of clear cell, papillary, or chromophobe renal cell carcinoma cases in a few large cohorts with comprehensive genomic characterization [22,23,24]. We have also previously reported SMARCB1 somatic mutations in 3 of 62 (5%) cases of high-grade renal cell carcinoma with unclassified histology and no clinical history of sickle cell trait/disease, but interestingly, all these 3 cases had retained SMARCB1 protein despite the presence of mutations [12]. Nonetheless, loss of SMARCB1 protein has been described in rare cases that display medullary morphology but occur in patients without hemoglobinopathies [10, 25, 26]. Meanwhile, a complete loss or decreased expression of SMARCB1, as well as a focal and weak staining pattern, have been reported in some cases of collecting duct carcinoma [27, 28]. It is unknown whether there were any underlying SMARCB1 molecular alterations in these non-medullary cases with a loss of SMARCB1 expression.
To interrogate the molecular mechanisms underlying the loss of SMARCB1 expression in renal medullary carcinoma, we assembled a cohort of 20 cases with detailed clinicopathologic information. We investigated the status of SMARCB1 locus using a 3-color fluorescence in situ hybridization (FISH) assay complemented by targeted next-generation sequencing platforms that detect somatic mutations in many cancer-related genes including SMARCB1. We further analyzed clinicopathologic correlations in tumor subsets with distinctive molecular alterations.
Materials and methods
Case selection and histologic assessment
Patients with a diagnosis of renal medullary carcinoma rendered at two institutions between 1996 and 2017 were retrospectively identified from the institutional databases, respectively. The study was approved by the respective institutional review board of each institution. All cases had been reviewed to confirm the pathologic diagnosis of renal medullary carcinoma, including corroborating the status of sickle cell trait or other hemoglobinopathies and performing additional immunohistochemical stains to exclude recently recognized entities that were not considered in the initial diagnosis.
All cases included in the current study (n = 20) had archival tissue available for at least one method of molecular analysis (FISH or sequencing). The clinical and follow-up information was obtained from prospectively maintained institutional databases or electronic medical records. Nine of the 20 cases were included in a previous study [6].
All available archival materials were reviewed, and the morphologic features of each case were recorded. These included growth patterns (reticular/yolk sac-like and cribriform, tubulopapillary, infiltrating tubules/cords/individual cells, and solid sheets), stromal changes, rhabdoid cytology, inflammatory infiltrates, and the presence or absence of drepanocytes, necrosis, and mucin. As reticular or yolk sac tumor-like growth and cribriform pattern often overlapped, these were combined as one architectural pattern group.
Immunohistochemical analysis of SMARCB1 and OCT4
Immunohistochemistry was performed on 4-µm-thick sections from representative formalin-fixed, paraffin-embedded tissue blocks utilizing mouse monoclonal antibodies SMARCB1/INI1 (Clone 25/BAF47, dilution 1:200, BD Bioscience) and OCT4 (Clone MRQ10, dilution 1:200, Cell Marque) and an automated system (Benchmark Ultra, Roche). SMARCB1/INI1 staining was scored as retained or lost when compared to internal positive control cells (endothelial/stromal cells and lymphocytes). OCT4 staining was scored as 0 (negative), 1 + (positive in <10% tumor cells), 2 + (positive in 10–50% tumor cells) or 3 + (positive in >50% tumor cells).
FISH analysis
FISH analysis was performed on paraffin sections (4 µm) using a 3-color probe mix, as previously described [6]. Briefly, bacterial artificial chromosome clones spanning 5′SMARCB1 (RP11-248J22 and RP11-1112A23, labeled as red), SMARCB1 (RP11-71G19, labeled as orange), and 3′SMARCB1 (RP11-80O7 and RP-11-76E8, labeled as green) were used as probes. Probe labeling, tissue processing, hybridization, post-hybridization washing, and fluorescence detection were performed according to standard laboratory procedures. Slides were scanned using a Zeiss Axioplan 2i epifluorescence microscope equipped with MetaSystems (Waltham, MA) imaging system. Metafer and VSlide modules within the system were used to generate virtual images of H&E and DAPI-stained sections. In all, corresponding H&E sections assisted in localizing tumor region.
To assess the quality of hybridization and possible intratumoral heterogeneity, the entire section was scanned under 63× objective. Following the initial scan, representative regions were imaged through the depth of the tissue (compressed/merged stack of 12 z-section images taken at 0.5-micron intervals), and at least 10 images per representative region captured. Signal counts (scoring) were performed on captured images. For each case, a minimum of 3 distinct regions (representative of the distinct/major signal pattern) were selected and 50 discrete nuclei were scored. To derive cut-off values (mean percent nuclei plus three standard deviations), five normal/benign renal tissue sections or a minimum of 50 nuclei were also scored in each case. The cut-off value was set at 10% for SMARCB1 translocation (clear split/separation of SMARCB1 in at least one allele), 50% for hemizygous loss [SMARCB1 (orange) = 1, 5′SMARCB1 (red) and/or 3′SMARCB1 (green) ≥ 1], and 10% for homozygous loss [SMARCB1 = 0, 5′SMARCB1 and/or 3′SMARCB1 ≥ 1].
Targeted sequencing analysis
All H&E slides were reviewed to select representative areas of the tumors with sufficient tumor content. DNA was extracted from the macro-dissected tumor and matched normal samples using QIAamp DNA FFPE Tissue Kit or EZ1 Advanced XL system (Qiagen) according to the manufacturer’s instructions. DNA samples of 9 cases were subjected to MSK-IMPACT, a hybridization capture-based NGS assay for targeted deep sequencing of all exons and selected introns of ≥410 or 468 cancer-related genes [29]. In cases with paired normal tissue/blood DNA samples, somatic mutations were called after private germline single-nucleotide variants detected in the paired normal sample were appropriately filtered out. The functional impact of detected mutations was categorized as oncogenic/likely oncogenic and variants of unknown significance using OncoKB (http://oncokb.org), a precision oncology knowledge base maintained at MSKCC [30]. The allele-specific copy number analysis of MSK-IMPACT data was conducted using open-source FACETS (Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing) tool [31].
Tumor tissue samples of 5 cases from Montefiore Medical Center were subjected to the NexCourse® Complete test (Genoptix, Carlsbad, CA), a capture-based next-generation sequencing platform that detected mutations in targeted regions of key genes recurrently mutated in various human cancers (https://genoptix.com/test-menu/nexcourse-complete).
High-resolution single nucleotide polymorphism (SNP) array analysis
Genome-wide DNA copy number alterations and allelic imbalances were analyzed by SNP-array using Affymetrix OncoScan FFPE Assay (Affymetrix, CA) according to the manufacturer’s guidelines as previously described [32]. Assay data were analyzed by OncoScan Console software (Affymetrix) and OncoScan Nexus Express software (BioDiscovery, CA, USA) using Affymetrix TuScan algorithm. All array data were also manually reviewed for subtle alterations not automatically detected by the software.
Statistical analysis
The statistical significance of clinicopathologic features between subsets of renal medullary carcinoma cases was determined using Fisher’s exact test or Mann–Whitney U test (age and tumor size). Statistical significance was defined as a P-value <0.05.
Results
Clinical features
The clinical features of the study cases are summarized in Table 1. All 20 patients had a clinically documented diagnosis of sickle cell trait. Of the 19 patients who self-reported race, 15 (79%) were black or African-American. The mean age of patients was 26 years (range 7–61 years), with a male to female ratio of 3:2. Patients most often presented with flank or abdominal pain (12/20, 60%) and/or gross hematuria (8/20, 40%). Initial symptoms in 4 patients (20%) were related to metastases, including shortness of breath resulting from lung or pleural metastases in 3 patients and skin nodules in 1. In only one case the renal mass was found incidentally through imaging work-up for unrelated complaints. The mean tumor size was 6.0 cm (range 2.7–8.9 cm), and the majority were right-sided (14/20, 70%). Metastases were detected in 14 (70%) patients at initial diagnosis. Nine patients (45%) underwent nephrectomy and the others had diagnostic biopsies either from the kidney (n = 2) or metastatic sites (n = 9). With a median follow-up time of 9 months (range 1–78 months), 75% (15/20) of patients had died of disease, 25% (5/20) were alive with disease, and 4 of these 5 patients were subsequently lost to follow-up.
Morphological and immunohistochemical features
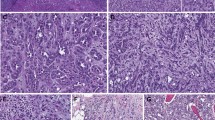
Among the 9 nephrectomies (Cases # 1–9) (Fig. 1), a range of architectural patterns was seen (Table 2). Reticular (or yolk sac tumor-like glands) and cribriform patterns represented the dominant architectural component in 6 (67%) cases, whereas the remaining 3 (33%) tumors mainly consisted of solid sheets or nests of cells. The pattern of infiltrating tubules, cords, or individual cells in a desmoplastic stroma was at least focally present in all cases (100%), but not as a dominant pattern. Tubulopapillary growth (including micropapillary pattern), as a minor component, was found in 4 (44%) cases.

Renal medullary carcinoma in nephrectomy specimens showed reticular/yolk sac tumor-like (a), cribriform (b), solid sheets (c), infiltrating tubules and individual cells in a desmoplastic stroma (d), and tubulopapillary (e) architectural patterns. Mucin and stromal myxoid changes are prominent in some cases (f). Insets, b neutrophil-rich inflammatory infiltrate, c rhabdoid feature
In comparison, core or small biopsies of the primary renal mass (n = 2) and metastatic sites (n = 9) (Fig. 2) revealed infiltrating tubules/solid cords/individual cells in a desmoplastic stroma as the dominant pattern in 5 of 11 cases (45%). The dominant pattern in 3 (27%) biopsy cases was solid (small or large nests), while the other 3 (27%) exhibited a predominantly reticular growth.

Renal medullary carcinoma revealed by biopsies at metastatic sites displayed infiltrating tubules/solid cords/individual cells in a desmoplastic and myxoid stroma (a), reticular/cribriform glands with cytoplasmic mucin in desmoplastic stroma (b), solid sheets/nests (b), and micropapillary features and individual cells (d). Rhabdoid cytologic feature was prominent in some cases (d)
The rhabdoid cytologic feature was extensive in 9 (50%) tumors and focal in 8 (40%) (Figs. 1c and 2d). Intraluminal or cytoplasmic mucin was identified in 6 (30%) cases (Figs. 1f and 2b). While desmoplastic stroma was present in all cases, stromal myxoid change was noted in 9 cases (45%) (Figs. 1f and 2a). Necrosis was present in 14 (70%) cases. The neutrophil-rich inflammatory infiltrate was present in 10 of 11 (89%) primary tumors (Fig. 1b) and 4 of 9 (44%) metastases. Although drepanocytes in small vessels were identified in all 9 (100%) nephrectomy specimens, it was difficult to be certain about their presence in 7 of 11 (64%) biopsy samples, mainly due to a paucity of small vessels with discernible red blood cells.
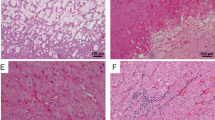
By immunohistochemistry (Fig. 3 and Table 2), all 20 cases showed a loss of nuclear expression of the SMARCB1 protein. OCT4 nuclear immunoreactivity was detected in 9 cases, including 6 (30%) with moderate to strong staining (2 + or 3 + ) and 3 with only focal staining (1 + ).

SMARCB1 protein expression was lost in tumor cells while the nuclear staining was retained in internal control cells (a). Nuclear immunoreactivity to OCT3/4 was observed in a subset of cases, some with strong positivity (b) and others with focal weak staining (c)
FISH and mutational analysis
All cases (n = 20) were analyzed by FISH (Table 3). Compared to the diploid pattern in the control (normal blood leukocytes) (Fig. 4a, b) or internal control cells (stromal and inflammatory cells), the majority of cells in 11 (55%) tumors showed the split of red-orange-green signals, indicating the presence of SMARCB1 rearrangement (Fig. 4c–f). This was accompanied by a concurrent hemizygous loss of SMARCB1 and neighboring regions in the other allele in the vast majority of cases (n = 10) (Fig. 4c–e). The remaining case (#6) was dominated by tumor cells with two sets of split green and red signals (Fig. 4f). Interestingly, the split signals in cases with SMARCB1 rearrangement showed various patterns, including one case with inversion (Fig. 4e), suggesting the presence of potentially different breakpoints within SMARCB1 and is in line with various fusion partners previously identified [21].

Three-color FISH analysis of SMARCB1 (22q11) and neighboring regions. Normal control blood leukocytes at interphase (a) and metaphase (b), representative renal medullary carcinoma cases with hemizygous loss (loss of one set of red-orange-green signals) and concurrent translocation (split of the other set of signals) (c, d), case #12 with hemizygous loss and concurrent inversion in the other allele (e), case #6 with two sets of split signals (f), representative cases with homozygous deletion (g–i), and a representative case showing diploid pattern without structural and copy number alteration (j). The arrowhead in (i) marks a tumor nucleus that completely lost all signals whereas the arrow marks the nucleus of an internal control cell
Six (30%) cases exhibited homozygous loss of SMARCB1 (orange) and, to a variable extent, the adjacent regions (red and green signals) (Fig. 4g–i). The remaining 3 (15%) cases showed a diploid pattern (Fig. 4j), without detectable structural or copy number alterations involving SMARCB1 locus despite the loss of protein expression.
Targeted sequencing was performed in 14 cases and all identified mutations are summarized (Supplementary Table 1). In one of the 3 FISH-negative cases, sequencing detected a somatic truncating mutation of SMARCB1 (Table 3). Aside from SMARCB1, somatic oncogenic or likely oncogenic mutations of TSC2, TP53, EP300, and JAK2 etc. were detected, but none of these was recurrent in the cohort. Importantly, in 9 of 14 (64%) cases, there were no somatic oncogenic mutations detected by our targeted sequencing panels, consistent with the notion that SMARCB1 protein loss is the crucial molecular alterations driving the tumor development.
The allele-specific copy number analysis using FACETS tool revealed copy number changes in 8 cases (Table 3 and Fig. 5a–c). Among these, 22q loss was the most frequent alterations. Interestingly, while 22q loss was consistently identified in cases with homozygous loss by FISH, the detection of a loss in 22q or SMARCB1 locus was less reliable in cases with hemizygous loss, presumably due to a high percentage of non-neoplastic cells (e.g., inflammatory and stromal cells) masking the low-level copy number changes. Beyond 22q, there was no consistent copy number alteration pattern identified. FACETS analysis did not identify SMARCB1 translocation.

Allele-specific copy number changes revealed by FACETS (fraction and copy number estimates from tumor sequencing) analysis showed flat genome (a), 22q loss (b), or additional gains and losses (c). The integer copy number (copy number call corrected for tumor purity and ploidy) is plotted on the y-axis. Diploid corresponds to n = 2. Chromosomes 1–22 are plotted on the x-axis. Black line – total copy number, red line – minor/B allele. a case #17, b case #18, c case #16. Genome-wide copy number changes in case #6 by SNP-array analysis (d). All chromosomes are color-coded. For copy number log-ratio (top panel), diploid corresponds to y = 0. For B-allele frequency (bottom panel), separation of dots from baseline indicates allelic imbalance. The arrow points to chromosome 22q. Pie chart summarizes the SMARCB1 alterations detected in 20 cases of renal medullary carcinoma using integrated analysis (e)
For case #6, to differentiate the possibility of two independent translocations vs. genomic doubling of one translocation event with concurrent hemizygous loss, we performed high-density SNP array analysis and found that this tumor predominantly consisted of a tetraploid clone, whereas chromosome 22q (including SMACRB1 locus) remained as diploid and showed loss of heterozygosity (Fig. 5d). These findings support that a genome-wide doubling occurred in this tumor after the SMARCB1 translocation and concurrent hemizygous loss. In this predominantly tetraploid genome, there were also additional losses of one or two copies of other chromosomes, including 15 as diploid and 3, 10, 12, and 16 as triploid (Table 3). These findings were further confirmed by FISH analysis using centromere probes of multiple chromosomes (data not shown).
Taken together, FISH and mutational analyses revealed mechanisms of biallelic inactivation of SMARCB1 in 17 of 20 (85%) RMC cases, either via concurrent hemizygous loss and translocation disrupting SMARCB1 or by homozygous loss (Fig. 5e). Among the remaining 3 cases, one had a SMARCB1 somatic mutation without accompanying copy number change detectable by FISH. In the other 2 cases, SMARCB1 protein loss was likely due to aberrations other than mutational, copy number, or structural alterations.
Correlation between clinicopathologic and molecular features
We further examined whether there were any clinicopathologic features of these cases associated with the different mechanisms of SMARCB1 loss, particularly for the two main molecular subsets, tumors with SMARCB1 translocation and hemizygous loss vs. those with homozygous loss (Table 4). Interestingly, among all the parameters assessed, the dominant architectural pattern (reticular and cribriform growth vs. solid growth) was the only feature exhibiting a significant correlation with these two molecular subgroups (p = 0.03). Tumors with SMARCB1 translocation were more likely to show reticular and cribriform pattern (64% vs 17%), whereas tumors with SMARCB1 homozygous loss were significantly enriched for a solid growth pattern (83% vs 9%).
Discussion
Renal medullary carcinoma is a rare but devastating subtype of renal cancer that is almost exclusively associated with sickle cell trait and rarely other hemoglobinopathies. While the biology of its pathogenesis largely remains unclear, the high prevalence of SMARCB1/INI1 protein loss in these cases examined by recent studies suggests it to be a critical molecular alteration that drives tumor development [14,15,16]. With the recent discovery of translocations disrupting SMARCB1 reported by Calderaro et al. [21] in 4 cases of renal medullary carcinoma, we sought to clarify the mechanisms of SMARCB1 loss in 20 well-characterized cases, to our knowledge the largest cohort of renal medullary carcinoma with molecular characterization. This cohort exhibited clinicopathologic features that are in line with previously reported series, and our molecular analysis emphasized the integration of different methods to elucidate alterations that might have evaded routine mutational or copy number analysis.
All cases in our study cohort indeed showed SMARCB1 protein loss. We demonstrate that distinct molecular mechanisms underlying the loss of SMARCB1 protein expression in renal medullary carcinoma, and biallelic inactivation of SMARCB1 occur in a large majority of cases. The combined translocation and hemizygous loss was the most common mechanism of SMARCB1 loss detected, accounting for 55% of the cohort and representing a relatively unique mechanism that has not been well described in other SMARCB1-deficient tumors. Meanwhile, homozygous deletion involving SMARCB1 and various neighboring regions on 22q did occur in 30% of patients in this cohort, all of whom had sickle cell trait and classic clinicopathologic presentation of renal medullary carcinoma. In comparison, somatic mutation of SMARCB1 was detected uncommonly, accounting for only 1 case (5%) in this cohort. Interestingly, the complete loss of SMARCB1 protein expression in about 15% of cases was not explained by translocation, copy number alterations, or mutations, suggesting the presence of additional molecular mechanisms, such as epigenetic and/or other regulatory mechanisms.
Moreover, there were no recurrent somatic oncogenic mutations detected in all other cancer-related genes included in the targeted next-generation sequencing panels utilized in our study. The non-recurrent somatic mutations such as TSC2, TP53, and EP300, while being pathogenic, more likely represent additional or later molecular alterations occurring in individual cases. We further elucidated copy number changes occurring in the remainder of the genome and identified additional gains and losses. We showed that genomic doubling can occur as a later event in renal medullary carcinoma, which potentially serves as a mechanism to increase the complexity of genomic aberrations. Taken together, our findings validate the central role of SMARCB1 inactivation in the pathogenesis of renal medullary carcinoma and clarify the different molecular mechanisms underlying this process.
Recent comprehensive molecular studies of other SMARCB1-deficient tumors such as atypical teratoid/rhabdoid tumors, extra-cranial malignant rhabdoid tumors, and epithelioid sarcoma, have revealed significantly different molecular characteristics and intertumoral heterogeneity among these tumors [33,34,35]. Johann et al. identified three distinct molecular subtypes of atypical teratoid/rhabdoid tumors, which not only show very different epigenetic features and gene expression patterns, but are also associated with differences in patient demographics, tumor location, and types of SMARCB1 alterations [34]. For example, broad SMARCB1 deletions were observed in 77% of the ATRT-TYR tumors, whereas focal SMARCB1 deletions were more prevalent in the ATRT-MYC subgroup (79%). It is interesting to note that a small percentage of atypical teratoid/rhabdoid tumors also lack detectable SMARCB1 DNA aberrations despite protein loss [34]. Previously, altered miRNA expression has been shown as an epigenetic mechanism to regulate SMARCB1 expression in epithelioid sarcoma [36, 37].
Although epigenetic and transcriptional data are very limited in renal medullary carcinoma, the findings from atypical teratoid/rhabdoid tumors raise an interesting question of whether there could be additional differences among renal medullary carcinomas with distinct molecular mechanisms of inactivating SMARCB1, particularly between the two main subgroups with either translocation or homozygous deletion. In the current cohort, while most clinicopathologic features were indistinguishable between these molecular subgroups, we did observe a significant difference in their dominant morphologic architectural patterns. The cases harboring translocation mainly displayed reticular or cribriform growth, the relatively distinctive features of renal medullary carcinoma when compared to collecting duct or fumarate hydratase-deficient renal cell carcinoma [13], whereas the cases with homozygous deletion predominantly showed solid sheets or nests. Additional studies are needed to further test this association and investigate the possibility of biologic or molecular differences among renal medullary carcinomas with distinct mechanisms inactivating SMARCB1. Our analysis also suggests that there are variations in both translocation patterns and the extent of homozygous deletion, which might add additional levels of complexity to the observed morphologic divergence and overlap between the two molecular subgroups. Moreover, one patient (case #9) in the cohort had an exceptionally long period of survival (78 months) before being lost to follow-up. This tumor lacked detectable structural or mutational aberrations but was morphologically indistinguishable from other cases.
Largely due to tissue constraints, we were not able to pursue fusion partners of SMARCB1 translocation or explore the alterations at transcriptional and epigenetic levels in the three subsets of renal medullary carcinoma with distinctive mechanisms of inactivating SMARCB1. Further collaborative investigation in this regard would be essential to shed light on its pathogenesis and discover therapeutic approaches for this aggressive disease.
The morphologic spectrum we observed in this cohort is consistent with what has been described for renal medullary carcinoma and highlights a few important issues for the differential diagnosis. First, in nephrectomy specimens, the combined reticular/yolk sac-like and cribriform patterns were found to be dominant in the majority (67%) of cases. The remaining cases were frequently dominated by solid sheets or nests, reflecting the potentially different mechanisms inactivating SMARCB1 based on our analysis. The infiltrating tubules, cords and individual cells in a desmoplastic stroma, a pattern commonly associated with collecting duct carcinoma, while universally present in all cases, did not present as a dominant pattern. In contrast, in biopsies, particularly core biopsies, probably due to the difficulty of appreciating certain architectural patterns (e.g., reticular), most cases appeared to fulfill the description of poorly differentiated adenocarcinoma and could easily lead to a diagnosis of collecting duct carcinoma. In a similar fashion, cases with solid growth pattern in small biopsies could closely mimic high-grade urothelial carcinoma. Although rhabdoid cytology can be a very helpful feature in small biopsies to raise a suspicion for renal medullary carcinoma, it was not apparent in a subset of cases, and may also occasionally present in metastatic carcinomas involving the kidney. Rhabdoid cells could also be mistaken as suggesting plasmacytoid features of urothelial carcinoma. Drepanocytes or sickled red blood cells, while almost always visible in nephrectomies and very helpful for considering medullary carcinoma, could be difficult to identify in biopsies mainly due to a paucity of evident vascular spaces in these small specimens. The neutrophil-rich inflammatory infiltrate was present in the vast majority of primary tumors but was much less notable at metastatic sites. Additionally, mucin, stromal myxoid or desmoplastic changes sometimes provided non-specific, yet helpful clues for considering a diagnosis of renal medullary carcinoma in suspicious clinical contexts. By immunohistochemistry, all the cases in our cohort showed SMARCB1 protein loss, whereas OCT4 staining was much more variable with only 30% cases with moderate to strong immunoreactivity.
Aside from collecting duct carcinoma and high-grade urothelial carcinoma of upper tract, other entities that enter in the differential diagnoses for renal medullary carcinoma include fumarate hydratase-deficient, ALK rearrangement-associated, high-grade unclassified renal cell carcinoma, metastatic poorly differentiated carcinoma secondarily involving kidney, and other SMARCB1-loss tumors. With the expanding histologic spectrum of hereditary leiomyomatosis renal cell carcinoma and fumarate hydratase-deficient renal cell carcinoma, we now recognize that these tumors can exhibit growth patterns indistinguishable from collecting duct carcinoma or show features mimicking renal medullary carcinoma, such as sieve-like or cribriform areas and mixed growth patterns [11, 13, 38]. Therefore, performing ancillary studies including fumarate hydratase and/or 2-succino-cysteine immunohistochemistry in cases raising concerns for these tumors is very important for distinguishing them from renal medullary carcinoma. In this setting, sickle cell trait can be a red herring and leads to an erroneous diagnosis. We excluded one such case in the re-review process of this study, a 27-year-old man with sickle cell trait but had a tumor exhibiting histologic features highly suspicious for fumarate hydratase-deficient renal cell carcinoma. This case was encountered before the pathologic features of hereditary leiomyomatosis renal cell carcinoma were recognized, and the additional studies we did showed diffuse immunopositivity for 2-succino-cysteine (nuclear and cytoplasmic staining) and a loss of fumarate hydratase, but retention of SMARCB1.
ALK rearrangement-associated RCC represents an emerging group of tumors that harbor ALK fusions. Several fusion partners have been identified, including VCL, TPM3, EML4, STRN, and HOOK1 [39,40,41,42,43,44]. Among these, tumors with VCL-ALK fusion have been identified in patients with sickle cell trait and consist of sheets of polygonal or spindle cells with eosinophilic cytoplasm, large vesicular nuclei with prominent nucleoli, frequent intracytoplasmic vacuoles, and lymphocytic inflammatory infiltrate [39, 40, 42]. These tumors have retained SMARCB1 expression and appear to be less aggressive than renal medullary carcinoma. But given the small number of reported cases, their biologic behavior and relationship with renal medullary carcinoma await further investigations. The identification of ALK-rearrangement is becoming increasingly important as there is now growing evidence that these tumors can respond to ALK-directed therapy, which otherwise would not be considered for patients with advanced renal cancer [45].
Naturally, excluding renal medullary carcinoma is also important before diagnosing a high-grade unclassified renal cell carcinoma with imitating histologic features. At metastatic sites, including PAX8 and SMARCB1 staining in cases with suspicious histomorphology and/or clinical features (presence of renal mass or sickle cell trait) usually will be very helpful to distinguish renal medullary carcinoma from other poorly differentiated tumors. In this scenario, distinguishing renal medullary carcinoma from SMARCB1-loss tumors originating from other sites could be very difficult and heavily dependent on clinical correlations and PAX8 stain in the presence of a renal mass. It is also worth noting that malignant rhabdoid tumor of the kidney can rarely occur in older children and may display PAX8 immunoreactivity [46, 47]. The clinicopathologic features of these cases would significantly overlap with those of renal medullary carcinomas in children, and it will be particularly diagnostically challenging if the tumor exhibits a solid growth pattern. Moreover, extremely rare cases of malignant rhabdoid tumor of the kidney have also been reported in adults [48]. In these rare settings, how to distinguish renal medullary carcinoma from malignant rhabdoid tumor of the kidney and the importance of sickle cell trait/hemoglobinopathy remain to be further clarified.
Together with recent studies of renal medullary carcinoma, our analysis strongly supports SMARCB1 protein loss as the main molecular feature and driving force underlying the development of this aggressive disease. However, given the increasingly detected SMARCB1 mutations and very rarely protein loss in other types of renal cell carcinoma, the roles of SMARCB1 alterations in specific biologic contexts of different renal cancer subtypes need to be investigated and compared. For instance, it will be very interesting to examine the mechanisms of SMARCB1 inactivation in unclassified renal cell carcinoma with medullary phenotype and rare tumors with loss of SMARCB1 protein by immunohistochemistry in patients without hemoglobinopathies [10, 26].
In summary, we performed an integrated analysis of 20 renal medullary carcinoma cases to delineate the molecular mechanisms underlying the loss of SMARCB1 expression and demonstrated biallelic inactivation of SMARCB1 in a large majority of cases. This most frequently develops via hemizygous loss and concurrent translocation disrupting SMARCB1 locus, but homozygous deletion of SMARCB1 also represents an important molecular mechanism in patients with sickle cell trait. Additionally, inactivation of SMARCB1 in renal medullary carcinoma may occur via somatic mutation and/or other yet to be defined mechanisms. We also identified an intriguing association between solid growth pattern and homozygous deletion whereas cases harboring translocation and hemizygous loss more commonly displayed reticular and cribriform architectures. These findings validate the central role of SMARCB1 inactivation and provide new insights into the pathogenesis of renal medullary carcinoma.
References
Davis CJ Jr., Mostofi FK, Sesterhenn IA. Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol. 1995;19:1–11.
Alvarez O, Rodriguez MM, Jordan L, Sarnaik S. Renal medullary carcinoma and sickle cell trait: A systematic review. Pedia Blood Cancer. 2015;62:1694–9.
Swartz MA, Karth J, Schneider DT, et al. Renal medullary carcinoma: clinical, pathologic, immunohistochemical, and genetic analysis with pathogenetic implications. Urology. 2002;60:1083–9.
Hakimi AA, Koi PT, Milhoua PM, et al. Renal medullary carcinoma: the Bronx experience. Urology. 2007;70:878–82.
Watanabe IC, Billis A, Guimaraes MS, et al. Renal medullary carcinoma: report of seven cases from Brazil. Mod Pathol. 2007;20:914–20.
Carlo MI, Chaim J, Patil S, et al. Genomic characterization of renal medullary carcinoma and treatment outcomes. Clin Genitourin Cancer. 2017;15:e987–94.
Shah AY, Karam JA, Malouf GG, et al. Management and outcomes of patients with renal medullary carcinoma: a multicentre collaborative study. BJU Int. 2017;120:782–92.
Amin MB, Merino MJ. Renal medullary carcinoma. In: Moch H, Humphrey PA, Ulbright TM, Reuter VE, editors. WHO Classification of Tumours of the Urinary System and Male Genital Organs. 4th edition. Lyon: IARC; 2016. p. 31–2.
Gupta R, Billis A, Shah RB, et al. Carcinoma of the collecting ducts of Bellini and renal medullary carcinoma: clinicopathologic analysis of 52 cases of rare aggressive subtypes of renal cell carcinoma with a focus on their interrelationship. Am J Surg Pathol. 2012;36:1265–78.
Amin MB, Smith SC, Agaimy A, et al. Collecting duct carcinoma versus renal medullary carcinoma: an appeal for nosologic and biological clarity. Am J Surg Pathol. 2014;38:871–4.
Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627–37.
Chen YB, Xu J, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131.
Ohe C, Smith SC, Sirohi D, et al. Reappraisal of morphologic differences between renal medullary carcinoma, collecting duct carcinoma, and fumarate hydratase-deficient renal cell carcinoma. Am J Surg Pathol. 2018;42:279–92.
Cheng JX, Tretiakova M, Gong C, et al. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol. 2008;21:647–52.
Calderaro J, Moroch J, Pierron G, et al. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology. 2012;61:428–35.
Liu Q, Galli S, Srinivasan R, et al. Renal medullary carcinoma: molecular, immunohistochemistry, and morphologic correlation. Am J Surg Pathol. 2013;37:368–74.
Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–92.
Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011;35:e47–63.
Agaimy A. The expanding family of SMARCB1(INI1)-deficient neoplasia: implications of phenotypic, biological, and molecular heterogeneity. Adv Anat Pathol. 2014;21:394–410.
Jackson EM, Sievert AJ, Gai X, et al. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res. 2009;15:1923–30.
Calderaro J, Masliah-Planchon J, Richer W, et al. Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas. Eur Urol. 2016;69:1055–61.
Ricketts CJ, De Cubas AA, Fan H, et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23:313–26.
Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–7.
Durinck S, Stawiski EW, Pavia-Jimenez A, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13–21.
O’Donnell PH, Jensen A, Posadas EM, et al. Renal medullary-like carcinoma in an adult without sickle cell hemoglobinopathy. Nat Rev Urol. 2010;7:110.
Sirohi D, Smith SC, Ohe C, et al. Renal cell carcinoma, unclassified with medullary phenotype: poorly differentiated adenocarcinomas overlapping with renal medullary carcinoma. Hum Pathol. 2017;67:134–45.
Elwood H, Chaux A, Schultz L, et al. Immunohistochemical analysis of SMARCB1/INI-1 expression in collecting duct carcinoma. Urology. 2011;78:474.e1-5.
Carvalho JC, Thomas DG, McHugh JB, Shah RB, Kunju LP. p63, CK7, PAX8 and INI-1: an optimal immunohistochemical panel to distinguish poorly differentiated urothelial cell carcinoma from high-grade tumours of the renal collecting system. Histopathology. 2012;60:597–608.
Cheng DT, Mitchell TN, Zehir A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–64.
Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;1:1–16.
Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131.
Ren Q, Wang L, Al-Ahmadie HA, et al. Distinct genomic copy number alterations distinguish mucinous tubular and spindle cell carcinoma of the kidney from papillary renal cell carcinoma with overlapping histologic features. Am J Surg Pathol. 2018;42:767–77.
Jamshidi F, Bashashati A, Shumansky K, et al. The genomic landscape of epithelioid sarcoma cell lines and tumours. J Pathol. 2016;238:63–73.
Johann PD, Erkek S, Zapatka M, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016;29:379–93.
Chun HE, Lim EL, Heravi-Moussavi A, et al. Genome-wide profiles of extra-cranial malignant rhabdoid tumors reveal heterogeneity and dysregulated developmental pathways. Cancer Cell. 2016;29:394–406.
Papp G, Krausz T, Stricker TP, Szendroi M, Sapi Z. SMARCB1 expression in epithelioid sarcoma is regulated by miR-206, miR-381, and miR-671-5p on Both mRNA and protein levels. Genes Chromosomes Cancer. 2014;53:168–76.
Sapi Z, Papp G, Szendroi M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671-5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA Study of 223 Soft Tissue Sarcomas. Genes Chromosomes Cancer. 2016;55:786–802.
Merino MJ, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–85.
Debelenko LV, Raimondi SC, Daw N, et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol. 2011;24:430–42.
Marino-Enriquez A, Ou WB, Weldon CB, Fletcher JA, Perez-Atayde AR. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer. 2011;50:146–53.
Sugawara E, Togashi Y, Kuroda N, et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer. 2012;118:4427–36.
Smith NE, Deyrup AT, Marino-Enriquez A, et al. VCL-ALK renal cell carcinoma in children with sickle-cell trait: the eighth sickle-cell nephropathy? Am J Surg Pathol. 2014;38:858–63.
Cajaiba MM, Jennings LJ, George D, Perlman EJ. Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1-ALK fusion transcript. Genes Chromosomes Cancer. 2016;55:814–7.
Kusano H, Togashi Y, Akiba J, et al. Two cases of renal cell carcinoma harboring a novel STRN-ALK Fusion Gene. Am J Surg Pathol. 2016;40:761–9.
Tao JJ, Wei G, Patel R, et al. ALK fusions in renal cell carcinoma: response to entrectinib. JCO Precis Oncol. 2018;2:1–8.
Weeks DA, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am J Surg Pathol. 1989;13:439–58.
Fan R. PAX immunoreactivity in poorly differentiated small round cell tumors of childhood. Fetal Pedia Pathol. 2014;33:244–52.
Podduturi V, Campa-Thompson MM, Zhou XJ, Guileyardo JM. Malignant rhabdoid tumor of the kidney arising in an adult patient. Proc (Bayl Univ Med Cent). 2014;27:239–41.
Acknowledgements
We gratefully acknowledge the members of the Molecular Diagnostics Service in the Department of Pathology, the Integrated Genomics Operation and Bioinformatics Core, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology of MSKCC. The study is supported by MSK Cancer Center Core Grant (P30 CA008748) and The Society of Memorial Sloan Kettering Research Grant (Y-BC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jia, L., Carlo, M.I., Khan, H. et al. Distinctive mechanisms underlie the loss of SMARCB1 protein expression in renal medullary carcinoma: morphologic and molecular analysis of 20 cases. Mod Pathol 32, 1329–1343 (2019). https://doi.org/10.1038/s41379-019-0273-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0273-1
This article is cited by
-
Haplotype-resolved germline and somatic alterations in renal medullary carcinomas
Genome Medicine (2021)
-
Molekularpathologie bei urologischen Tumoren
Der Pathologe (2021)
-
New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia
Modern Pathology (2021)
