
Abstract
Mesenchymal neoplasms of the uterus (corpus and cervix) encompass a heterogeneous group of tumors with differing morphologies, immunophenotypes and molecular alterations. With the advent of modern molecular techniques, such as next generation sequencing, newly defined genetic abnormalities are being reported in this group of neoplasms. Herein we report the clinicopathological and molecular features of a series of 13 spindle cell sarcomas of the uterus and vagina (10 cervix, 2 uterine corpus, 1 vagina) with morphology resembling fibrosarcoma. After targeted RNA-sequencing, dual FISH fusion and array-CGH analysis, 7 of 13 tumors exhibited NTRK rearrangements (6 TPM3-NTRK1 and 1 EML4-NTRK3) and 3 a COL1A1-PDGFB fusion; in the other 3 neoplasms, all of which were positive with S100 (2 diffuse, 1 focal), we identified no rearrangement. All the NTRK fusion-positive sarcomas were located in the cervix and exhibited diffuse staining with Trk while all the other neoplasms were negative. CD34 was diffusely positive in all 3 of the COL1A1-PDGFB fusion sarcomas. The latter molecular abnormality is identical to that commonly found in dermatofibrosarcoma protuberans and has not been reported previously in uterine mesenchymal neoplasms. We suggest that uterine sarcomas with a morphology resembling fibrosarcoma (and in which leiomyosarcoma and the known molecularly confirmed high-grade endometrial stromal sarcomas have been excluded) can be divided into 3 groups:- an NTRK fusion group, a COL1A1-PDGFB fusion group and a group containing neither of these molecular abnormalities which, on the basis of positive staining with S100, could be tentatively classified as malignant peripheral nerve sheath tumor, although additional molecular studies may identify specific genetic alterations necessitating a nomenclature change. We suggest a diagnostic algorithm when reporting such neoplasms. Identification of these newly described fusion-associated sarcomas is important given the potential for targeted treatments.
Similar content being viewed by others

Introduction
Mesenchymal neoplasms of the uterus (corpus and cervix) encompass a heterogeneous group of tumors with differing morphologies and immunophenotypes. The most common malignant mesenchymal neoplasms are leiomyosarcoma, low-grade and high-grade endometrial stromal sarcoma (YWHAE or BCOR rearranged), and undifferentiated sarcoma [1]. A variety of other malignant mesenchymal neoplasms occur rarely within the uterus, including malignant peripheral nerve sheath tumors which are most common in the cervix [2,3,4]. In 2011, 2 of us (TAL, AMM) coauthored a report of 3 cases of a rare malignant cervical mesenchymal neoplasm which was termed endocervical fibroblastic malignant peripheral nerve sheath tumor (neurofibrosarcoma) [5]; these cervical neoplasms were composed of fascicles of spindle cells arranged in herringbone, fascicular, and storiform patterns and were positive with S100 and CD34 [5].
Recently a series of 4 uterine mesenchymal neoplasms harboring NTRK (Neurotropic Tyrosine Receptor kinase) rearrangements was reported [6]. These neoplasms were described as having morphological features resembling fibrosarcoma and exhibited brisk mitotic activity and sometimes focal necrosis; immunohistochemically, there was variable expression of smooth muscle actin and S100, while SOX10, estrogen receptor (ER), progesterone receptor (PR), desmin, and CD34 were negative [6]. The 4 neoplasms expressed Trk (Tropomyosin Related Kinase) and harbored NTRK rearrangements (RBPMS-NTRK3, TPR-NTRK1, LMNA-NTRK1, and TPM3-NTRK1) [6]. The authors noted that endocervical fibroblastic malignant peripheral nerve sheath tumors bore some morphological and immunohistochemical resemblance to the neoplasms they reported and speculated that the former may represent NTRK fusion-positive uterine sarcomas.
Herein we report the clinicopathological and molecular features of a series of 13 spindle cell sarcomas of the uterus and vagina (10 cervix, 2 corpus, 1 vagina) with features resembling fibrosarcoma. We investigated these for NTRK abnormalities and show that NTRK rearrangements are common in these neoplasms. We also report the COL1A1-PDGFB fusion in a subset of these tumors; as far as we are aware, this fusion has not previously been reported in uterine mesenchymal neoplasms.
Materials and methods
Case selection
Thirteen tumors were derived from the in-house material and consultation practice of the pathology departments to which the authors are affiliated. The cases had all been diagnosed as malignant peripheral nerve sheath tumor or as undifferentiated sarcoma composed of uniform cells and resembling fibrosarcoma. The cases included the 3 originally reported endocervical fibroblastic malignant peripheral nerve sheath tumors (neurofibrosarcoma) [5]. Ten tumors were located in the cervix, 2 in the corpus and 1 in vagina. For two cases (10 and 12), we analyzed the primary tumor and the recurrence. Leiomyosarcomas, endometrial stromal sarcomas (low-grade and high-grade) and undifferentiated sarcomas composed predominantly of pleomorphic cells were not included in the study. All available slides were reviewed by two of the authors (SC and WGM); in consultation cases, often all of the slides were not available for examination.
Immunohistochemistry
Immunohistochemistry for Trk was performed on 4 micron thick sections. The primary antibody was a rabbit monoclonal anti- Trk (pan) covering Trk A, B and C (clone A7H6R, 92991 S, Ozyme, Cell Signaling). A dilution of 1/50 was used with 52 min incubation on a Benchmark ULTRA (Roche-Ventana, Tucson, AZ) and detection Kit Optiview DAB IHC (reference: 760-500). Heat-induced antigen retrieval was performed using Cell Ventana Conditioning buffer (CC1 standard) for 64 min. An infantile fibrosarcoma with a known NTRK fusion served as a positive external control.
A variety of other immunohistochemical stains were also undertaken. Most of these were performed at the various institutions during the original reporting of the neoplasms while others were undertaken during the preparation of this manuscript.
RNA Extraction and RNA-Sequencing
RNA was extracted from 14 macrodissected formalin fixed paraffin embedded tumor blocks according to the protocol Maxwell® RSC RNA formalin fixed paraffin embedded Kit (Promega®); in 1 case (case 13), sequencing was not performed. Tumor cell percentage varied from 20% to 80% (mean value 72%). Total RNA (200 to 250 ng) was reverse transcribed to cDNA. Libraries were prepared using the CTL FusionPlex Assay for Illumina Platform (ArcherDx®) following the manufacturer’s Protocol [7] and sequenced using MiSeq (Illumina, San Diego, CA).
Data were analyzed using the CTL Target Region File and vendor supplied software (Archer Analysis, version 5.0). A minimum of 5 reads with 3 unique sequencing start sites that cross the breakpoints was set as the cutoff value to indicate strong evidence of fusions. The percentage of fusion reads is calculated as following: only reads spanning the breakpoint are considered to support the fusion/isoform. The percentage is calculated in reference to the total number of reads intersecting the genomic locations on either side of the primary breakpoint, restricted to RNA reads for RNA workflows. Paired reads where both reads completely cover only one of the genes are not considered as supporting a gene fusion. The cutoff is defined as 10%.
Reverse transcriptase PCR (RT-PCR) and sanger sequencing for chimeric transcript validation
From 100 ng to 1 µg RNA were reverse transcribed with the expand reverse transcriptase (Roche) with the specific complementary reverse PCR primers (Table 1). PCR products were then amplified with the Platinum™ Taq DNA Polymerase (Invitrogen™) with the thermal cycling profile of 95 °C for 10 min, then 35 cycles of 95 °C for 30 s, 58 °C for 45 s, 72 °C for 45 s, with a final extension at 72 °C for 10 min. Direct sequencing was performed with the same primers using the Big-Dye DyeDeoxy terminator cycle sequencing kit (Applied Biosystems, Foster City, CA). Sequencing reactions were carried out on the ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA).
DNA extraction and array-comparative genomic hybridization (CGH) analysis
Genomic DNA was extracted from formalin fixed paraffin embedded tissues using QIAmp®DSP DNA formalin fixed paraffin embedded tissue kit according to the manufacturer’s protocol for DNA isolation (Qiagen®) after RNAse digestion. A cutoff of 50% of cellularity in tumor samples was set for the analysis. DNA was hybridized onto 8 × 60 K whole-genome arrays (G4450A; Agilent® Technologies) according to the manufacturer’s protocol. Microarray Scanner and images were analyzed by Feature Extraction V10.7.3.1 followed by Agilent® Cytogenomic software 4.0.3.12. The ADM-2 algorithm of the CGH Analytics v4.0.76 software (Agilent® Technologies) was used to identify the DNA copy number abnormalities at the probe level. A low-level copy number gain was defined as a log 2 ratio > 0.25 and a copy number loss was defined as a log 2 ratio < −0.25. A high-level gain or amplification was defined as a log 2 ratio > 1.5 and a homozygous deletion was suspected when the ratio was <−1.5. The range for Derivative Log ratio spread cutoff was fixed to 0.50.
The genomic index was calculated for each profile as follows: Genomic Index = A²/C, where A is the total number of alterations (segmental gains and losses) and C is the number of involved chromosomes [8, 9].
Fluorescence in situ hybridization (FISH)
A COL1A1-PDGFB FISH double fusion was performed in order to validate the observation in 2 cases in our series of unbalanced breaks on chromosomes 17 and 22 (involving the genes COL1A1 and PDGFB) by array-CGH analysis as well in 4 other cases (1, 2, 7 and 13).
FISH assay was performed on 4 micron whole tissue sections using a commercially available COL1A1/PDGFB dual-fusion probe (ZytoLight® SPEC COL1A1/PDGFB Dual Color Dual Fusion Probe, Zytovision) and the Histology FISH accessory kit (DAKO) according to the manufacturer’s instructions. Nuclei were scored for non-translocated patterns (split orange and green signals) and translocated patterns (at least one fusion signal) using a Nikon Eclipse 80i fluorescent microscope with appropriate filters (Nikon). For each case, 50 tumor nuclei were counted, and a come-together signal (orange and green merged signals) in more than 30% of the tumor nuclei was considered a positive result. Pictures were captured using a Pathscan Combi (Excilone).

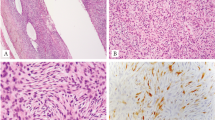
NTRK-rearranged uterine sarcomas. Morphologic features of NTRK1 rearranged tumors. Fibroblastic appearance with spindle cells exhibiting moderate nuclear atypia (a) (case 4). There is diffuse cytoplasmic immunoreactivity with Trk (b) (case 4). Prominent inflammatory infiltrate and scattered symplastic nuclei (c) (case 8). There is diffuse cytoplasmic immunoreactivity with Trk with a negative stromal background (case 8) (d). CD34 (e) and S100 (f) are diffusely positive (case 5). Sanger sequencing of the transcript fusions detected by RNA-sequencing with the ArcherDX® CTL panel: EML4 exon 2-NTRK3 exon 14 (g) (case 5), TPM3 exon 6-NTRK1exon 10 (3,4,8,9,11 and 12), TPM3 partial exon4-NTRK1 partial exon 10). Arrows indicate the point of the junction. Array-CGH genomic profile of a NTRK1-rearranged tumor (h) (case 4). Note the quite simple genomic profile showing rare genomic events and the homozygous CDKN2A loss (arrow)
Results
The clinical and pathological features are presented in Table 2 and the immunohistochemical, RNA-sequencing and genomic data in Table 3.
RNA-sequencing results and sanger sequencing
Seven of the 13 tumors harbored a NTRK rearrangement (6 TPM3-NTRK1 and 1 EML4-NTRK3). In 5 tumors (cases 3, 4, 8, 9, 12), the rearrangement was recurrent affecting exon 6 of TPM3 gene and exon 10 of NTRK1 gene. In 1 tumor (case 11), the fusion was located between exon 4 of TPM3 and exon 10 of NTRK1. The EML4-NTRK3 fusion involved exon 2 of EML4 gene and exon 14 of the NTRK3 gene (case 5). The gene annotation was performed according to the hg19 assembly with the following gene reference: EML4 NM_001145076.1, NTRK3 NM_001012338.2, NTRK1 NM_002529.3 and TPM3 NM_001278188.1. The specific fusion transcript was confirmed by Sanger sequencing in all positive cases (Fig. 1g).

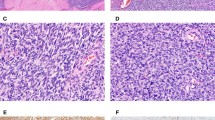
COL1A1-PDGFB-rearranged uterine sarcomas (dermatofibrosarcoma protuberans). Cellular tumors composed of relatively bland spindle-shaped cells with pronounced herringbone pattern of growth (a, b) (case 6). COL1A1-PDGFB dual fusion FISH. Note the presence of two orange/green fusion signals (c) (case 10). Diffuse CD34 immunostaining (d) (case 13). Array-CGH genomic profile of a COL1A1-PDGFB rearranged tumor. Note the unbalanced break-points (arrows) on CH17q (COL1A1) and 22q (PDGFB) (e) (case 10)
The TPM3-NTRK1 fusion transcript (consisting of the TPM3 exon 6 fusion with the NTRK1 exon 10) was also detected in case 10 but in a small proportion (6%) and only in the primary tumor and not in the recurrence. The fusion proportion was below the 10% cutoff defined by the supplier. The TPM3-NTRK1 fusion transcript was confirmed by Sanger sequencing. However, it was considered not relevant since it was only present in the primary tumor and the percentage of the fusion transcript for the positive tumors ranged from 56 to 90%.
Case 7 was not interpretable with ARCHERDX®CTL panel RNA-sequencing. Therefore the tumor was analyzed by Sanger sequencing for TPM3 (exon 6)-NTRK1 (exon 10), TPM3 (exon 4)-NTRK1 (exon 10) and EMLA (exon 2)-NTRK3 (exon 14) fusion transcripts and no specific fusion transcript nor translocation were detected. Case 13 was only tested by Sanger sequencing for the same primers and no fusion was detected. In all cases, β2microglobulin sequence serving as a positive control, was obtained. In those cases where the primary and recurrent tumors were analyzed, an identical molecular abnormality was found in both neoplasms (with the exception of TPM3-NTRK1 fusion in case 10, as discussed above).
Array-CGH genomic profile and FISH dual fusion results
Among the 14 available array-CGH profiles, 11 tumors were interpretable (Table 3). The tumors showed low rearranged genomic profiles (mean Genomic Index = 11.7; range from 3 to 21.4). No significant differences were observed in the genomic complexity between the profiles of the NTRK rearranged tumors and the other neoplasms (p = 0.7). The array-CGH analysis of the NTRK rearranged tumors did not show any unbalanced rearrangement of NTRK1 or NTRK3. On the contrary, 2 tumors (cases 6 and 10, primary tumor and recurrence in case 10) showed intrachromosomal breaks involving COL1A1 on chromosome 17q21.33 and PDGFB on chromosome 22q13.1 (Fig. 3e). FISH dual fusion was performed on these 2 tumors (3 samples) with unbalanced 17-22 rearrangement and also on case 13 which morphologically exhibited a pronounced herringbone and storiform architecture and lacked NTRK-rearrangements. This analysis showed COL1A1-PDGFB fusion in 56%, 86% and 74% of the tumor cells (cases 6, 10 and 13 respectively) (Fig. 3c).

Malignant Peripheral Nerve Sheath Tumors. Spindle cell tumor infiltrating adipose tissue with mild nuclear atypia and focal collections of ganglion-like cells (a, b) (case 7). Spindle cell tumor with a focal lobular architecture but elsewhere with infiltrative borders (c) (case 1).Cellular tumor composed of spindled and epithelioid cells with focal cytoplasmic vacuolization (d) (case 2). Spindle cell tumor with mild nuclear atypia but significant mitotic activity (case 2) (e)
Interestingly among the 12 interpretable tumors at CGH analysis, 9 showed a loss of CDKN2A gene (chr 9p21;3), 5 homozygous (cases 1,4,5,9 and the recurrence of case 12) and 4 heterozygous (case 3, 7, 8 and the primary tumor of case 12) (Table 3). The losses were localized, involving short chromosomic regions (between 0.1.and 1.6 Mb) (Fig. 2h).
Trk Immunohistochemistry
Among the 13 tumors, there was diffuse cytoplasmic Trk positivity in 7 (in case 12, both the primary and recurrent tumor were diffusely positive); no nuclear immunoreactivity was observed. All positive tumors harbored a NTRK rearrangement.
The single tumor (case 10) which harbored a TPM3-NTRK1 fusion at low frequency (6%) was negative for Trk. As discussed, the rearrangement was detected in low percentage in the primary tumor only and was absent in the recurrence and the same tumor harbored the COL1A1/PDGF translocation.
The other immunohistochemical results are discussed below.
Comparative morphologic, immunohistochemical and molecular data
Taking account of the RNA-sequencing, CGH analysis, morphology and immunohistochemistry, we identified 3 subgroups of “fibroblastic” spindle cell sarcomas of the uterus, namely a subgroup of NTRK-rearranged neoplasms, a subgroup harboring COL1A1-PDGFB fusions and a subgroup without any known rearrangement that could be tentatively classified as malignant peripheral nerve sheath tumor given that all were S100 positive (see below) and negative for other melanoma markers (HMB45, Melan A, SOX10), except for 1 case where Melan A and SOX10 were focally positive (discussed below). However, it is likely that other molecular abnormalities may be identified in this group of neoplasms which would necessitate a nomenclature change.
NTRK-rearranged sarcomas
In our series, the 7 tumors harboring NTRK-rearrangements were all located in the cervix. Patients with NTRK-rearranged tumors were significantly younger than the other patients (p = 0.012), the mean age at diagnosis being 31 years (median: 30 years, range 23 to 44 years). They generally presented at early stage (4 at stage IA, 1 at stage IB, 1 at stage IIA, 1 stage unknown) (FIGO 2009 staging system for uterine sarcomas). Follow-up was available for all patients except one and 4 of 6 were alive with no evidence of disease (mean follow-up = 39 months, range 2 to 108 months) and 2 were alive with disease (32 and 52 months follow-up). No correlation was observed between the NTRK status and clinical outcome when comparing patients alive with alive with no evidence of disease versus patients alive with disease or dead of disease (p = 0.24).
The primary tumors ranged in size from 2.5 to 12 cm in those cases where this information was available. When the sections included tumor borders (4 of 7 cases), the neoplasms could be seen to be infiltrating. The neoplasms usually had a predominantly diffuse patternless architecture but in some cases there was a focal or diffuse herringbone pattern with intersecting fascicles of cells. The tumors were predominantly composed of spindle-shaped cells with ovoid nuclei, inapparent nucleoli and scant cytoplasm. In a few cases, small collections of tumor cells with a more epithelioid appearance were present and/or there was focal cytoplasmic vacuolization. Atypia was generally mild to moderate with scattered markedly atypical symplastic-like cells in two tumors and diffuse marked nuclear atypia in the recurrence in case 12. The degree of mitotic activity was highly variable ranging from 1 to 50 per 10 High Power Fields and necrosis was present in 4 of 7 tumors. In 3 of 8 tumors, a focal hemangiopericytoma-like vasculature was present. A prominent lymphocytic infiltrate was observed in 5 of 7 tumors. No lymphovascular invasion was seen. In 1 case (case 3), the tumor infiltrated around pre-existing normal endocervical glands.
Immunohistochemically these tumors were all diffusely positive with CD34 and S100, except for 1 case each where CD34 and S100 exhibited focal immunoreactivity. Trk was diffusely and strongly expressed in all tumors with cytoplasmic positivity. Desmin, ER and PR were negative in all cases with the exception of scattered PR positive cells in case 8. All cases were essentially BCOR negative except for scattered positive nuclei (less than 5%) in some cases. Cyclin D1 was positive in all cases (percentage of positive nuclei ranging from 20 to 90%).
This subgroup showed relatively simple genomic profiles without unbalanced intrachromosomal breaks with a mean Genomic Index of 10.84 (range 2 to 21.3). Among the interpretable profiles (7/8 samples), all NTRK-tumors harbored CDKN2A loss (4 homozygous and 3 heterozygous, from 0.1 to 1.6 Mb).
Figure 1 illustrates the morphological, immunohistochemical and molecular features of some of these cases.
COL1A1-PDGFB rearranged sarcomas
Three tumors (cases 6, 10 and 13) harbored the COL1A1-PDGFB fusion. Two were cervical tumors and one was located in the uterine corpus. The patients were aged 48, 60 and 82 years. The tumors presented at stage IB, IIIB and IB respectively (Table 2). At follow-up, 1 patient was dead of disease after 60 months and 1 was alive with alive with no evidence of disease (10 months follow-up); the third case is recent with no significant follow-up. One of the patients (case 10) had been diagnosed with a Dermatofibrosarcoma Protuberans on her back approximately a year before the cervical lesion was discovered; the lesion on the back had been completely excised and we believe that it is most likely that the cervical tumor represents a separate independent lesion rather than a metastasis.
The tumors measured 5.8, 8.2 and 12 cm. In the 2 tumors with evaluable margins, the borders were infiltrating. Morphologically the tumors were cellular and exhibited a prominent storiform and herringbone architecture and were composed of uniform cells with ovoid to spindle-shaped nuclei, scant cytoplasm and indistinct borders. There was mild to moderate nuclear atypia and mitotic activity ranged from 8 to 20 mitoses/10 High Power Fields. A lymphocytic infiltrate was absent and there was no lymphovascular invasion. Only 1 case exhibited necrosis (case 6).
All tumors diffusely expressed CD34 and were negative for ER, PR, desmin, S100 and Trk when these markers were performed. All cases were essentially BCOR negative except for scattered positive nuclei (less than 5%) in some cases. Cyclin D1 was positive in all cases (percentage of positive nuclei ranging from 10 to 90%).
The two genomic profiles analyzed showed moderately rearranged profiles (Genomic Index = 9.14 and 16.2) with unbalanced 17-22 rearrangements. No CDKN2A deletion was detected.
Figure 2 illustrates the morphological, immunohistochemical and molecular features of some of these cases.
Fibroblastic sarcomas with features of malignant peripheral nerve sheath tumor
Three tumors (cases 1, 2, 7) did not harbor any known rearrangement (at Archer® sequencing and after COL1A1-PDGFB dual FISH). Although we have tentatively classified these as malignant peripheral nerve sheath tumor predominantly on the basis of S100 positivity and complete loss of staining with H3K27me3 in 2 cases (see below), we recognize that there is limited evidence for this and this classification is likely to change, especially if alternative molecular changes are identified in these neoplasms. None of these patients had a history of neurofibromatosis and in no case was origin from a nerve trunk identified.
The first tumor (case 1) occurred in a 32 year old woman with a 5.5 cm cervical mass which comprised a highly cellular spindle cell neoplasm which was focally well circumscribed with a lobular appearance but elsewhere had infiltrative borders. There was moderate nuclear atypia and 10 mitoses/10 High Power Fields. No lymphovascular invasion was seen. The tumor was ER, PR, desmin, CD34 and Trk negative. S100 was diffusely positive and SOX10 and melan A focally positive but HMB45 and MITF were negative. H3K27me3 was focally positive. Less than 5% of nuclei were BCOR positive and 80% of nuclei were cyclin D1 positive. No recurrent KIT, BRAF or NRAS mutations were detected and no NF1 deletion was detected (data not shown). Despite the immunophenotype raising the possibility of a melanocytic neoplasm, the genomic profile was more in favor of a malignant peripheral nerve sheath tumor (see discussion). The tumor recurred with peritoneal and bone metastases 6 months after the primary surgery and the patient was alive 19 months later.
The second tumor (case 2) occurred in a 47 year old woman with a 5 cm mass involving the uterine corpus and lower uterine segment. Metastases to the adnexa and upper abdomen were present at diagnosis and the patient died 11 months after presentation. The tumor was highly cellular and had no particular architectural arrangement and was composed of moderately atypical spindle-shaped cells with vacuolated cytoplasm. There were 50 mitoses/ per 10 High Power Fields. No lymphovascular invasion was seen.The tumor diffusely expressed CD34 and S100 was focally positive. ER, PR, desmin, h-caldesmon, Trk, HMB45, melan A and SOX10 were negative. H3K27me3 was totally negative. Less than 5% of nuclei were BCOR positive and 90% of nuclei were cyclin D1 positive.
The third tumor (case 7) occurred in a 50 year old woman who had undergone a prior hysterectomy for benign reasons. She presented with a mass centered on the vagina which infiltrated the bladder, rectum and other pelvic tissues. She underwent a total pelvectomy. The patient died of disease 34 months following surgery. The tumor had no particular architectural arrangement, exhibited an infiltrative pattern of growth with entrapment of adipose tissue around the periphery and was composed of bland spindle-shaped cells with focal collections of ganglion-like cells with large nuclei, prominent nucleoli and abundant eosinophilic cytoplasm. There was low mitotic activity (1 per 10 High Power Fields) and no necrosis or lymphovascular invasion. There was a prominent lymphocytic infiltrate. S100 was diffusely positive while CD34, HMB45, Melan A, ER, PR, desmin and Trk were negative. H3K27me3 was totally negative. BCOR was negative and 80% of nuclei were cyclin D1 positive.
Two of the cases (cases 2 and 7) showed very simple genomic profiles with a low Genomic Index (12.5 and 5.3 respectively). The other case (case 1) exhibited more intrachromosomal breaks (Genomic Index 21.4). In 2 of the cases, a loss of CDKN2A was detected (homozygous in case 1 and heterozygous in case 7).
Figure 3 illustrates the morphological, immunohistochemical and molecular features of some of these cases.
Discussion
A wide range of sarcomas occur within the uterus (corpus and cervix), the most common being leiomyosarcoma, endometrial stromal sarcoma (low-grade and high-grade), and undifferentiated uterine sarcoma. A variety of more uncommon sarcomas have also been reported as primary uterine neoplasms, including rhabdomyosarcoma, angiosarcoma, malignant peripheral nerve sheath tumor, alveolar soft part sarcoma, liposarcoma, malignant solitary fibrous tumor, and Ewing sarcoma. It is clear that the full spectrum of uterine sarcomas has not yet been fully characterized and that so-called undifferentiated uterine sarcomas comprise a morphologically and molecularly heterogeneous group of neoplasms. Modern molecular techniques, such as next generation sequencing, have resulted in the description of new categories of uterine sarcoma, including two which have been classified as high-grade endometrial stromal sarcoma, those associated with YWHAE and BCOR gene rearrangements [10,11,12,13,14]. Recently, Chiang and coworkers reported a series of 4 uterine mesenchymal neoplasms resembling fibrosarcoma, expressing Trk and harboring NTRK rearrangements (RBPMS-NTRK3, TPR-NTRK1, LMNA-NTRK1 and TPM3-NTRK1) [6]. All tumors exhibited brisk mitotic activity with an aggressive clinical outcome in two of four patients [6]. Immunophenotypically all tumors were positive for S100 and negative for CD34, ER, and PR [6]. The authors speculated that so-called endocervical fibroblastic malignant peripheral nerve sheath tumors may represent NTRK fusion-positive uterine sarcomas on the basis of shared morphology and immunophenotype.
In this study, we report the clinicopathological and molecular features of a series of 13 spindle cell sarcomas of the uterus and vagina with morphological features resembling fibrosarcoma, including the 3 originally reported cases of endocervical fibroblastic malignant peripheral nerve sheath tumor and neoplasms diagnosed as malignant peripheral nerve sheath tumor. In reporting these tumors, we propose a diagnostic algorithm when faced with a uterine sarcoma with features resembling fibrosarcoma. The tumors we report were typically infiltrative and composed of spindle-shaped cells exhibiting mild or moderate nuclear atypia, variable but often prominent mitotic activity and a diffuse, herringbone and/ or storiform architecture. The majority (11/13) of the tumors were CD34-positive, S100 was variable, while ER, PR and desmin were typically negative [5].
After transcriptomic and genomic analysis, we identified three subgroups of neoplasms: a subgroup harboring NTRK-transcript fusions, a subgroup associated with COL1A1-PDGFB fusion and a subgroup without any identified rearrangements. The first group of NTRK-rearranged sarcomas, all of which were located within the cervix, corresponds to the tumors recently reported by Chiang et al. [6] and our findings expand on their publication. Three of the neoplasms we report (2 with a cervical and 1 with a corpus location) harbored a COL1A1-PDGFB fusion which, as far as we are aware, has never been reported in a uterine neoplasm. The final group comprised 3 S100-positive neoplasms which we currently tentatively suggest to categorize as malignant peripheral nerve sheath tumors (see discussion below).
As discussed, as in the original publication [6], all NTRK-associated sarcomas arose in the uterine cervix and generally presented at an early stage. The mean age (31 years) was significantly lower than in the other tumors we report. The tumors recurred in 2 of 6 patients with follow-up and all 6 patients with available follow-up were alive when last seen (2 alive with disease and 4 alive with no evidence of disease). These tumors were generally similar morphologically and immunophenotypically to the other neoplasms we report and we did not identify any specific features which definitively assist in distinguishing these from the other neoplasms. All tumors were positive for CD34 and S100 (usually diffusely) while desmin, ER and PR were negative (except for focal PR positivity in 1 case). The immunophenotype is slightly different from the cases reported by Chiang et al., all of which were CD34 negative [6].
The only feature that reliably identified the NTRK rearranged tumors was the Trk positivity with all cases being diffusely positive (except for 1- see below). All of the other neoplasms were Trk negative. This is in keeping with the findings of Hechtman et al. who found Trk expression to be highly concordant with the fusion status detected in tumors of varying histotypes (colorectal, lung, appendiceal and gallbladder adenocarcinomas, secretory-type salivary and breast carcinomas, glioblastomas, melanomas and soft tissue sarcomas) [15]. As discussed, the only NTRK rearranged case which was negative for Trk immunohistochemistry harbored a TPM3-NTRK1 fusion at low frequency (confirmed by Sanger sequencing). The translocation was detected in the primary tumor but not in the recurrence (case 10) and the tumor harbored a COL1A1-PDGFB translocation in 74% of the tumor cells. This suggests that the driver genomic event is the COL1A1-PDGFB translocation which was present in both the primary and recurrent tumor. We speculate that theTPM3-NTRK1 fusion represents a subclonal population which was lost during tumor progression.
Given the immunohistochemical results, Trk immunohistochemistry represents a useful diagnostic tool that reliably identifies NTRK-rearranged uterine sarcomas. We suggest to screen all uterine (cervical and corpus) “fibroblastic” spindle cell sarcomas without smooth muscle or endometrial stromal differentiation with Trk. Given the sensitivity and specificity of this antibody for NTRK rearrangement [15], positive staining, especially when diffuse, should be followed by molecular testing to prove the presence of a NTRK rearrangement. However, we recognize that the specificity of this marker for NTRK rearranged neoplasms has not been fully investigated since no study has looked at the full range of uterine mesenchymal neoplasms. In the study of Chiang et al., 2 of 97 uterine leiomyosarcomas showed strong and diffuse pan-Trk expression while 4 of 97 showed diffuse weak TrkA immunoreactivity; this study was performed on tissue microarrays [6].
The Trk A, B and C proteins are encoded by NTRK1, NTRK2 and NTRK3 respectively [16]. The fusion juxtaposes the kinase domain-containing 3’ region of NTRK with the 5’ region of NTRKs gene partner and promotes oncogenesis by constitutive cell proliferation [17]. NTRK fusions are relatively rare in tumors but have been identified in < 5% of lung cancers, between 0.5–2% of colorectal cancers, 12% of papillary thyroid carcinomas, 40% of pediatric and 3% of adult brain tumors, 16% of Spitzoid melanomas [18] and 1% of adult soft tissue sarcomas. They have also been identified in 91% of congenital fibrosarcomas and 92–100% of secretory breast carcinomas [17]. Identification of NTRK fusions in cancer is important because of a potential effective therapeutic strategy given the durable response to treatment with TRK inhibitors [17].
Interestingly, among the interpretable genomic profiles, all the NTRK-rearranged sarcomas in our series harbored a CDKN2A deletion (4 homozygous and 3 heterozygous) and the losses affected short chromosomal segments (from 0.1 to 1.6 Mb). CDKN2A deletion has been reported in 50% of NTRK-rearranged sarcomas, in 14% of soft tissue sarcomas [19] and in 50% of soft tissue malignant peripheral nerve sheath tumor [20] but in these studies no information was provided regarding the homozygous or heterozygous status of the loss and the biological implications of this deletion. Further investigations are needed in order to understand the role and the effects of CDKN2A loss in these sarcomas. No CDKN2A loss was detected in the COL1A1-PDGFB rearranged sarcomas. In 2 of the 3 malignant peripheral nerve sheath tumors, a loss of CDKN2A was detected (homozygous in 1 case 1 and heterozygous in 1).
Among the 13 tumors we studied, we identified 3 cases with a COL1A1-PDGFB fusion. This molecular abnormality has not, as far as we are aware, been reported in uterine neoplasms but is characteristic of dermatofibrosarcoma protuberans [21,22,23] and pediatric giant cell fibroblastoma [24]. These 3 tumors occurred in older patients (48 to 82 years), 2 were located in the cervix and one in the corpus. Morphologically they were cellular with storiform and herringbone architecture and significant mitotic activity. All exhibited diffuse positivity with CD34 and were negative for S100, ER, PR, desmin and Trk. Dermatofibrosarcoma protuberans, the most common dermal sarcoma [25], is a low-grade slow growing tumor with low risk of distant metastases but a significant rate of local recurrence and which may undergo fibrosarcomatous transformation [25]. The tumor characteristically harbors a translocation t(17;22)(q22;q13) resulting in the chimeric fusion transcript COL1A1-PDGFB [25]. The trunk and proximal extremities are the most common locations [26,27,28] but this tumor also occurs on the vulva [29]. Morphologically dermatofibrosarcoma protuberans is composed of spindle-shaped cells arranged in a storiform and herringbone architecture and immunohistochemically there is diffuse expression of CD34 and S100 is negative. COL1A1-PDGFB FISH is a useful tool in confirming the diagnosis [22]. While the 3 tumors we report with COL1A1-PDGFB translocation could be regarded as dermatofibrosarcoma of the uterus, we prefer not to use this designation given the significant mitotic activity. We prefer to categorize these as COL1A1-PDGFB translocation associated fibrosarcomas. Moreover, it should be remembered that different tumor types may exhibit the same molecular abnormality, for example YWHAE translocations are found in high-grade endometrial stromal sarcomas and in some clear cell sarcomas of the kidney[10,11,12, 30, 31]. An interesting observation is that in 1 of our cases harboring a COL1A1-PDGFB fusion (case 13), 1 of the tissue blocks was CD34 negative while the others were diffusely positive; CD34 can be focally lost in some cases of dermatofibrosarcoma protuberans, especially in areas of fibrosarcomatous transformation.
The other 3 tumors in our series did not harbor any known rearrangement. Two of them were highly cellular with brisk mitotic activity. These neoplasms were diffusely positive with S100 and generally negative with other melanocytic markers, although SOX10 and melan A were focally positive in 1 case. Although it is difficult to exclude spindle cell malignant melanoma, especially in the case which was focally positive with SOX10 and melan A, we believe these are currently tentatively best categorized as malignant peripheral nerve sheath tumors; no recurrent KIT, BRAF or NRAS mutations (molecular abnormalities which are characteristic of malignant melanoma) were detected in the case positive for SOX10 and melan A. However, none of these patients had a history of neurofibromatosis and in no case could origin from a nerve trunk be demonstrated which would have assisted in classifying these neoplasms as malignant peripheral nerve sheath tumors. One of the cases contained a population of ganglion-like cells which may be seen in other “neural” neoplasms, such as ganglioneuroma [32] and gangliocytic paraganglioma [33]. H3K27me3 was completely negative in 2 of these cases and focally positive in the other. While complete loss of H3K27me3 is common in malignant peripheral nerve sheath tumors and could offer some support to this diagnosis [34], a significant number of malignant melanomas also exhibit complete loss. For example, in 1 study, a complete loss of H3K27me3 was found in 37% of all melanomas and partial loss (‘mosaic’ pattern) in 19% of malignant peripheral nerve sheath tumors and in 51% of melanomas. As such, complete immunohistochemical loss of H3K27me3 is not specific for malignant peripheral nerve sheath tumor and cannot be used reliably in distinguishing this from melanoma [35].
The prognosis of this group was poor; one patient died of disease 11 months after the diagnosis (case 2) and another (case 1) recurred early with local and distant metastases; this patient is alive with disease after 19 months. The third case was a primary vaginal tumor which involved the bladder and rectum. Although the numbers are small, this suggests that these neoplasms have a particular propensity for aggressive behavior. As stated previously, while we currently suggest to tentatively classify these neoplasms as malignant peripheral nerve sheath tumor, it is arguable whether this is an appropriate designation and the nomenclature may change especially if other molecular alterations are identified in this neoplasms.
The differential diagnosis of spindle cell sarcomas of the uterus composed of a uniform population of cells is wide and includes leiomyosarcoma, high-grade endometrial stromal sarcoma, undifferentiated uterine sarcoma (uniform type) and a variety of other uncommon neoplasms including inflammatory myofibroblastic tumor, malignant solitary fibrous tumor [36] and spindle cell malignant melanoma. To that list should be added NTRK-rearranged sarcoma, COL1A1-PDGFB fusion-associated fibrosarcoma and malignant peripheral nerve sheath tumor. The differential diagnosis will not be discussed in detail but leiomyosarcoma typically expresses smooth muscle markers including desmin [37], high-grade endometrial stromal sarcomas exhibit high expression of cyclin D1 and BCOR (see below) and the majority of cases harbor YWHAE or BCOR rearrangements [10, 11, 38, 39].
As discussed, high-grade endometrial stromal sarcomas typically exhibit diffuse expression of cyclin D1 and BCOR. All our cases were BCOR negative except for scattered positive nuclei (less than 5%) in some cases. Cyclin D1 was positive in all of our cases (percentage of positive nuclei ranging from 10 to 90%). This illustrates that cyclin D1 is not a reliable marker in distinguishing YWHAE or BCOR rearranged high-grade endometrial stromal sarcoma from NTRK or COL1A1-PDGFB rearranged tumors. This is not surprising since cyclin D1 is a cell cycle related marker rather than a marker of a specific line of differentiation. One of us (WGM) has also observed diffuse cyclin D1 immunoreactivity in a small number of other uterine mesenchymal neoplasms.
Recent studies have emphasized that inflammatory myofibroblastic tumors occur within the uterus and are likely to be misdiagnosed, especially as leiomyomatous neoplasms [40,41,42,43]. Inflammatory myofibroblastic tumor is ALK positive and is characterized by genetic fusion of ALK with a variety of gene partners [40,41,42,43]. Undifferentiated uterine sarcoma is a diagnosis of exclusion and it is likely that cases of NTRK-rearranged sarcoma and COL1A1-PDGFB fusion-associated fibrosarcoma are currently encompassed into that category [13, 44]. Given the recent identification of YWHAE and BCOR translocated high-grade endometrial stromal sarcomas and the new entities we describe, it is clear that the category of undifferentiated uterine sarcoma represents a heterogeneous group of neoplasms which will likely significantly decrease with the identification of new molecular entities using modern molecular techniques. We suggest a diagnostic algorithm for uterine spindle cell sarcomas composed of uniform cells resembling fibrosarcoma (Fig. 4); it is stressed that other neoplasms, for example malignant solitary fibrous tumor, may also enter into the differential diagnosis.

Diagnostic algorithm for spindle cell sarcomas of the uterus. Although the typical immunophenotypes are listed, in some cases there may be exceptions to these typical staining patterns and other neoplasms may enter into the differential diagnosis
In summary, we report the clinicopathological and molecular features of a series of 13 spindle cell sarcomas of the uterus with features resembling fibrosarcoma. We show that NTRK rearrangements are common in these neoplasms and describe a novel COL1A1-PDGFB fusion which has not previously been reported in uterine mesenchymal neoplasms. Identification of the latter 2 neoplasms is clinically important since these tumors could potentially respond to targeted therapies such as the PDGFβR, KIT, and ABL inhibitor imatinib [45] and anti NTRK inhibitors [17].
References
Oliva E. CM, Carinelli SG, et al. Endometrial stromal and related tumors. In: Kurman RJ CM, Herrington CS, Young RH, editor.WHO classification of tumors of female reproductive organs. Lyon, IARC; 2014. p. 141–7.
Sangiorgio V, Zanagnolo V, Aletti G, Bocciolone L, Bruni S, Landoni F, et al. Fibroblastic malignant peripheral nerve sheath tumour of the uterine cervix: report of a case and literature review with emphasis on possible differential diagnosis. Int J Gynecol Pathol. 2018;37:497–503.
Rodriguez AO, Truskinovsky AM, Kasrazadeh M, Leiserowitz GS. Case report: Malignant peripheral nerve sheath tumor of the uterine cervix treated with radical vaginal trachelectomy. Gynecol Oncol. 2006;100:201–4.
Bernstein HB, Broman JH, Apicelli A, Kredentser DC. Primary malignant schwannoma of the uterine cervix: a case report and literature review. Gynecol Oncol. 1999;74:288–92.
Mills AM, Karamchandani JR, Vogel H, Longacre TA. Endocervical fibroblastic malignant peripheral nerve sheath tumor (neurofibrosarcoma): report of a novel entity possibly related to endocervical CD34 fibrocytes. Am J Surg Pathol. 2011;35:404–12.
Chiang S, Cotzia P, Hyman DM, Drilon A, Tap WD, Zhang L, et al. NTRK Fusions Define a Novel Uterine Sarcoma Subtype With Features of Fibrosarcoma. Am J Surg Pathol. 2018;42:791–8.
Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84.
Croce S, Ribeiro A, Brulard C, Noel JC, Amant F, Stoeckle E, et al. Uterine smooth muscle tumor analysis by comparative genomic hybridization: a useful diagnostic tool in challenging lesions. Mod Pathol. 2015;28:1001–10.
Cuppens T, Moisse M, Depreeuw J, Annibali D, Colas E, Gil-Moreno A, et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int J Cancer. 2018;142:1230–43.
Lee CH, Marino-Enriquez A, Ou W, Zhu M, Ali RH, Chiang S, et al. The clinicopathologic features of YWHAE-FAM22 endometrial stromal sarcomas: a histologically high-grade and clinically aggressive tumor. Am J Surg Pathol. 2012;36:641–53.
Lee CH, Ou WB, Marino-Enriquez A, Zhu M, Mayeda M, Wang Y, et al. 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc Natl Acad Sci USA. 2012;109:929–34.
Croce S, Hostein I, Ribeiro A, Garbay D, Velasco V, Stoeckle E, et al. YWHAE rearrangement identified by FISH and RT-PCR in endometrial stromal sarcomas: genetic and pathological correlations. Mod Pathol. 2013;26:1390–400.
Hoang L, Chiang S, Lee CH. Endometrial stromal sarcomas and related neoplasms: new developments and diagnostic considerations. Pathology. 2018;50:162–77.
Hoang LN, Aneja A, Conlon N, Delair DF, Middha S, Benayed R, et al. Novel high-grade endometrial stromal sarcoma: a morphologic mimicker of myxoid leiomyosarcoma. Am J Surg Pathol. 2017;41:12–24.
Hechtman JF, Benayed R, Hyman DM, Drilon A, Zehir A, Frosina D, et al. Pan-Trk immunohistochemistry is an efficient and reliable screen for the detection of NTRK fusions. Am J Surg Pathol. 2017;41:1547–51.
Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–60.
Kheder ES, Hong DS. Emerging targeted therapy for tumors with NTRK fusion proteins. Clin Cancer Res. 2018.
Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S, et al. An Oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015;5:1049–57.
Carroll SL. The challenge of cancer genomics in rare nervous system neoplasms: malignant peripheral nerve sheath tumors as a paradigm for cross-species comparative oncogenomics. Am J Pathol. 2016;186:464–77.
Pedeutour F, Simon MP, Minoletti F, Barcelo G, Terrier-Lacombe MJ, Combemale P, et al. Translocation, t(17;22)(q22; q13), in dermatofibrosarcoma protuberans: a new tumor-associated chromosome rearrangement. Cytogenet Cell Genet. 1996;72:171–4.
Karanian M, Perot G, Coindre JM, Chibon F, Pedeutour F, Neuville A. Fluorescence in situ hybridization analysis is a helpful test for the diagnosis of dermatofibrosarcoma protuberans. Mod Pathol. 2015;28:230–7.
Simon MP, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre JM, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997;15:95–8.
Maire G, Pedeutour F, Coindre JM. COL1A1-PDGFB gene fusion demonstrates a common histogenetic origin for dermatofibrosarcoma protuberans and its granular cell variant. Am J Surg Pathol. 2002;26:932–7.
Thway K, Noujaim J, Jones RL, Fisher C. Dermatofibrosarcoma protuberans: pathology, genetics, and potential therapeutic strategies. Ann Diagn Pathol. 2016;25:64–71.
Bowne WB, Antonescu CR, Leung DH, Katz SC, Hawkins WG, Woodruff JM, et al. Dermatofibrosarcoma protuberans: a clinicopathologic analysis of patients treated and followed at a single institution. Cancer . 2000;88:2711–20.
Garcia JJ, Folpe AL. The impact of advances in molecular genetic pathology on the classification, diagnosis and treatment of selected soft tissue tumors of the head and neck. Head Neck Pathol. 2010;4:70–6.
Chang CK, Jacobs IA, Salti GI. Outcomes of surgery for dermatofibrosarcoma protuberans. Eur J Surg Oncol. 2004;30:341–5.
Edelweiss M, Malpica A. Dermatofibrosarcoma protuberans of the vulva: a clinicopathologic and immunohistochemical study of 13 cases. Am J Surg Pathol. 2010;34:393–400.
Kao YC, Sung YS, Zhang L, Huang SC, Argani P, Chung CT, et al. Recurrent BCOR internal tandem duplication and YWHAE-NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol. 2016;40:1009–20.
O’Meara E, Stack D, Lee CH, Garvin AJ, Morris T, Argani P, et al. Characterization of the chromosomal translocation t(10;17)(q22;p13) in clear cell sarcoma of kidney. J Pathol. 2012;227:72–80.
Scheithauer BW WJ, Erlandson RA. Atlas of tumor pathology: tumors of the peripheral nervous system. 3rd, ed. (AFIP), fascicle 24, Bethesda, MA, 1999.
Okubo Y, Yoshioka E, Suzuki M, Washimi K, Kawachi K, Kameda Y, et al. Diagnosis, pathological findings, and clinical management of gangliocytic paraganglioma: a systematic review. Front Oncol. 2018;8:291.
Pekmezci M, Cuevas-Ocampo AK, Perry A, Horvai AE. Significance of H3K27me3 loss in the diagnosis of malignant peripheral nerve sheath tumors. Mod Pathol. 2017;30:1710–9.
Le Guellec S, Macagno N, Velasco V, Lamant L, Lae M, Filleron T, et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: a study of 387 cases including mimicking lesions. Mod Pathol. 2017;30:1677–87.
Yang EJ, Howitt BE, Fletcher CDM, Nucci MR. Solitary fibrous tumour of the female genital tract: a clinicopathological analysis of 25 cases. Histopathology. 2018;72:749–59.
Oliva E. Practical issues in uterine pathology from banal to bewildering: the remarkable spectrum of smooth muscle neoplasia. Mod Pathol. 2016;29:S104–20.
Lee CH, Ali RH, Rouzbahman M, Marino-Enriquez A, Zhu M, Guo X, et al. Cyclin D1 as a diagnostic immunomarker for endometrial stromal sarcoma with YWHAE-FAM22 rearrangement. Am J Surg Pathol. 2012;36:1562–70.
Chiang S, Lee CH, Stewart CJR, Oliva E, Hoang LN, Ali RH, et al. BCOR is a robust diagnostic immunohistochemical marker of genetically diverse high-grade endometrial stromal sarcoma, including tumors exhibiting variant morphology. Mod Pathol. 2017;30:1251–61.
Haimes JD, Stewart CJR, Kudlow BA, Culver BP, Meng B, Koay E, et al. Uterine Inflammatory Myofibroblastic Tumors Frequently Harbor ALK Fusions With IGFBP5 and THBS1. Am J Surg Pathol. 2017;41:773–80.
Bennett JA, Nardi V, Rouzbahman M, Morales-Oyarvide V, Nielsen GP, Oliva E. Inflammatory myofibroblastic tumor of the uterus: a clinicopathological, immunohistochemical, and molecular analysis of 13 cases highlighting their broad morphologic spectrum. Mod Pathol. 2017;30:1489–503.
Pickett JL, Chou A, Andrici JA, Clarkson A, Sioson L, Sheen A, et al. Inflammatory myofibroblastic tumors of the female genital tract are under-recognized: a low threshold for alk immunohistochemistry is required. Am J Surg Pathol. 2017;41:1433–42.
Parra-Herran C, Quick CM, Howitt BE, Dal Cin P, Quade BJ, Nucci MR. Inflammatory myofibroblastic tumor of the uterus: clinical and pathologic review of 10 cases including a subset with aggressive clinical course. Am J Surg Pathol. 2015;39:157–68.
Kurihara S, Oda Y, Ohishi Y, Iwasa A, Takahira T, Kaneki E, et al. Endometrial stromal sarcomas and related high-grade sarcomas: immunohistochemical and molecular genetic study of 31 cases. Am J Surg Pathol. 2008;32:1228–38.
Rutkowski P, Debiec-Rychter M. Current treatment options for dermatofibrosarcoma protuberans. Expert Rev Anticancer Ther. 2015;15:901–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Croce, S., Hostein, I., Longacre, T.A. et al. Uterine and vaginal sarcomas resembling fibrosarcoma: a clinicopathological and molecular analysis of 13 cases showing common NTRK-rearrangements and the description of a COL1A1-PDGFB fusion novel to uterine neoplasms. Mod Pathol 32, 1008–1022 (2019). https://doi.org/10.1038/s41379-018-0184-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0184-6
This article is cited by
-
Adult NTRK-rearranged spindle cell neoplasms of the viscera: with an emphasis on rare locations and heterologous elements
Modern Pathology (2022)
-
Targeted RNA expression profiling identifies high-grade endometrial stromal sarcoma as a clinically relevant molecular subtype of uterine sarcoma
Modern Pathology (2021)
-
Re-evaluating tumors of purported specialized prostatic stromal origin reveals molecular heterogeneity, including non-recurring gene fusions characteristic of uterine and soft tissue sarcoma subtypes
Modern Pathology (2021)
-
Mesenchymal tumors of the gastrointestinal tract with NTRK rearrangements: a clinicopathological, immunophenotypic, and molecular study of eight cases, emphasizing their distinction from gastrointestinal stromal tumor (GIST)
Modern Pathology (2021)
