
Abstract
Lipofibromatosis is a rare pediatric soft tissue tumor with predilection for the hands and feet. Previously considered to represent “infantile fibromatosis”, lipofibromatosis has distinctive morphological features, with mature adipose tissue, short fascicles of bland fibroblastic cells, and lipoblast-like cells. Very little is known about the genetic underpinnings of lipofibromatosis. Prompted by our finding of the FN1-EGF gene fusion, previously shown to be a characteristic feature of calcifying aponeurotic fibroma (CAF), in a morphologically typical case of lipofibromatosis that recurred showing features of CAF, we studied a cohort of 20 cases of lipofibromatosis for this and other genetic events. The cohort was composed of 14 males and 6 females (median age 3 years; range 1 month–14 years). All primary tumors showed classical lipofibromatosis morphology. Follow-up disclosed three local recurrences, two of which contained calcifying aponeurotic fibroma-like nodular calcifications in addition to areas of classic lipofibromatosis, and no metastases. By FISH and RNA sequencing, four cases were positive for FN1-EGF and one case each showed an EGR1-GRIA1, TPR-ROS1, SPARC-PDGFRB, FN1-TGFA, EGFR-BRAF, VCL-RET, or HBEGF-RBM27 fusion. FN1-EGF was the only recurrent fusion, suggesting that some cases of “lipofibromatosis” may represent calcifying aponeurotic fibroma lacking hallmark calcifications. Several of the genes involved in fusions (BRAF, EGFR, PDGFRB, RET, and ROS1) encode receptor tyrosine kinases (RTK), or ligands to the RTK EGFR (EGF, HBEGF, TGFA), suggesting a shared deregulation of the PI3K–AKT–mTOR pathway in a large subset of lipofibromatosis cases.
Similar content being viewed by others

Introduction
Lipofibromatosis, originally described by Stout as part of the spectrum of “infantile fibromatosis”, is a rare soft tissue tumor that occurs mainly in children [1, 2]. In 2000, Fetsch and colleagues [3] coined the term “lipofibromatosis” in their seminal description of 45 cases culled from the archives of the Armed Forces Institute of Pathology, emphasizing their unique clinicopathological features, including the frequent presence of abundant adipose tissue. Lipofibromatosis occurs more often in males and preferentially involves the hands and feet, although it can occur in other locations such as the trunk, and head and neck [3,4,5]. By magnetic resonance imaging (MRI), lipofibromatosis lacks specific features and appears as a soft tissue mass with variable proportions of adipocytic and solid components [6]. Morphologically, lipofibromatosis displays a distinctive admixture of mature adipose tissue and fascicles of bland, small, cuboidal to spindled cells. Lipofibromatosis lacks metastatic potential, but may recur locally in up to one-third of the cases [3, 7].
The morphologic features of lipofibromatosis overlap with other pediatric fibroblastic/myofibroblastic mesenchymal neoplasms, including calcifying aponeurotic fibroma [8], the recently described “lipofibromatosis-like neural tumor” [9] and fibrous hamartoma of infancy [10]. In most cases, this differential diagnosis can be resolved by clinical correlation and careful morphological evaluation, but there are some cases in which this distinction is quite challenging, particularly in the setting of limited biopsies or in tumors occurring in unusual locations. Thus, there is continued interest in the identification of “disease-specific” genetic alterations in lipofibromatosis. Unlike calcifying aponeurotic fibroma, lipofibromatosis-like neural tumor and fibrous hamartoma of infancy, which are associated with FN1-EGF [11], NTRK1 fusions [9], and EGFR exon 20 alterations [12], respectively, a specific genetic event has not been identified to date in lipofibromatosis; information about the genetic events underlying lipofibromatosis is limited to a single karyotypic study of one case, showing a three-way translocation t(4;9;6)(q21;q22;q2?4) of unknown pathogenetic significance [13].
We recently encountered an unusual case (case 14 in the present study), which initially presented as a morphologically classical example of lipofibromatosis in the finger of 2-year-old girl, and recurred 3 years later with typical morphological features of calcifying aponeurotic fibroma, including nodular calcification (Fig. 1). Subsequent genetic study of this case showed FN1-EGF. Prompted by these observations, we investigated the molecular genetic features of a cohort of typical lipofibromatosis cases with the goals of better characterizing the genetic features of this lesion and ascertaining its relationship, if any, to other pediatric fibroblastic tumors.

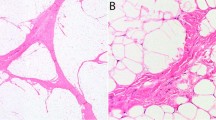
This case of lipofibromatosis occurred in the finger of a one -year-old female, and showed only typical morphological features in the primary tumor (a, b). In contrast, a local recurrence at 3 years showed higher cellularity, a pronounced fascicular growth pattern, and vaguely chondroid areas with multinucleated giant cells and nodular calcification, reminiscent of calcifying aponeurotic fibroma (c–e)
Materials and methods
Case selection
This study was approved by the Institutional Review Boards at all participating institutions. Institutional and consultation archives were queried from 1990 to 2017 for pediatric cases (patients 0–18 years of age) diagnosed as lipofibromatosis. All available routinely stained slides and immunohistochemical studies for 35 cases were re-reviewed by two soft tissue pathologists (AA and ALF). Upon re-review, 15 cases either had insufficient material or were felt to better correspond to other entities (e.g., fibrous hamartoma of infancy) and were excluded, leaving a final study population of 20 cases (17 primary tumors, 3 local recurrences). Clinical data including age, gender, anatomic location, and follow-up were obtained by reviewing medical records.
Fluorescence in situ hybridization
Interphase fluorescence in situ hybridization (FISH) with a break-apart probe (BAP) for the FN1 gene and/or a combination of probes for FN1 and EGF to detect fusion signals were performed as described [11]; the home-made 5′ FN1 probe was replaced with a commercial 5′ FN1 probe (CytoTest, Rockville, MD, USA) at BAP FISH in cases 2 (primary and recurrent tumor), 4, 8, and 11, and at FN1-EGF fusion FISH in cases 1 and 8. Cases were scored as negative if <35% of the nuclei showed split signals with a BAP for FN1 or <30% fusion signals with probes for FN1 and EGF. To verify SPARC-PDGFRB and TPR-ROS1 fusions detected by RNA-Seq, commercial BAPs for PDGFRB (Vysis, Abbott Park, IL, USA) and ROS1 (ZytoVision, Bremerhaven, Germany) were used. For confirmation of the FN1-TGFA fusion, home-made bacterial artificial chromosome (BAC) probes for the 3′-part of TGFA were combined with the commercial 5′ FN1 probe; the BAC probes were obtained from the BACPAC Resource Center (http:/bacpac.chori.org; Supplementary Table 2). On average 107 (range 57–190) nuclei were analyzed per hybridization.
RNA sequencing
For 16 tumor samples from 15 patients, RNA sequencing (RNA-Seq) was attempted on RNA extracted from FFPE blocks using Qiagen’s RNeasy FFPE Kit (Qiagen, Valencia, CA, USA), as described [14]. The RNA DV200 values varied between 28 and 68 (Table 2) and mRNA libraries were prepared from 20 to 400 ng of RNA, depending on the DV200 value, using the capturing chemistry of the TruSeq RNA Access Library Prep Kit (Illumina, San Diego, USA). Paired-end 85 nt reads were generated from the mRNA libraries on a NextSeq 500 (Illumina). Sectioning, RNA extraction, library preparation and sequencing were performed as described [14].
ChimeraScan and FusionCatcher, using default settings, were used to identify candidate fusion transcripts from the sequence data [15, 16]. The GRCh37/hg19 build was used as the human reference genome.
RT-PCR
RT-PCR and sequencing to confirm EGR-GRIA1, FN1-TGFA, HBEGF-RBM27, SPARC-PDGFRB, and TPR-ROS1 fusions detected at RNA-Seq were performed as described [17]. Primers are listed in Supplementary Table 1.
Results
Clinicopathologic features
Table 1 summarizes the clinicopathologic findings for the 20 studied cases. The tumors occurred in 14 males and 6 females (male: female ratio = 2:1) with a median age of 3 years (range: 1 month to 14 years), and involved the subcutaneous tissues of the forearm/arm (n = 5; 25%), fingers/hand (n = 4; 20%), leg/foot (n = 4; 20%), trunk/buttock (n = 4; 20%), neck (n = 2; 10%), and an unknown location (n = 1; 5%). All patients presented with painless, slowly growing subcutaneous masses.
Follow-up information was available for 14 patients with a range of 21 to 168 months (median 57 months). Local recurrences were observed in 3 patients, 32–72 months after resection with only one patient developing local recurrences twice. No distant metastases occurred. At the time of last follow-up, all patients were alive without evidence of disease.
Morphologically, all 17 primary tumors showed classic features of lipofibromatosis, with a variable component of mature adipose tissue, and a somewhat haphazard proliferation of bland fibroblastic cells (Fig. 2). The lesional cells ranged from cuboidal to spindled in shape, and contained scant eosinophilic cytoplasm, small, regular, normochromatic nuclei, and inapparent nucleoli. Mitotic activity was low (0 to 3 mitotic figures per 10 high power fields) and necrosis was absent. A subset of the fibroblastic spindled cells showed intracytoplasmic vacuolization, resembling uni- or occasionally bi-vacuolated lipoblasts. In addition to typical areas of lipofibromatosis, one case (case 7) showed areas with moderately higher cellularity, a more pronounced fascicular pattern of growth, and infiltration of underlying skeletal muscle (Fig. 3). By definition, no case showed calcification or nodules of primitive myxoid mesenchyme.

Lipofibromatosis (LPF) classically shows variable component of mature adipose tissue, and haphazard proliferation of bland fibroblastic cells. a, b LPF, presenting as a mass in the foot of a 3-year male. This tumor was positive for FN1-TGFA gene fusion by molecular methods. c, d LPF, presenting as a mass in the hand of a 4-year-old boy with foci of lipoblast-like cells. This tumor was positive for HBEGF-RBM27 gene fusion by molecular methods. e, f Lipofibromatosis, presenting as a mass in the forearm of a 2-year-old female. This tumor was positive for EGR1-GRIA1 gene fusions by molecular methods. g, h Lipofibromatosis, presenting as a mass in the foot of a 1-year-old male. This tumor was positive for FN1-EGF gene fusions by molecular methods

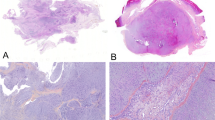
This case of lipofibromatosis involved the arm of a two year old female, and showed in addition to typical areas of lipofibromatosis (a, b) areas with moderately higher cellularity, fascicular pattern of growth, and infiltration of underlying skeletal muscle (c, d)
Two recurrent tumors (cases 12 and 14) showed morphological features of calcifying aponeurotic fibroma, with much diminished fat, cellular, “fibromatosis-like” fibroblastic fascicles and scattered islands of calcification, surrounded by epithelioid cells (Fig. 4). The third recurrent tumor continued to display typical morphological features of lipofibromatosis.

This case of lipofibromatosis occurring in a forearm of one month old boy showed only typical morphological features in the primary tumor (a). In contrast, a local recurrence at 3 years showed areas reminiscent of calcifying aponeurotic fibroma (b, c)
By immunohistochemistry, the spindled cells showed variable expression of smooth muscle actin (in a myofibroblastic “tram-track” pattern) and CD34. S100 protein was present only in the adipocytic component, without expression in spindled cells. Other tested markers were negative. A ROS1 immunostain, performed in case 7, was negative.
FISH
FISH for FN1 rearrangement and/or FN1-EGF fusion was performed on 14 cases, with positive results in 4 (29%) cases. Three cases, none of which could be analyzed at RNA-Seq, were positive for the FN1-EGF fusion (Fig. 5a), and the FN1-TGFA fusion in case 13 detected at RNA-Seq was found also by FISH. In addition, the involvement of PDGFRB and ROS1 in fusions was verified by FISH (Table 2).

a Interphase FISH with probes for 5′ FN1 (green) and 3′ EGF (red) showing fused red and green signals, indicative of an FN1-EGF fusion in case 6. b Chromatogram illustrating the TPR-ROS1 fusion transcript in case 7. c Schematic view of the tyrosine kinases involved in gene fusions in lipofibromatosis. Lollipops indicate the breakpoints, which consistently were close to the kinase domains. d Increased expression of the distal part of GRIA1 as a result of the fusion of EGR1 exon 1 with GRIA1 exon 3 in case 3
RNA-Seq
RNA-Seq identified in-frame fusion transcripts in 9 of 16 (56%) samples from 15 patients (Table 2). The only recurrent fusion was between VCL, encoding the cytoskeletal protein vinculin, and RET, encoding a receptor tyrosine kinase (RTK); the VCL-RET fusion was found in two separate samples from case 18. Also two other fusions involving RTK-encoding genes as 3′-partners were found in one case each: SPARC-PDGFRB and TPR-ROS1 (Fig. 5b). In all three patients with RTK fusions, the kinase domain was included in the predicted fusion protein (Fig. 5c). Three cases displayed fusions (FN1-EGF, FN1-TGFA, and HBEGF-RBM27, respectively) involving genes that encode ligands (epidermal growth factor, transforming growth factor alpha, and heparin-binding EGF-like growth factor, respectively) for the epidermal growth factor receptor (EGFR). Also the EGFR gene itself was involved in a fusion in one case, with BRAF as 3′-partner. Finally, one case had an EGR1-GRIA1 fusion, involving the genes encoding the C2H2 zinc-finger early growth response protein and the glutamate ionotropic receptor AMPA-type subunit 1, upregulating the expression of GRIA1 (Fig. 5d; Supplementary Figure 1).
Discussion
To the best of our knowledge, this represents the first molecular genetic study of lipofibromatosis. Our combined FISH and RNA-Seq analyses have identified an unprecedented variety of gene fusions in these distinctive pediatric tumors, with 8 different fusions found in 11 of the 20 tumors (55%) analyzed. Further adding to the genetic heterogeneity of lipofibromatosis, only one of the 11 fusion-related genes (ROS1 in 6q22) we have identified in lipofibromatosis maps to any of the three breakpoints in the previously reported, cytogenetically abnormal case reported by Kenney et al. [13]. Although several other soft tissue tumor types, such as inflammatory myofibroblastic tumor and benign fibrous histiocytoma, are known to display many different gene fusions, their repertoire is typically much smaller, with one specific fusion accounting for the majority of cases [18]. In contrast, in the present study, FN1-EGF was the only recurrent fusion, detected in four cases. While the observed genetic heterogeneity might raise concerns about the reliability and significance of our results, it should also be emphasized that almost all fusions detected by RNA-seq were corroborated by repeated RNA-seq, RT-PCR, and/or FISH; the only exception, because of lack of suitable material, was an FN1-EGF fusion that, however, was highly expressed and had breakpoints similar to those reported before in calcifying aponeurotic fibroma [11]. With regards to the two FN1-EGF fusions detected only by FISH, both cases showed concordant results with fusion probes and a BAP probe for FN1. Thus, we are confident that our results do not represent technical artifacts.
As this study utilized only FFPE tissue, more detailed experiments to elucidate the functional outcome of each fusion or to investigate potentially shared molecular pathways could not be performed. However, from the genes and breakpoints involved in the detected fusions several plausible pathogenetic mechanisms can be inferred, suggesting that the development of lipofibromatosis may be attributed to deregulation of the PI3K–AKT–mTOR pathway in a substantial proportion of the cases (Fig. 6).

Predicted shared pathways among fusion proteins in lipofibromatosis. The detected fusions involved either ligands (EGF, HBEGF, TGFA) to the EGF receptor (EGFR), EGFR itself, or other receptor tyrosine kinases (ROS1, RET, PDGFRB) that all are known to activate the PI3K–AKT–mTOR pathway. The black boxes indicate the kinase domains. Also the two proteins involved in the EGR1-GRIA1 fusion have been implicated in EGFR signaling
Three of the fusions (FN1-EGF, FN1-TGFA, and HBEGF-RBM27) involve genes that encode ligands for the epidermal growth factor receptor (EGFR): EGF, TGFA, and HBEGF, respectively [19]. FN1-EGF fusions with similar breakpoints have been reported before in calcifying aponeurotic fibroma [11], and the likely pathogenetic mechanism is that the fusion results in increased expression of the part of EGF that contains the mature EGF peptide. A similar scenario could be envisioned for the FN1-TGFA fusion, which is predicted to result in a chimeric protein that retains the mature EGF-like domain of TGFA, encoded by exons 3–4, but fused with a slightly smaller portion (only the first 20 exons, compared to the first 23–42 exons in calcifying aponeurotic fibroma) of the amino-terminal part of FN1. The functional outcome of HBEGF-RBM27 is more difficult to predict. The EGF-like domain of HBEGF, encoded by exons 3 and 4 (aa 104–44), is only partly (first three exons, aa 1–133) included in the predicted fusion, and the fusion partner, RNA-binding motif protein 27 (RBM27), does not contribute any transmembrane domain. Thus, if and how the HBEGF-RBM27 fusion affects EGFR signaling remains to be elucidated. The fusion involving the EGFR gene (the EGFR-BRAF chimera in case 19) adds further support for an important role of distorted EGFR signaling in lipofibromatosis. In this detected fusion, the last exon of EGFR was replaced by the last nine exons of BRAF. Thus, in the predicted chimeric protein, the kinase domain of EGFR would be retained, with an added kinase domain from BRAF added to its carboxy-terminal, intracellular portion.
A second subgroup of detected fusion transcripts in lipofibromatosis is arguably composed of the three cases with fusions involving receptor tyrosine kinases (RTK) as 3′-partners. Such fusions are commonly observed in a variety of neoplasms, and all 3 genes involved in the present study (ROS1, PDGFRB, and RET) have been reported as 3′-partners in fusions before, including in soft tissue tumors [18, 20]. Furthermore, the breakpoints in the RTK-encoding genes were in all three cases at, or very close to, the beginning of the respective kinase domains (Fig. 5c). Further supporting the pathogenetic significance of these fusions, two of the three 5′-partners, TPR and VCL, have been found as 5′-partners with kinase-encoding genes in other neoplasms [18]. Also the third 5′-partner, SPARC, fits well as a potent driver of kinase expression in the detected SPARC-PDGFRB fusion; SPARC encodes a matricellular protein (also known as osteonectin) that regulates cell–matrix interactions and is abundantly expressed in various mesenchymal tissues [21].
Although neither the multifaceted effects of RTK signaling nor the individual characteristics of the different EGFR ligands or RTKs (EGFR, ROS1, RET, and PDGFRB) involved should be neglected, it is of interest to note that activation of these RTKs all have one downstream pathway in common, namely PI3K–AKT–mTOR [22,23,24,25,26]. Thus, the pathogenesis of lipofibromatosis might be less heterogeneous than expected from the plethora of gene fusions.
The only fusion detected by RNA-Seq that did not directly involve an RTK or an RTK ligand is EGR1-GRIA1, in which the first exon of EGR1 was fused in-frame with exon 3 of GRIA1. Early growth response-1 (EGR1) is a zinc-finger transcription factor belonging to the group of immediate-early response proteins [27] and glutamate ionotropic receptor AMPA-type subunit 1 (GRIA1) encodes a transmembranous protein that is important for mediating excitatory signals in the central nervous system [28]. As neither EGR1 nor GRIA1 has been described as a partner in any recurrent neoplasia-associated gene fusion [18], the EGR1-GRIA1 fusion detected in case 3 might represent a passenger mutation, although both the fusion transcript and GRIA1 were highly expressed (Fig. 5d, Supplementary Figure 1). However, GRIA proteins have attracted increasing attention in tumor biology due to the finding of increased expression in many tumor types, including sarcomas [29]. Furthermore, it has been shown that Ca2+ signaling through AMPA receptors, including GRIA1, results in phosphorylation and activation of AKT1 in glioblastoma [30] and that glutamate promotes cell growth by increasing the levels of EGFR and phosphorylated AKT1 in a glioblastoma cell line [31]. In addition, EGR1 is highly expressed in mesenchymal stem cells, in which its transcription is upregulated by EGF [32]. Thus, it cannot be excluded that also the EGR1-GRIA1 fusion might have an impact on the PI3K–AKT–mTOR pathway (Fig. 6).
Lipofibromatosis would thus seem to join a growing list of pediatric fibroblastic/myofibroblastic tumors displaying genetic rearrangements resulting in direct or indirect RTK activation. Notably, our findings overlap with those of Agaram et al. in their recent description of lipofibromatosis-like neural tumors [9]. In that study, 10 of 14 cases had fusions affecting the RTK NTRK1, and one of the 2 cases that were studied by RNA-seq had the same 5′-partner (TPR) as one of the cases in the present study. Furthermore, using FISH, the authors showed that 2 of 4 NTRK1-negative tumors had either ALK or ROS1 fusions instead. Importantly, they also investigated 25 cases of classical lipofibromatosis by FISH, without detecting any NTRK1-rearrangement [9], in agreement with the absence of NTRK1 fusions in the present study. A more extensively studied tumor type is inflammatory myofibroblastic tumor, in which roughly 85% of cases show fusions involving RTK-encoding genes. Most commonly, they affect ALK, but PDGFRB, ROS1, or NTRK3 may serve as alternate 3′-partners [33,34,35,36,37]. The 5′-partners vary extensively, and for ALK alone, more than 10 different fusion partners have been described [34, 35, 38]. The RTK-activating fusions have provided means to use specific RTK-inhibitors in inoperable/metastatic inflammatory myofibroblastic tumor cases [39]. Thus, lipofibromatosis, lipofibromatosis-like neural tumor, and inflammatory myofibroblastic tumor all display fusions resulting in activation of several different RTKs. Other examples include fibrous hamartoma of infancy, in which a duplication of exon 20 of EGFR has been recently described [12] and calcifying aponeurotic fibroma, which shares the FN1-EGF fusion with some cases of lipofibromatosis [11].
Our finding of the FN1-EGF fusion in morphologically typical cases of lipofibromatosis which recurred as calcifying aponeurotic fibromas strongly suggests that some cases of “lipofibromatosis” represent instead “early” examples of calcifying aponeurotic fibroma in which the adipocytic component is more pronounced, and diagnostic calcifications are not yet apparent. However, it should be kept in mind that there are several examples of identical gene fusions occurring in distinct, unrelated soft tissue tumors [18]. Whether FN1-EGF fusion-positive cases of “lipofibromatosis” indeed are variants of CAF or whether they differ clinically from cases with other fusions will have to be investigated in larger series.
In summary, the results of our study of lipofibromatosis suggest that this tumor may represent a morphological pattern common to a variety of locally recurring pediatric tumors of the extremities, rather than a single specific entity. Despite stereotypical morphology, lipofibromatosis is genetically heterogeneous, with 8 different fusions identified in 11 of 20 cases. The morphological features of recurrent lesions, and the presence of FN1-EGF or related FN1-TGFA fusions, suggest that some cases of lipofibromatosis likely represent “early” or “non-calcified” calcifying aponeurotic fibromas. The presence of alterations involving RTK-encoding genes also suggests a link to the recently described “lipofibromatosis-like neural tumor”.
References
Miettinen MM, Fetsch JF, Zambrano E Lipofibromatosis. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds). WHO classification of tumors of soft tissue and bone. 4th ed. Lyon: International Agency for Research on Cancer (IARC); 2013, pp 74.
Stout AP. The fibromatoses and fibrosarcoma. Bull Hosp Jt Dis. 1951;12:126–30.
Fetsch JF, Miettinen M, Laskin WB, Michal M, Enzinger FM. A clinicopathologic study of 45 pediatric soft tissue tumors with an admixture of adipose tissue and fibroblastic elements, and a proposal for classification as lipofibromatosis. Am J Surg Pathol. 2000;24:1491–500.
Taran K, Woszczyk M, Kobos J. Lipofibromatosis presenting as a neck mass in eight-years old boy—a case report. Pol J Pathol. 2008;59:217–20.
Lao QY, Sun M, Yu L, Wang J. [Lipofibromatosis: a clinicopathological analysis of eight cases]. Zhonghua Bing Li Xue Za Zhi. 2018;47:186–91.
Vogel D, Righi A, Kreshak J, Dei Tos AP, Merlino B, Brunocilla E, et al. Lipofibromatosis: magnetic resonance imaging features and pathological correlation in three cases. Skelet Radiol. 2014;43:633–9.
Boos MD, Chikwava KR, Dormans JP, Chauvin NA, Jen M. Lipofibromatosis: an institutional and literature review of an uncommon entity. Pediatr Dermatol. 2014;31:298–304.
Fetsch JF, Miettinen M. Calcifying aponeurotic fibroma: a clinicopathologic study of 22 cases arising in uncommon sites. Hum Pathol. 1998;29:1504–10.
Agaram NP, Zhang L, Sung Y-S, Chen C-L, Chung CT, Antonescu CR, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol. 2016;40:1407–16.
Al-Ibraheemi A, Martinez A, Weiss SW, Kozakewich HP, Perez-Atayde AR, Tran H, et al. Fibrous hamartoma of infancy: a clinicopathologic study of 145 cases, including 2 with sarcomatous features. Mod Pathol. 2017;30:474–85.
Puls F, Hofvander J, Magnusson L, Nilsson J, Haywood E, Sumathi VP, et al. FN1-EGF gene fusions are recurrent in calcifying aponeurotic fibroma. J Pathol. 2016;238:502–7.
Park JY, Cohen C, Lopez D, Ramos E, Wagenfuehr J, Rakheja D. EGFR Exon 20 insertion/duplication mutations characterize fibrous hamartoma of infancy. Am J Surg Pathol. 2016;40:1713–8.
Kenney B, Richkind KE, Friedlaender G, Zambrano E. Chromosomal rearrangements in lipofibromatosis. Cancer Genet Cytogenet. 2007;179:136–9.
Walther C, Hofvander J, Nilsson J, Magnusson L, Domanski HA, Gisselsson D, et al. Gene fusion detection in formalin-fixed paraffin-embedded benign fibrous histiocytomas using fluorescence in situ hybridization and RNA sequencing. Lab Invest. 2015;95:1071–6.
Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903–4.
Nicorici DSM, Edgren H, Kangaspeska S, Murumagi A, Kallioniemi O, Virtanen S, et al. FusionCatcher—a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv. 2014.
Walther C, Tayebwa J, Lilljebjörn H, Magnusson L, Nilsson J, Vult von Steyern F, et al. A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J Pathol. 2014;232:534–40.
Mertens F, Antonescu CR, Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer. 2016;55:291–310.
Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–17.
Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846.
Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19:816–27.
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45.
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62.
Gharibi B, Ghuman MS, Hughes FJ. Akt- and Erk-mediated regulation of proliferation and differentiation during PDGFRbeta-induced MSC self-renewal. J Cell Mol Med. 2012;16:2789–801.
Manfredi GI, Dicitore A, Gaudenzi G, Caraglia M, Persani L, Vitale G. PI3K/Akt/mTOR signaling in medullary thyroid cancer: a promising molecular target for cancer therapy. Endocrine. 2015;48:363–70.
Pal P, Khan Z. ROS1-1. J Clin Pathol. 2017;70:1001–9.
Bhattacharyya S, Wu M, Fang F, Toutellotte W, Feghali-Bostwick C, Varga J. Early growth response transcription factors: key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix Biol. 2011;30:235–42.
Greger IH, Watson JF, Cull-Candy SG. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron. 2017;94:713–30.
Ribeiro MP, Custodio JB, Santos AE. Ionotropic glutamate receptor antagonists and cancer therapy: time to think out of the box? Cancer Chemother Pharmacol. 2017;79:219–25.
Ishiuchi S, Yoshida Y, Sugawara K, Aihara M, Ohtani T, Watanabe T, et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. 2007;27:7987–8001.
Schunemann DP, Grivicich I, Regner A, Leal LF, de Araújo DR, Jotz GP, et al. Glutamate promotes cell growth by EGFR signaling on U-87MG human glioblastoma cell line. Pathol Oncol Res. 2010;16:285–93.
Tamama K, Barbeau DJ. Early growth response genes signaling supports strong paracrine capability of mesenchymal stem cells. Stem Cells Int. 2012;2012:428403.
Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59:2776–80.
Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–95.
Antonescu CR, Suurmeijer AJH, Zhang L, Sung Y-S, Jungbluth AA, Travis WD, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. 2015;39:957–67.
Alassiri AH, Ali RH, Shen Y, Lum A, Strahlendorf C, Deyell R, et al. ETV6-NTRK3 Is expressed in a subset of ALK-negative inflammatory myofibroblastic tumors. Am J Surg Pathol. 2016;40:1051–61.
Yamamoto H, Yoshida A, Taguchi K, Kohashi K, Hatanaka Y, Yamashita A, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology. 2016;69:72–83.
Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000;157:377–84.
Mossé YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A Children’s Oncology Group Study. J Clin Oncol. 2017;35:3215–21.
Acknowledgements
This study was supported by grants to FM from the Swedish Childhood Cancer Foundation, the Swedish Cancer Society, and Governmental Funding of Clinical research within the National Health Service.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Al-Ibraheemi, A., Folpe, A.L., Perez-Atayde, A.R. et al. Aberrant receptor tyrosine kinase signaling in lipofibromatosis: a clinicopathological and molecular genetic study of 20 cases. Mod Pathol 32, 423–434 (2019). https://doi.org/10.1038/s41379-018-0150-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0150-3
This article is cited by
-
Giant dorsal lipofibromatosis in an infant: a case report
BMC Pediatrics (2022)
-
Calcified chondroid mesenchymal neoplasms with FN1-receptor tyrosine kinase gene fusions including FGFR2, FGFR1, MERTK, NTRK1, and TEK: a molecular and clinicopathologic analysis
Modern Pathology (2021)
-
What is new in pericytomatous, myoid, and myofibroblastic tumors?
Virchows Archiv (2020)
-
What is new about the molecular genetics in matrix-producing soft tissue tumors? -The contributions to pathogenetic understanding and diagnostic classification
Virchows Archiv (2020)
-
Kindliche Tumoren mit Spindelzellmorphologie
Der Pathologe (2019)

{kind=link}