
Abstract
The spectrum and evolution of proliferation rates in stage IV lung carcinoids is poorly defined. In particular, there are limited data on the prevalence and characteristics of tumors exceeding the standard upper proliferative criteria—as defined largely based on early-stage carcinoids—in metastatic setting. Sixty-six patients with stage IV lung carcinoids were identified, and all evaluable samples (n = 132; mean 2 samples per patient) were analyzed for mitotic counts and Ki-67 rate. Clinicopathologic and genomic features associated with elevated proliferation rates (>10 mitoses per 2 mm2 and/or >20% hot-spot Ki-67), and evolution of proliferation rates in serial specimens were analyzed. We found that mitoses and/or Ki-67 exceeded the standard criteria in 35 of 132 (27%) samples, primarily (31/35 cases) at metastatic sites. Although neuroendocrine neoplasms with >10 mitoses per 2 mm2 are currently regarded as de facto neuroendocrine carcinomas, the notion that these cases are part of the spectrum of carcinoids was supported by (1) well-differentiated morphology, (2) conventional proliferation rates in other samples from same patient, (3) genetic characteristics, including the lack of RB1/TP53 alterations in all tested samples (n = 19), and (4) median overall survival of 2.7 years, compared to <1 year survival of stage IV neuroendocrine carcinomas in the historic cohorts. In patients with matched primary/metastatic specimens (48 pairs), escalation of mitoses or Ki-67 by ≥10 points was observed in 35% of metastatic samples; clonal relationship in one pair with marked proliferative progression was confirmed by next-generation sequencing. Notably, escalation of proliferation rate was documented in a subset of metastases arising from resected typical carcinoids, emphasizing that the diagnosis of typical carcinoid in primary tumor does not assure low proliferation rate at metastatic sites. In conclusion, stage IV lung carcinoids frequently exceed the standard proliferative criteria established for primary tumors, and commonly exhibit proliferative escalation at metastatic sites. Despite the overlap of proliferation rates, these tumors show fundamental morphologic, genomic and clinical differences from neuroendocrine carcinomas, and should be classified separately from those tumors. Awareness of the increased proliferative spectrum in metastatic carcinoids is critical for their accurate diagnosis. Further studies are warranted to explore the impact of proliferation indices on prognosis and therapeutic responses of patients with metastatic carcinoids.
Similar content being viewed by others

Introduction
Lung carcinoids account for 25–30% of well-differentiated neuroendocrine tumors in the body [1, 2]. Unlike neuroendocrine tumors of the pancreas and small intestine, which are commonly metastatic at presentation [3], lung carcinoids are overwhelmingly an early-stage disease, with stage IV spread being relatively uncommon (<5–10% of patients) [4]. Consequently, metastatic lung carcinoids are substantially less well studied than metastatic enteropancreatic neuroendocrine tumors. In particular, most studies on proliferation metrics in lung carcinoids have been based on the more common early-stage tumors, whereas the spectrum and evolution of proliferation rates in metastatic carcinoids remain poorly defined.
According to the thoracic World Health Organization (WHO) classification, lung carcinoids are categorized as typical (<2 mitoses per 2 mm2 and no necrosis) and atypical (2–10 mitoses per 2 mm2 and/or necrosis), corresponding to low-grade (grade 1) and intermediate-grade (grade 2), respectively [2, 5]. These categories were developed for resected primary tumors, with the goal to predict the risk of postsurgical recurrence/metastasis, whereas the role and applicability of these categories in metastatic setting is unclear. Conversely, the distinction of carcinoids from small cell and large cell neuroendocrine carcinomas—a biologically distinct group of highly aggressive neuroendocrine neoplasms—is critical in both early and advanced stage disease, since the clinical management of these tumor types is markedly different in both settings. Aside from morphologic differences, neuroendocrine carcinomas are defined in the WHO by mitotic counts of >10 per 2 mm2, although actual mitotic counts in these tumors typically substantially exceed this threshold [1, 2]. Unlike the classification of enteropancreatic neuroendocrine neoplasms, which incorporates both mitotic counts and Ki-67 proliferation index [6], Ki-67 is currently not included in the classification of lung neuroendocrine neoplasms given the unresolved question regarding the added value of Ki-67 relative to mitotic counts in resected primary tumors [7, 8]. Conversely, the value of Ki-67 is nonresection specimens is widely recognized, given that Ki-67 rates are largely proportional to mitotic counts but can be more readily evaluated in small samples [9]. In particular, the use of Ki-67 is widely recommended for the distinction of carcinoids from small cell or large cell neuroendocrine carcinomas in crushed biopsies and cytology specimens, where mitotic counts cannot be accurately performed [2, 10, 11]. In this setting, the WHO recommends a Ki-67 threshold of 20% as the upper limit for carcinoid tumors [2]. Ki-67 of 20% has also been used until recently as the cut-point between neuroendocrine tumors and carcinomas at enteropancreatic sites [6].
The challenging area with the above criteria concerns the classification of tumors that meet the morphologic criteria of atypical carcinoids, but exceed the proliferative limit of 10 mitoses per 2 mm2 or Ki-67 index of 20%. In enteropancreatic organs, tumors of this type have been a subject of numerous recent investigations [12,13,14,15,16,17,18]. In the pancreas, well-differentiated neuroendocrine tumors that exceed the grade 2 proliferative criteria are common, and they recently became formally recognized by the WHO classification as “grade 3 neuroendocrine tumors” [19], which contrasts with their prior classification as neuroendocrine carcinomas [6]. In contrast, until recently, there has been virtually a void of information regarding the analogous tumors in the lung. Currently, per WHO classification, tumors with morphology of atypical carcinoids but mitotic rate of >10 per mm2 are regarded as de facto large cell neuroendocrine carcinomas, which carries the implication of highly aggressive behavior, and treatment with platinum-based chemotherapy and recently immunotherapy regimens [20]. In a recent study, our group reported on molecular features of two such “carcinoid-like large cell neuroendocrine carcinomas”—tumors with morphology of atypical carcinoids but mitotic counts (and Ki-67) exceeding the above thresholds, though remaining below the levels typically seen in most neuroendocrine carcinomas—and demonstrated that they exhibited genomic characteristics of carcinoid tumors rather than carcinomas [21]. Subsequently, Quinn et al. [22] described 12 additional tumors of this type, and showed that their clinicopathologic features were more akin to carcinoids than neuroendocrine carcinomas, including the more indolent clinical behavior and limited chemosensitivity. In fact, to our knowledge, the first well-documented example of carcinoid-type lung tumor exceeding the standard mitotic criteria was reported by Megyesi et al. [23]. In addition, several studies have documented instances of >20% Ki-67 rates in lung carcinoids [7], although clinicopathologic characteristics of such tumors are also not well established.
Importantly, the proliferative criteria for lung carcinoids have been established based entirely on early-stage tumors, and the aforementioned studies describing rare variants exceeding the standard proliferative criteria have also been based entirely or primarily on surgically resected primary tumors. While they provide evidence for the existence of variants with elevated proliferation rates, there remains limited information regarding their prevalence or clinicopathologic and molecular characteristics. This particularly applies to the uncommon stage IV setting, where one may anticipate an increased prevalence of samples with increased proliferation rates, as supported by our anecdotal clinical experience leading up to this study. Furthermore, there are only limited data on the evolution of proliferation rates during metastatic progression of lung carcinoids, and applicability of the dichotomous classification of typical vs. atypical carcinoids in metastatic setting. To address these questions, we performed a comprehensive analysis of proliferation rate via mitotic counts and Ki-67 immunohistochemistry for all evaluable samples from 66 patients with stage IV lung carcinoids, focusing on detailed clinicopathologic and genomic features of tumors exceeding the current proliferative criteria for atypical carcinoids, and on the evolution of proliferation rates in serial specimens.
Methods
Study design
Patients diagnosed with stage IV lung carcinoids at our institution were identified (n = 66). The cases were diagnosed predominantly during a 6-year period (2011–2016). Only samples with slides and/or blocks available for rereview were included in the study. In patients with >1 sample, diagnosis of carcinoid tumor in any one sample was sufficient for the inclusion in the study. Electronic medical records were reviewed to collect demographic, smoking, and radiological information. Slides from all available relevant specimens were collected and analyzed as described below. The study was performed with the approval of the Institutional Review Board of Memorial Sloan Kettering Cancer Center, New York.
Assessment mitotic counts
Mitotic counts were performed manually using an Olympus BX51 microscope (Olympus Optical Co., Tokyo, Japan). For this microscope, the diameter of a single high-power field (HPF)—a field of view with a 40× objective—is 0.55 mm (0.238 mm2), and 2 mm2 area corresponds to 8.4 HPFs. Thus, mitotic counts were recorded per 8.4 HPFs. Only definite mitotic figures were included, avoiding apoptotic or pyknotic cells. For resection specimens, mitotic figures were counted in at least 5 sets of 2 mm2 counts, and recorded as an average count per 2 mm2. For biopsies, the counts were performed in the entire evaluable tissue. For biopsies containing less than 2 mm2 of tumor (n = 16), a projected mitotic count per 2 mm2 was calculated based on counts in the evaluable tissue. Biopsies with <1 mm2 of tumor or with significant crush artifact were excluded from mitotic counts.
Immunohistochemistry
Immunohistochemistry was performed using standard methods on Ventana Discovery XT Automated Stainer (Ventana Medical Systems Inc., Tucson, AZ, USA). Ki-67 antibodies included MIB1 clone for histologic specimens (Dako; 1:200 dilution), and Ki-67 30-9 clone for cytology cell blocks (Ventana; prediluted) based on the prior data that MIB1 reactivity is inhibited by methanol-based fixatives in some cytologic collection media [24]. Rb immunohistochemistry was performed with antibody clone 13A10 (Leica; 1:50 dilution). Loss of Rb was defined as complete loss of nuclear staining in the presence of retained expression in nontumor cells (stromal, endothelial, or benign epithelial cells).
Assessment of Ki-67 index
The Ki-67 index was assessed by counting a percentage of cells with positive-nuclear labeling relative to total number of counted cells. For each specimen, the distribution of Ki-67 was assessed at scanning magnification. For samples with homogeneous Ki-67 (same rate throughout the evaluable tissue; n = 19), a single Ki-67 value was recorded based on the counts of all cells in a representative 20× field or at least 500 cells in dispersed biopsies or fine needle aspiration cell blocks. In specimens with heterogeneous Ki-67 areas (n = 89), a hot-spot and a weighted average (average) Ki-67 values were recorded. For hot-spot Ki-67, the highest Ki-67 rate in a 20× field was recorded [6, 25]. Average Ki-67 rate was calculated as follows: [% surfacea × Ki-67 ratea] + [% surfaceb × Ki-67 rateb] + [% surfacec × Ki-67 ratec]. For example, a tumor with Ki-67 rate of 2% occupying 80% of the tumor, and 10% Ki-67 rate occupying 20% of the tumor, would have average Ki-67 of (2% × 0.8) + (10% × 0.2) = 3.6%.
Next-generation sequencing
Targeted next-generation sequencing was performed using the hybridization capture-based MSK-IMPACT platform (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets assay), as previously described [26]. Briefly, DNA extracted from formalin-fixed paraffin embedded tumor samples was sequenced on an Illumina HiSeq2500. Patient’s peripheral blood DNA was used as a reference to ensure the somatic nature of the variant calls.
Results
Patient and sample characteristics
Patients with stage IV lung carcinoids (n = 66) had the following characteristics: mean age 62 (range: 25–84), 64% female and 50% never smoker (Table 1). Most patients (71%) had stage IV disease at presentation, whereas 29% developed stage IV following the resection of early-stage tumors. Patients had on average 2 tissue samples through the course of disease (range: 1–5), with a total number of samples for the entire cohort of 132. Sample were derived from metastatic sites (n = 93) and primary tumors (n = 39), and included surgical resections/excisions (n = 53), biopsies (n = 66), and fine needle aspirates (n = 13). Of resected primary tumors (n = 20), 16 were atypical and 4 were typical carcinoids. Analyses performed included mitotic counts on 117 samples (excluded were fine needle aspirates and biopsies with severe crush artifact or minute size, precluding mitotic counts; n = 15) and Ki-67 quantification on 108 samples (excluded from Ki-67 assessment were samples for which paraffin blocks were unavailable or insufficient for immunohistochemistry; n = 24); 93 samples had both mitotic and Ki-67 rates assessed.
Metastatic site distributions
The most common sites of distant metastases represented in pathologic samples comprised liver (33% of metastatic samples), skin (11%), bone (10%), and brain (6%). Based on the review of computed tomography scans, the most common sites involved radiologically included liver (74% of patients), bone (56%), and brain (32%) (Table 2).
Spectrum of mitotic counts and Ki-67 rates
Mitotic counts, performed on 117 samples, averaged 4.8 per mm2 (range: 0–44 per 2 mm2), and tended to be higher at metastatic than primary sites (mean 5.6 vs. 3.0 mitoses per 2 mm2, respectively; p = 0.06; Fig. 1a, b). Mitoses exceeding 10 per 2 mm2 were identified in 19 of 81 (23%) metastatic samples, of which 3 exceeded 20 per 2 mm2.

Distribution of mitotic counts and Ki-67 rates in stage IV carcinoids at primary and metastatic sites: dot-pots (a, c, e; bars show mean and standard deviation) and histograms (b, d, f)
Ki-67, quantified on 108 samples, revealed the mean (range) for hot-spot and average values of 15.9% (0.5–60%) and 8.6% (0–49%), respectively. Overall, Ki-67 was significantly higher in metastatic than primary samples for both hot-spot (p = 0.02) and average (p = 0.01) values (Fig. 1c–f). Hot-spot Ki-67 rate exceeding 20% was seen in 13% (4/31) of primary and 27% (21/77) of metastatic samples. Average Ki-67 rate exceeding 20% was seen in only 9 samples (8%), all from metastatic sites.
Samples with elevated proliferation rate: clinicopathologic and molecular characteristics
As summarized in Table 3, elevated proliferation rates—defined as >10 mitoses per 2 mm2 or >20% hot-spot Ki-67—were identified in 35 of 132 (27%) samples, with at least one highly proliferative sample found in 28 of 66 (42%) patients. This included 16 samples with elevated Ki-67 only, 10 samples with elevated mitoses only (for 6 of 10 latter samples Ki-67 was unavailable), and 9 samples with elevation of both mitoses and Ki-67. In the majority of cases, elevated mitotic counts were in the range of >10–20 per 2 mm2, and elevated Ki-67 rates were in the range of >20–40% (see Fig. 1), with only 6 samples—all metastatic—surpassing these ranges (detailed summary in Supplemental Fig. 1). Notably, in nearly all cases (22/25), Ki-67 elevation above 20% was regional, in that Ki-67 hot-spots were present in the background of lower-proliferative areas of 1–10% Ki-67 (Fig. 2); the three samples with homogeneously elevated Ki-67 were small biopsies.

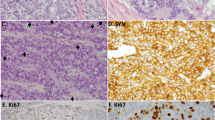
Well-differentiated morphology characteristic of the majority of carcinoids with elevated proliferation rate. Liver metastasis (a–c) and ovarian metastasis (d–f) demonstrating nested/trabecular architecture and bland, monotonous plasmacytoid cytomorphology with low-nuclear/cytoplasmic ratios (a, d) but elevated mitotic counts (arrows) and Ki-67 of >20% in hot-spot areas (b, e) in the background of low-proliferative Ki-67 areas (c, f)
Histologically, the majority (28/35) of samples with elevated proliferation rates exhibited entirely well-differentiated neuroendocrine tumor morphology, characterized by nested/trabecular architecture, bland uniform usually plasmacytoid cytology, and low-nuclear/cytoplasmic ratios (Fig. 2). In a minority of cases (7/35), areas of histologic disorganization were noted, manifesting as increased cell crowding, higher nuclear/cytoplasmic ratios, presence of intra-nuclear inclusions, increased prominence of nucleoli, and/or loss of nested/trabecular architecture with sheet-like or single cell/cord-like infiltration. In four cases, such areas were juxtaposed with regions of conventional carcinoid morphology (as illustrated in Fig. 3), and in three other cases, metastatic biopsies were composed entirely of cells with increased cell crowding, whereas well-differentiated carcinoid morphology was evident in samples from the primary tumors (as illustrated in Fig. 4a–d). None of the cases exhibited geographic/confluent necrosis typical of small cell or large cell neuroendocrine carcinomas (except for one case of 15 cm ovarian metastasis that showed large zones of infarct-like necrosis, which was likely related to torsion/ischemic changes). Conversely, focal/punctuate (comedo-like) necrosis—a characteristic feature of atypical carcinoids—was seen in 22/35 (63%) samples.

Examples of histologic disorganization and hypercellularity in a minority of carcinoids with elevated proliferation rates. a–f Illustrate a liver metastasectomy specimen with well-demarcated areas of increased proliferation associated with increased cell crowding and higher nuclear/cytoplasmic ratios (d, f) in the background of conventional nested/organoid carcinoid morphology with low mitotic and Ki-67 rates (c, e). This was the only specimen in this series where areas of increased proliferation were sharply demarcated from low-proliferative areas by H&E. g, h Illustrate specimens from lung (g) and liver (h) in which focal areas of singe cell/cord-like infiltration were intermixed with areas of conventional carcinoid morphology. Arrows in d highlight mitotic figures. Despite increased cellularity and loss of organoid architecture, none of the cases exhibited geographic/confluent necrosis; only limited comedo-like necrosis was present (h, asterisks)

Histopathologic features and next-generation sequencing in a case showing marked proliferative progression and increased cell crowding at metastatic site (patient 8 in Fig. 6a). H&E section from the endobronchial biopsy (a) shows nested pattern and bland nuclei with no identifiable mitoses and uniformly low- Ki-67 index (<5%; panel c). Core biopsy from supraclavicular metastasis (b, d) shows elevated mitotic counts (arrows; 11 per 2 mm2), high hot-spot Ki-67 of 40–60% (seen in the majority of the specimen; d—left), but with focal low-proliferative regions (d—right); H&E section shows increased cell crowing with the loss of organoid morphology (b). By next-generation sequencing, primary and metastatic samples harbored identical somatic KDM5C E646A mutation and CDKN1B deletion (e)
Highly proliferative samples were found primarily at metastatic sites (31/35; 89%), including liver (n = 7), brain (n = 5), bone (n = 3), and other sites (n = 16). Interestingly, elevated proliferation rates were over-represented in brain metastases compared to liver (P = 0.008) and bone (P = 0.043) specimens (Fig. 5).

Prevalence of samples with elevated proliferation rate by metastatic site. *P < 0.05; **P < 0.005
Patients with highly proliferative samples were predominantly women (64%) and never-smoker (64%). Survival analysis, performed with a median follow-up of 2.4 years (range: 0.05–11.9 years), revealed that after stage IV diagnosis, these patients had a median overall survival of 2.7 years, which was comparable to that of patients without the evidence of elevated proliferation rates. One-year and 3-year overall survivals were 92 and 49%, respectively (Table 3).
Review of next-generation sequencing results (n = 9) and analysis of Rb expression by immunohistochemistry (10 additional cases) was performed on a total of 19 samples with elevated proliferation rates (Table 3). No genomic alterations in RB1 or TP53 were identified, and none of the samples exhibited the loss of Rb expression. Instead, 5 of 9 cases tested by next-generation sequencing revealed alterations in MEN1 and other chromatin modifiers (ARID1A, ARID1B, and KDM5C), as seen in carcinoids. Also, in contrast to neuroendocrine carcinomas, these tumors had a low-mutation burden (averaging 1.5 mutations per Mb; range: 0.9–3.9).
Evolution of proliferation rate in patients with >1 sample
Evolution of proliferation rates was assessed for 40 patients who had more than 1 evaluable sample, which included 32 patients with samples from both primary and metastatic tumors (48 metastatic/primary tumor pairs) and 8 patients with >1 metastatic sample. The heat-map depicting mitotic and Ki-67 indices in serial samples highlights the remarkable heterogeneity of mitotic and Ki-67 rates in different samples through the course of disease (Fig. 6a), including the co-occurrence of samples with elevated proliferation rates (red and orange fields) and samples with conventional proliferation rates in the same patient.

Evolution of proliferation rate in stage IV carcinoid patients with >1 evaluable sample. In a heat-map (a), each row represents a single patient and columns represent the values for primary (P) and metastatic samples (M, # in chronological order). White spaces represent absence of value or specimen. “R” indicates resection specimens. Flow chart (b) summarizes proliferation rate changes in matched metastatic (Met) vs. primary samples. Specimen types in each group are summarized in a table below the flow chart. Res = resections/excisions, small = nonresection (biopsy/fine needle aspirate) specimens. c Graphic representation of evolution and heterogeneity of proliferation rates in multiple samples. Bx biopsy, met metastasis, Res resection
Comparison of proliferation rates in matched metastatic vs. primary samples (n = 48 pairs) revealed that 17 (35%) exhibited an increase in mitoses and/or hot-spot Ki-67 by ≥10 points, of which 9 samples showed a marked (≥20 point) elevation at metastatic sites. Conversely, ≥10 and ≥20 point decrease in proliferation rate was observed in only 5 and 1 metastatic samples, respectively (all biopsies; Fig. 6b). Eleven matched pairs comprised resections of both primary and metastatic samples, allowing the greatest accuracy of proliferation rate comparison; of those, 3 metastatic samples (27%) exhibited the escalation of proliferation rate by ≥10 points, whereas the rest showed similar proliferation rates.
Of patients with matched primary and metastatic samples, 5 had the diagnosis of typical carcinoid in resected primary tumors (Fig. 6a, Supplemental Fig. 2). Metastatic samples from 2 of these patients (patient ID 2 and 12 in Fig. 6a) revealed mitotic count increase from 0 to 3.2 and 1.7 to 5 per 2 mm2, respectively, thus evolving to the criteria of atypical carcinoids. In both patients, Ki-67 rate also became markedly elevated (up to 40% in hot-spot areas). The third patient (patient ID 10) did not show a detectable elevation of mitoses, but the hot-spot Ki-67 increased from 5 to 15%. For the remaining two patients (patient ID 11 and 13), proliferation values remained low in the evaluable metastatic samples. Overall, 3 of 5 patients with the diagnosis of typical carcinoid in resected primary tumors developed increased mitotic and/or Ki-67 rates in metastatic samples, some with marked Ki-67 elevation.
Next-generation sequencing of matched primary and metastatic samples showing marked progression of proliferation rate
Given the strikingly increased proliferation rate in some metastatic samples, it could be hypothesized that such metastases were arising from unrelated primary tumors, although in none of those patients was there a suspicion of another primary tumor on clinicoradiologic grounds. Nevertheless, to address this definitely, we performed next-generation sequencing on both primary and metastatic samples from one such patient (Fig. 4). The patient was a 70-year-old woman, a never smoker, whose bronchoscopic biopsy of a lung mass revealed a carcinoid tumor with undetectable mitotic rate and Ki-67 of <5%, whereas a biopsy of supraclavicular lymph node revealed a metastatic neuroendocrine neoplasm with a Ki-67 index of 40–60% in the majority of the specimen but with focal areas of low (5%) Ki-67. Next-generation sequencing on both samples revealed matching somatic mutations in KDM5C gene (p.E646A) and matching deletions of CDKN1B, supporting their clonal relationship. Neither specimen harbored RB1 or TP53 alterations (Fig. 4e).
Mitotic counts vs. Ki-67 as a function of specimen type, and challenges of classifying stage IV carcinoids into dichotomous typical vs atypical categories
As a secondary analysis, we compared the distribution of mitotic counts versus Ki-67 rate as a function of specimen type in samples for which both measurements were available (n = 93). As shown in Fig. 7a, overall, mitotic counts and Ki-67 showed a good correlation (Pearson r = 0.59). However, by specimen type, low/undetectable mitotic counts (<2 per 2 mm2) were significantly over-represented in biopsies compared to resections (60% vs. 22%, respectively; p = 0.0003), whereas the distribution of Ki-67 rates did not vary significantly as a function of specimen type (Fig. 7b).

Comparison of mitotic counts versus hot-spot Ki-67 index in the same sample (n = 93) as a function of specimen type. a Scatter dot plots. Black dots represent resection specimens, gray—biopsy specimens. Bars indicate a mean. b Summary for the distribution of mitotic counts vs. Ki-67 index by specimen type highlighting over-representation of low-mitotic counts (<2 per 2 mm2) in biopsies compared to resections (60% vs. 22%, P = 0.0003)
WHO classification was developed for resected primary carcinoids, whereas its applicability to stage IV setting is not well established; the challenges are illustrated in Supplemental Table 1. Aside from the lack of a category for samples with mitoses of >10 per 2 mm2, other issues included frequent biopsies with low-/undetectable-mitotic counts, in which typical carcinoids cannot be distinguished from low-end of atypical carcinoids [2, 25], and frequent detection of mitotic counts both below and above 2 mm2 in different samples from the same patient (42% of patients with >1 sample).
Discussion
To our knowledge, this is the largest series to date to examine the spectrum and evolution of proliferation rates in stage IV lung carcinoids. The major finding in this study is that 27% of samples in this setting exceed the current ceiling criteria of 10 mitoses per 2 mm2 and/or Ki-67 rate of 20%, as defined largely based on early-stage tumors. The second major finding is that escalation of proliferation rate is a common feature of metastatic progression in lung carcinoids, with a 10-point or more increase of mitoses and/or Ki-67 observed in 35% of metastatic samples compared to matched primary tumors. These findings have implication for classification, differential diagnosis, and potentially clinical management of metastatic carcinoids.
Implications for classification
Currently, mitotic counts are considered to be widely separated in carcinoids and neuroendocrine carcinomas, and are thus regarded as a strict criterion for the diagnosis of these tumor types in the WHO classification [2]. While the ceiling rate of 10 mitoses for 2 mm2 is indeed largely suitable in early-stage carcinoids—with evidence of only rare exceptions recently emerging (see below)—it is clear from the current study that this mitotic threshold is not infrequently exceeded, albeit usually only mildly, in metastatic carcinoids (23% of metastatic samples). How such samples should be classified, and whether they should be regarded as a de facto large cell neuroendocrine carcinoma, is unclear in the current classification system. Similarly unclear is how to classify neuroendocrine neoplasms with well-differentiated morphology of carcinoid tumors, but Ki-67 rate exceeding the 20% threshold commonly regarded as a ceiling rate for the separation of carcinoids from neuroendocrine carcinomas in small specimens [2, 9,10,11], which in this series accounted for 27% of metastatic samples. Several lines of evidence provide support that samples with elevated mitotic and/or Ki-67 rates described herein represent carcinoids with elevated proliferation rate rather than neuroendocrine carcinomas:
First, based on histologic considerations, samples with increased proliferation rates exhibited well-differentiated morphology (bland, uniform, usually plasmacytoid cells in nested/trabecular arrangements, and consistently lacking geographic necrosis), characteristic of carcinoids/well-differentiated neuroendocrine tumors (as illustrated in Fig. 2). Likewise, morphology of the analogous early-stage tumors with elevated mitotic counts in the study by Quinn et al. [22] was described as “readily identifiable as carcinoid tumors”.
Second, in most cases, elevated proliferation rates were found in patients who had additional samples during the course of disease showing carcinoids with conventional proliferation rates. In most instances, elevated proliferation rates were found in metastatic samples, while matched primary tumors specimensif available—demonstrated conventional proliferation rates. In one such patient (illustrated in Fig. 4), we confirmed using next-generation sequencing that a tumor pair of this type was clonally related, providing direct evidence for the evolution of highly proliferative metastasis from the lower-proliferative lung carcinoids. In addition, co-existent low-proliferative zones evident in most samples also support the concept of increased proliferation rates arising in the background of conventional carcinoids.
Third, exploratory genomic analysis on a subset of highly proliferative samples revealed the presence of MEN1 and other chromatin-modifier gene mutations, low-mutation burden, and the lack of RB1 or TP53 gene alterations, in line with genomic findings expected for carcinoid tumors [27], and sharply contrasting the genomic profiles of neuroendocrine carcinomas, which show consistent mutations in TP53, frequent alterations in RB1, and high-tumor mutation burden [21, 28, 29]. Similar findings were recently reported by our group for two early-stage tumors of this type [21]. In addition, carcinoid-like genomic profiles for tumors with morphologic features resembling atypical carcinoids but elevated mitotic counts, were reported in the abstract form by Vivero and Sholl [30], where such tumors were designated as “borderline neuroendocrine carcinomas”. Together, prior and current genomic observations support that these tumors are molecularly akin to carcinoids rather than neuroendocrine carcinomas. Nevertheless, further molecular studies are needed to determine the genomic or epigenomic basis of their deregulated proliferation.
Fourth, on the clinical side, survival analysis revealed a more protracted clinical course for the patients with highly proliferative samples (median survival 2.7 years after stage IV diagnosis) compared to <12 months median survival in the historic cohorts for stage IV large cell [31] and small cell [32, 33] neuroendocrine carcinomas. Similarly, analogous patients in the study by Quinn et al. [22] had substantially longer survivals than expected for neuroendocrine carcinomas. Furthermore, unlike neuroendocrine carcinomas, no responses to platinum-based agents were observed in four patients treated for recurrent disease in that study [22]. Demographically, our patients were predominantly never-smokers, which contrasts with consistent smoking histories in patients with neuroendocrine carcinomas.
Lastly, it is important to note that in the majority of cases with increased proliferation rates, mitotic and Ki-67 thresholds were surpassed only mildly, remaining largely below 20 per 2 mm2 for mitotic counts and at or below 40% for hot-spot Ki-67 rate. Such proliferation rates are generally uncommon for neuroendocrine carcinomas, which typically feature median mitotic counts of 70–80 per 2 mm2 and Ki-67 rates of >50–100% [2]. Nevertheless, some neuroendocrine carcinomas, particularly of large cell type, do exhibit lower proliferation rates, overlapping with those observed in this series (see below) [1, 2, 5, 21].
Taken together, the above histologic, molecular, and clinical considerations support the conclusion that samples with elevated proliferation rates do not represent bona fide neuroendocrine carcinomas, despite the overlap in the proliferation rates with those tumors. These observations closely parallel those in multiple recent studies on enteropancreatic neuroendocrine tumors, which described tumors meeting the proliferative criteria of neuroendocrine carcinomas, yet exhibiting morphologic, molecular, and clinical characteristics akin to grade 1–2 neuroendocrine tumors, leading to a recent recommendation to classify such tumors as grade 3 neuroendocrine tumors rather than carcinomas [12,13,14,15,16,17,18,19]. Our findings echo this recommendation for lung tumors.
Although the current study was based on stage IV tumors, it is becoming increasingly recognized that carcinoids with mitotic counts exceeding 10 per 2 mm2 can also be rarely encountered in resected, early-stage tumors [21,22,23]. Indirectly, the expanded range of mitotic count observed in stage IV setting also supports the notion that 10 mitoses per 2 mm2 is not the absolute ceiling rate for lung carcinoids in general, although it does encompass the vast majority rates in the primary tumors. Further studies in early-stage cohorts will be needed to address how such rare tumors are best incorporated into the WHO classification relative to the existing criteria.
Evolution of proliferation rate
The second major finding in this study concerns the evolution of proliferation rate in metastatic carcinoids. We document an increase in either mitoses and/or Ki-67 by ≥10 points in 35% of metastatic samples relative to the rate in the primary tumors. However, we emphasize that escalation was not universal, and in most metastatic samples evident proliferation rate remained comparable to that in the primary tumor. These findings are in line with clinical observations that some metastatic carcinoids display indolent growth, while others exhibit more rapid progression [34]. We acknowledge that the data on the proliferative dynamics may be limited by the inherent possibility of under-estimation of proliferation rate in small specimens; however, a subset of these observations was made in paired resection specimens, showing a similar rate of escalation. Interestingly, proliferative progression may be the hallmark of metastatic progression in neuroendocrine tumors of various sites, since this phenomenon has been well-documented in pancreatic and intestinal neuroendocrine tumors [35,36,37,38]. We are aware of only a single report describing escalation of Ki-67 in three cases of metastatic lung carcinoids [35], as well as a case report describing variability of proliferation rate in different samples causing a “diagnostic puzzle” in a patient with metastatic carcinoid [39]. However, to our knowledge, this is the first documentation of a high prevalence of this phenomenon in stage IV carcinoids.
Of particular interest are our observations related to proliferation rates in metastases arising from typical carcinoids, as diagnosed in resected primary tumors prior to the development of stage IV disease. Of five such patients, metastatic samples exhibited proliferative escalation in three cases, resulting in the increase of mitotic counts to the criteria of atypical carcinoids in two patients, which was also accompanied by marked elevation of Ki-67 rate. Overall, distant metastases arising from resected typical carcinoids are rare (~3%) [40, 41]. To our knowledge, this is the first documentation that when distant metastases do occur, typical carcinoids have the capacity to develop elevated—and in some instances substantially elevated—proliferation rates. These data indicate that proliferation rate is a dynamic variable during metastatic progression, and the diagnosis of typical carcinoid in the primary tumor does not assure low- proliferation rate in metastases. This has potential clinical implications as the current management of stage IV carcinoids is increasingly guided by tumor aggressiveness, and takes into account the diagnosis of typical vs atypical carcinoid [42, 43]. These data stress the importance of ample sampling of metastatic sites, as well as correlation with clinicoradiologic rate of disease progression in rare patients with metastases arising from typical carcinoids.
The above findings also have clear implications for the classification of lung carcinoids in stage IV setting. The findings of progression and variability of proliferation rates at metastatic sites indicate that the dichotomous classification of carcinoids into typical vs. atypical—as defined for resected early-stage tumors [5]—is not applicable in metastatic setting. This issue is further compounded by the challenges in detecting minute differences in mitotic counts that separate typical and low-end of atypical carcinoids in biopsies—the dominant specimen type in metastatic setting. Thus, rather than classifying metastatic carcinoids into typical and atypical, we suggest that their classification should be restricted to general tumor type as “metastatic carcinoid”, followed by a comment on the evident mitotic and/or Ki-67 rate in individual sample(s). We emphasize, however, that the clinical utility of proliferation metrics in the management of stage IV lung carcinoids requires clinical validation.
Practical diagnostic implications
From the diagnostic perspective, given that the majority of highly-proliferative samples displayed the characteristic well-differentiated carcinoid morphology, and only mildly elevated proliferation rates, their separation from neuroendocrine carcinomas should not present a major diagnostic challenge in routine practice, aside from the current dilemma related to their nomenclature. Nevertheless, it can be readily anticipated that carcinoids with elevated proliferation rates may present a major diagnostic challenge in the setting of a crushed, poorly preserved biopsy. Presence of crush artifact is a hallmark feature of neuroendocrine carcinomas, especially small cell carcinoma; however, it can also be encountered in biopsied carcinoids. In this setting, Ki-67 is currently a routine method for distinguishing carcinoids (<20% Ki-67) and neuroendocrine carcinomas (generally >50% Ki-67) [2, 9,10,11], although there is no recommendation for the interpretation of cases with in-between “gray zone” rates. Our findings do not dispute the utility of Ki-67 at the ends of proliferation spectra, but they emphasize that the intermediate rates are commonly encountered in stage IV carcinoids, and could present a pitfall for over-diagnosis as neuroendocrine carcinomas. This particularly applies to a minority of cases reaching the hot-spot Ki67 rates of > 40–60%, which could be most susceptible to mis-interpretation. Thus, Ki-67 rates of >20–60% in small or crushed biopsies of neuroendocrine neoplasms should be interpreted with caution, and should not be regarded as unqualified evidence of neuroendocrine carcinoma.
Another potential diagnostic challenge, even in well-preserved specimens, could arise for the distinction of highly-proliferative carcinoids and large cell neuroendocrine carcinomas, given that these tumors share nested/organoid architecture and some large cell neuroendocrine carcinomas have fairly monotonous cytomorphology. Our findings emphasize that proliferation rates in the “gray zone” cannot be used as the exclusive criterion for distinguishing these tumors, and other features must be taken into consideration. Generally, large cell neuroendocrine carcinomas display more prominent nucleoli and overall greater cytologic atypia than carcinoids. Furthermore, geographic/confluent necrosis is a consistent feature of large cell neuroendocrine carcinomas, whereas necrosis in all cases in this series was limited to small punctuate, comedo-like areas. Lastly, regional rather than homogeneous pattern of Ki-67 elevation, in which Ki-67 hot-spots coexist with low-proliferative areas, was seen in the majority of highly-proliferative carcinoids in this series, whereas neuroendocrine carcinomas typically lack significant low-proliferative zones. Thus, heterogeneous elevation of Ki-67 may serve as a clue to carcinoids with elevated proliferation rates over neuroendocrine carcinomas. The heterogeneous nature of Ki-67 elevation in lung carcinoids was recently highlighted by Marchiò et al. [44], and is also a well-known feature of enteropancreatic neuroendocrine tumors [6, 45]. Nevertheless, it is possible that in a small biopsy sample, the distinction of a highly proliferative carcinoid from large cell neuroendocrine carcinoma or crushed small cell carcinoma may be extremely difficult. In this setting, additional immunohistochemical markers or molecular studies may be helpful, as demonstrated in pancreatic tumors, where Rb, p53, and several other markers have been shown to aid in the separation of grade 3 pancreatic neuroendocrine tumors from carcinomas [46, 47]. Further studies will be needed to explore the utility of additional ancillary studies for this differential diagnosis in lung tumors. Preliminarily, our findings support that genomic studies and/or documentation of Rb loss or aberrant p53 expression by immunohistochemistry could aid in separating neuroendocrine carcinomas from highly-proliferative carcinoids.
Also noteworthy are the areas of unusual morphology (increased cell crowding, loss of organoid architecture, and single-cell infiltration), observed in a minority of carcinoids with increased proliferation rates. Such areas may be a reflection of disordered differentiation, and have been noted in some atypical carcinoids in several prior publications [48, 49]. Of cases with altered morphology, it appears that none fully fit the concept of full-fledged “transformation”, as exemplified by small cell transformation of EGFR-mutated lung adenocarcinoma, or the well-established phenomena of transformation of low-grade lymphoma or glioma, where secondary tumors exhibit full morphologic phenotype of the high-grade counterparts. Rather, the findings here may best fit the concept of a more moderate histologic progression, as known to occur in various metastatic tumors [50, 51]. In particular, most cases here exhibited substantially lower proliferation rates than conventional neuroendocrine carcinomas, and none showed evidence of geographic necrosis, or acquisition of TP53/RB1 genomic alterations. We note that although few instances of apparently full histologic transformation from carcinoids to overt secondary neuroendocrine carcinomas have been documented in the thymus, accompanied by acquisition of TP53 mutation in one case [52,53,54], the possibility of such phenomenon occurring in lung carcinoids will require further study.
Issues related to the assessment of Ki-67 and mitotic counts in stage IV carcinoids
Currently, there is no recommendation in thoracic pathology literature to routinely document Ki-67 values in lung carcinoids, aside from diagnostic role of Ki-67 in distinguishing carcinoids from neuroendocrine carcinomas in crushed biopsies [2, 55]. Conversely, recent National Comprehensive Cancer Network (NCCN) [42] and European Neuroendocrine Tumor Society (ENETS) [43] guidelines do incorporate Ki-67 and mitotic indices in the algorithms for systemic management of patients with stage IV lung carcinoids, although these recommendations are largely adopted from the clinical approach to enteropancreatic neuroendocrine tumors [6, 56]. Thus, while clinical validation is clearly warranted, the use of proliferation metrics in metastatic lung carcinoids is likely to become more routine in pathology practice, as supported by a recent survey indicating that most oncologists use Ki-67 results to make management decisions for pulmonary carcinoids [57]. With increased use, the awareness of the expanded Ki-67 and mitotic spectrum in metastatic carcinoids will become increasingly important to avoid their overdiagnosis as neuroendocrine carcinomas.
The method of scoring of Ki-67 as hot-spot vs. average value, as well as other analytical and postanalytical variables, are currently not standardized for lung carcinoids given the aforementioned lack of consensus on its prognostic or predictive utility. Hot-spot values were emphasized in this study in order to highlight potential diagnostic pitfall that can be presented by sampling of such areas in small biopsies. Furthermore, hot-spot approach has been recommended a general approach for neuroendocrine tumors of various organs in the recent expert consensus proposal [58].
The data on concurrent analysis of mitoses and Ki-67 in a large number of samples allowed us to illustrate the clear advantage of Ki-67 for gauging of proliferative status of carcinoid tumors in small biopsies. This concept is not novel, but we are not aware of prior studies to directly demonstrate that Ki-67 is substantially less affected by specimen size than mitotic counts. Although the role for Ki-67 in grading/classification of resected, early-stage carcinoids is currently debated, our data support that proliferative assessment of small biopsies—dominating in stage IV setting—requires Ki-67 marker.
Our description of a high prevalence of elevated mitoses and Ki-67 in stage IV lung carcinoids is novel, even though many prior studies have assessed these parameters in pulmonary carcinoids. Importantly, prior studies were based predominantly or entirely on early-stage tumors [2]. For example, in a recent large series of early-stage carcinoids, only 1.7% of all cases (5.9% of atypical carcinoids) displayed hot-spot Ki-67 rate of ≥25% [44]. Thus, the high prevalence of samples with elevated proliferation rates in this series largely reflects the unique nature of this cohort, consisting entirely of stage IV tumors.
We note that this study was focused on proliferation rate in metastatic carcinoids, specifically with the goal to describe the subset of cases exceeding the current proliferative criteria and potential diagnostic or classification issues that could arise for such cases. However, the detailed analysis of correlation of proliferation rates with prognosis and treatment outcomes, as well as the analysis of other factors potentially associated with aggressiveness of metastatic carcinoids will require future study.
Metastatic site distribution
In terms of the distribution of metastatic sites, our series are in line with prior studies showing that the most common sites of distant metastases for lung carcinoids include liver, bone and brain [59,60,61]. We also noted frequent involvement of unusual sites, such as skin, breast, eye, and ovary. Our finding that increased proliferation rates are over-represented in brain metastases is intriguing; future studies will be needed to explore the potential biological factors contributing to this predilection. Overall, the high rate of brain metastases in patients with stage IV lung carcinoids (32% of patients in this series) has been also documented in prior studies [59,60,61], but this remains an under-recognized phenomenon in clinical practice.
Conclusions
This is one of the largest studies to date to examine the spectrum and evolution of proliferation rates in stage IV lung carcinoids. It reveals that in the metastatic setting, lung carcinoids frequently exceed the mitotic and Ki-67 ceiling criteria currently recognized in the WHO classification for primary tumors, and commonly exhibit proliferative escalation at metastatic sites. These findings add to the emerging concept of elevated (grade 3) proliferation rate in well-differentiated neuroendocrine tumors of enteropancreatic sites. Awareness of increased proliferative range in metastatic carcinoids is critical for avoiding their over-diagnosis as neuroendocrine carcinomas. Further studies are warranted to assess the utility of Rb and p53 immunohistochemistry and/or molecular testing in separating problematic cases, and to explore the impact of proliferation indices on prognosis and therapeutic responses of patients with metastatic carcinoids.
References
Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med. 2010;134:1628–38.
Beasley MB, Brambilla E, Chirieac LR, et al. Carcinoid tumour. In: Travis WD, Brambilla E, Burke AP, et al. (editors). WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. Lyon: IARC Press; 2015. p. 73–77.
Frilling A, Modlin IM, Kidd M, et al. Recommendations for management of patients with neuroendocrine liver metastases. Lancet Oncol. 2014;15:e8–21.
Abdel-Rahman O. Modified staging system for pulmonary carcinoids on the basis of lung cancer TNM system. Clin Transl Oncol. 2018;20:670–77.
Travis WD, Rush W, Flieder DB, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22:934–44.
Klimstra DS. Pathologic classification of neuroendocrine neoplasms. Hematol Oncol Clin North Am. 2016;30:1–19.
Pelosi G, Rindi G, Travis WD, et al. Ki-67 antigen in lung neuroendocrine tumors: unraveling a role in clinical practice. J Thorac Oncol. 2014;9:273–84.
Rindi G, Klersy C, Inzani F, et al. Grading the neuroendocrine tumors of the lung: an evidence-based proposal. Endocr Relat Cancer. 2014;21:1–16.
Fabbri A, Cossa M, Sonzogni A, et al. Ki-67 labeling index of neuroendocrine tumors of the lung has a high level of correspondence between biopsy samples and surgical specimens when strict counting guidelines are applied. Virchows Arch. 2017;470:153–64.
Lin O, Olgac S, Green I, et al. Immunohistochemical staining of cytologic smears with MIB-1 helps distinguish low-grade from high-grade neuroendocrine neoplasms. Am J Clin Pathol. 2003;120:209–16.
Pelosi G, Rodriguez J, Viale G, et al. Typical and atypical pulmonary carcinoid tumor overdiagnosed as small-cell carcinoma on biopsy specimens: a major pitfall in the management of lung cancer patients. Am J Surg Pathol. 2005;29:179–87.
Velayoudom-Cephise FL, Duvillard P, Foucan L, et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr Relat Cancer. 2013;20:649–57.
Basturk O, Yang Z, Tang LH, et al. The high-grade (WHO G3) pancreatic neuroendocrine tumor category is morphologically and biologically heterogenous and includes both well differentiated and poorly differentiated neoplasms. Am J Surg Pathol. 2015;39:683–90.
Heetfeld M, Chougnet CN, Olsen IH, et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2015;22:657–64.
Tang LH, Untch BR, Reidy DL, et al. Well-differentiated neuroendocrine tumors with a morphologically apparent high-grade component: a pathway distinct from poorly differentiated neuroendocrine carcinomas. Clin Cancer Res. 2016;22:1011–7.
Milione M, Maisonneuve P, Spada F, et al. The clinicopathologic heterogeneity of grade 3 gastroenteropancreatic neuroendocrine neoplasms: morphological differentiation and proliferation identify different prognostic categories. Neuroendocrinology. 2017;104:85–93.
Raj N, Valentino E, Capanu M, et al. Treatment response and outcomes of grade 3 pancreatic neuroendocrine neoplasms based on morphology: well differentiated versus poorly differentiated. Pancreas. 2017;46:296–301.
Hijioka S, Hosoda W, Matsuo K, et al. Rb loss and KRAS mutation are predictors of the response to platinum-based chemotherapy in pancreatic neuroendocrine neoplasm with grade 3: a Japanese Multicenter Pancreatic NEN-G3 Study. Clin Cancer Res. 2017;23:4625–32.
Klöppel GCA, Hruban RH, et al. Neoplasms of the neuroendocrine pancreas, introduction. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J (editors). WHO Classification of Tumours of Endocrine Organs. Lyon: IARC; 2017.
Hann CL, Weu MA, Rekhtman N, et al. Small-cell and neuroendocrine tumors of the lung. In: DeVita VT, Rosenberg SA, Lawrence TS (editors). DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology. 11th Edition. Philadelphia: Wolters Kluwer, 2018, p. 671–99.
Rekhtman N, Pietanza MC, Hellmann MD, et al. Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin Cancer Res. 2016;22:3618–29.
Quinn AM, Chaturvedi A, Nonaka D. High-grade neuroendocrine carcinoma of the lung with carcinoid morphology: a study of 12 cases. Am J Surg Pathol. 2017;41:263–70.
Megyesi M, Berta M, Khoor A. Endobronchial large cell neuroendocrine carcinoma. Pathol Oncol Res. 2003;9:198–200.
Konno F, Jungbluth A, Frosina D, et al. Drastic loss of MIB1/Ki67 immunoreactivity in CytoLyt-fixed cell blocks. Lab Invest. 2015;95(Suppl. 1):95A.
Caplin ME, Baudin E, Ferolla P, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26:1604–20.
Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–64.
Fernandez-Cuesta L, Peifer M, Lu X, et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat Commun. 2014;5:3518.
George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53.
George J, Walter V, Peifer M, et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat Commun. 2018;9:1048.
Vivero M, Scholl LM. “Borderline” neuroendocrine carcinomas of the lung are clinically and genomically distinct from large cell neuroendocrine carcinoma. Mod Pathol. 2016;29(suppl 2):485A.
Naidoo J, Santos-Zabala ML, Iyriboz T, et al. Large cell neuroendocrine carcinoma of the lung: clinico-pathologic features, treatment, and outcomes. Clin Lung Cancer. 2016;17:e121–9.
Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24:4539–44.
Nicholson AG, Chansky K, Crowley J, et al. The International Association for the Study of Lung Cancer Lung Cancer Staging Project: Proposals for the Revision of the Clinical and Pathologic Staging of Small Cell Lung Cancer in the ForthcomingEighth Edition of the TNM Classification for Lung Cancer. J Thorac Oncol. 2016;11:300–11.
Wolin EM. Advances in the diagnosis and management of well-differentiated and intermediate-differentiated neuroendocrine tumors of the lung. Chest. 2017;151:1141–46.
Singh S, Hallet J, Rowsell C, et al. Variability of Ki67 labeling index in multiple neuroendocrine tumors specimens over the course of the disease. Eur J Surg Oncol. 2014;40:1517–22.
Shi C, Gonzalez RS, Zhao Z, et al. Liver metastases of small intestine neuroendocrine tumors: Ki-67 heterogeneity and World Health Organization grade discordance with primary tumors. Am J Clin Pathol. 2015;143:398–404.
Grillo F, Albertelli M, Brisigotti MP, et al. Grade increases in gastroenteropancreatic neuroendocrine tumor metastases compared to the primary tumor. Neuroendocrinology. 2016;103:452–9.
Panzuto F, Cicchese N, Partelli S, et al. Impact of Ki67 re-assessment at time of disease progression in patients with pancreatic neuroendocrine neoplasms. PLoS One. 2017;12:e0179445.
Derks JL, Speel EJ, Thunnissen E, et al. Neuroendocrine cancer of the lung: a diagnostic puzzle. J Thorac Oncol. 2016;11:e35–8.
Lou F, Sarkaria I, Pietanza C, et al. Recurrence of pulmonary carcinoid tumors after resection: implications for postoperative surveillance. Ann Thorac Surg. 2013;96:1156–62.
Garcia-Yuste M, Matilla JM, Canizares MA, et al. Surgical treatment of low and intermediate grade lung net. J Thorac Dis. 2017;9:S1435–41.
National Comprehensive Cancer Network (NCCN) Guidelines Version 2.2018: Neuroendocrine and Adrenal Tumors.
Pavel M, O’Toole D, Costa F, et al. ENETS Consensus Guidelines Update for the Management of Distant Metastatic Disease of Intestinal, Pancreatic, Bronchial Neuroendocrine Neoplasms (NEN) and NEN of Unknown Primary Site. Neuroendocrinology. 2016;103:172–85.
Marchio C, Gatti G, Massa F, et al. Distinctive pathological and clinical features of lung carcinoids with high proliferation index. Virchows Arch. 2017;471:713–20.
Yang Z, Tang LH, Klimstra DS. Effect of tumor heterogeneity on the assessment of Ki67 labeling index in well-differentiated neuroendocrine tumors metastatic to the liver: implications for prognostic stratification. Am J Surg Pathol. 2011;35:853–60.
Yachida S, Vakiani E, White CM, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36:173–84.
Tang LH, Basturk O, Sue JJ, et al. A Practical Approach to the Classification of WHO Grade 3 (G3) Well-differentiated Neuroendocrine Tumor (WD-NET) and Poorly Differentiated Neuroendocrine Carcinoma (PD-NEC) of the pancreas. Am J Surg Pathol. 2016;40:1192–202.
Tsuta K, Raso MG, Kalhor N, et al. Histologic features of low- and intermediate-grade neuroendocrine carcinoma (typical and atypical carcinoid tumors) of the lung. Lung Cancer. 2011;71:34–41.
Beasley MB, Thunnissen FB, Brambilla E, et al. Pulmonary atypical carcinoid: predictors of survival in 106 cases. Hum Pathol. 2000;31:1255–65.
Wu JM, Fackler MJ, Halushka MK, et al. Heterogeneity of breast cancer metastases: comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin Cancer Res. 2008;14:1938–46.
Cserni G. Tumour histological grade may progress between primary and recurrent invasive mammary carcinoma. J Clin Pathol. 2002;55:293–7.
Wick MR, Scheithauer BW. Oat-cell carcinoma of the thymus. Cancer. 1982;49:1652–7.
Moran CA, Suster S. Thymic neuroendocrine carcinomas with combined features ranging from well-differentiated (carcinoid) to small cell carcinoma. A clinicopathologic and immunohistochemical study of 11 cases. Am J Clin Pathol. 2000;113:345–50.
Fabbri A, Cossa M, Sonzogni A, et al. Thymus neuroendocrine tumors with CTNNB1 gene mutations, disarrayed ss-catenin expression, and dual intra-tumor Ki-67 labeling index compartmentalization challenge the concept of secondary high-grade neuroendocrine tumor: a paradigm shift. Virchows Arch. 2017;471:31–47.
Yi ES, Lee GK. Updates on selected topics in lung cancers: air space invasion in adenocarcinoma and Ki-67 staining in carcinoid tumors. Arch Pathol Lab Med. 2018;142:947–51.
Klimstra DS, Modlin IR, Adsay NV, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am J Surg Pathol. 2010;34:300–13.
Marchevsky AM, Hendifar A, Walts AE. The use of Ki-67 labeling index to grade pulmonary well-differentiated neuroendocrine neoplasms: current best evidence. Mod Pathol. 2018;31:1523–31.
Rindi G, Klimstra DS, Abedi-Ardekani B, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol. 2018;31:1770–86.
Hlatky R, Suki D, Sawaya R. Carcinoid metastasis to the brain. Cancer. 2004;101:2605–13.
Chong CR, Wirth LJ, Nishino M, et al. Chemotherapy for locally advanced and metastatic pulmonary carcinoid tumors. Lung Cancer. 2014;86:241–6.
Riihimaki M, Hemminki A, Sundquist K, et al. The epidemiology of metastases in neuroendocrine tumors. Int J Cancer. 2016;139:2679–86.
Acknowledgements
This study was supported in part by the grant from the Fiona and Stanley Druckenmiller Center for Lung Cancer Research (to NR). This research was made possible by infrastructure support by the NIH/NCI Cancer Center Support Grant P30 CA008748.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no relevant conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally as co-first authors: Natasha Rekhtman, Patrice Desmeules
Supplementary information
Rights and permissions
About this article

Cite this article
Rekhtman, N., Desmeules, P., Litvak, A.M. et al. Stage IV lung carcinoids: spectrum and evolution of proliferation rate, focusing on variants with elevated proliferation indices. Mod Pathol 32, 1106–1122 (2019). https://doi.org/10.1038/s41379-019-0248-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0248-2
This article is cited by
-
Neuroendocrine neoplasms of the lung and gastrointestinal system: convergent biology and a path to better therapies
Nature Reviews Clinical Oncology (2023)
-
Diagnostic, Prognostic, and Predictive Role of Ki67 Proliferative Index in Neuroendocrine and Endocrine Neoplasms: Past, Present, and Future
Endocrine Pathology (2023)
-
Lung neuroendocrine neoplasms: recent progress and persistent challenges
Modern Pathology (2022)
-
An analysis of 130 neuroendocrine tumors G3 regarding prevalence, origin, metastasis, and diagnostic features
Virchows Archiv (2022)
-
Challenges in High-grade Neuroendocrine Neoplasms and Mixed Neuroendocrine/Non-neuroendocrine Neoplasms
Endocrine Pathology (2021)
