
Abstract
Diagnosing soft tissue tumors is challenging, even on ample incisional biopsies or resection specimens. There are more than 100 distinct types of soft tissue neoplasms, including more than 80 benign and intermediate mesenchymal tumors and around 40 soft tissue sarcomas. Accurate diagnosis relies first upon recognition of characteristic histologic and cytologic features, including architecture, stromal characteristics, vascular patterns, and dominant cytology; these features may not be represented or apparent in limited core needle biopsy or fine needle aspiration specimens. Once a differential diagnosis is established, application of immunohistochemistry and cytogenetic or molecular diagnostic assays (especially fluorescence in situ hybridization) is used in an attempt to reach a specific diagnosis. In recent years, the diagnostic armamentarium for soft tissue tumors has expanded dramatically, following the discovery of molecular alterations that underlie the pathogenesis of soft tissue tumors. These include new diagnostic immunohistochemical markers that serve as useful surrogates for molecular genetic alterations. Availability of such markers has improved our ability to render accurate and specific diagnoses based on limited biopsy samples. In this review, examples of recently developed markers for the diagnosis of selected soft tissue tumor types will be discussed, including solitary fibrous tumor (STAT6), malignant peripheral nerve sheath tumor (H3K27me3), epithelioid hemangioendothelioma (CAMTA1), dedifferentiated liposarcoma (MDM2), and CIC-DUX4 sarcoma (WT1 and ETV4).
Similar content being viewed by others

Introduction
There are more than 100 different mesenchymal neoplasms of soft tissue, including approximately 80 distinct types of benign and intermediate soft tissue tumors and approximately 40 distinct types of soft tissue sarcomas [1]. Soft tissue sarcomas are rare (representing 1% of malignant neoplasms) but remarkably diverse. Owing to their rarity and diversity, diagnosing soft tissue tumors is challenging, even on ample incisional biopsies or surgical resection specimens. The World Health Organization (WHO) Classification is organized based on lineage; however, for many types of soft tissue tumors, the line of differentiation is not obvious, and some tumor types are of uncertain lineage [1]. Furthermore, tumors of diverse lineages can show similar histologic appearances, and distinguishing among benign, intermediate, and malignant mesenchymal neoplasms can be difficult.
Many (especially deep-seated) soft tissue tumors are first sampled by core needle biopsy or fine needle aspiration [2, 3]. In such limited samples, accurate diagnosis is particularly challenging [4, 5]. It is important to consider what information is needed from these limited biopsies (Table 1). First and most importantly, distinguishing benign from malignant is critical; this distinction is needed for surgical planning (marginal or simple excision vs. wide excision) and for consideration of neoadjuvant therapy. Histologic grade is also important information: in many practices, neoadjuvant radiation therapy is reserved for intermediate or high-grade sarcomas. In some cases, a specific diagnosis is important: [1] for surgical planning, in terms of the extent of resection (e.g., myxofibrosarcoma and epithelioid sarcoma require particularly wide margins), or for medical oncology (e.g., Ewing sarcoma, synovial sarcoma, and some rhabdomyosarcomas require specific chemotherapy protocols). However, each of these tasks may be difficult, and in many cases, impossible, based on a limited biopsy [4, 5]. Soft tissue tumors often exhibit architectural, stromal, and vascular features that allow pathologists to render a specific diagnosis; limited biopsies are often insufficient to sample or recognize these findings. Sarcoma grading includes “tumor differentiation” (which is essentially the diagnostic category), mitotic count, and tumor necrosis; [6] even if it is possible to achieve the diagnosis of a specific sarcoma type, the latter two features are unlikely to be representative of the tumor as a whole in a limited biopsy specimen [7]. Applying these features in a limited biopsy without regard to heterogeneity will often result in a “low grade” (grade 1) designation, whereas the sarcoma may in fact be higher grade. Attention to the radiologic findings (i.e., if there is evidence of necrosis) can be helpful for an integrated assessment of the most likely sarcoma grade. In addition, some specific sarcoma types are ungradable (many of which are translocation-associated); if a specific diagnosis for such a sarcoma cannot be rendered on a limited biopsy, grading would (inappropriately) be attempted. Other sarcoma types are high grade by definition (including many round cell sarcomas); this group of sarcomas may be easier to recognize in a core or fine needle aspiration biopsy, even if a specific diagnostic category cannot be achieved.
Ancillary diagnostic methods
Conventional immunohistochemistry attempts to identify tumor cell lineage. This approach certainly has value for soft tissue tumor pathology, but in many cases, the findings with such markers are insufficient to achieve a specific diagnosis. Fortunately, many new, much more specific immunohistochemical markers have been developed based on molecular genetic alterations (Table 2) [8]. In addition, molecular diagnostic techniques perform very well on core biopsies, cell blocks, and cytologic smears and touch preparations, including fluorescence in situ hybridization (FISH), reverse transcriptase-polymerase chain reaction (RT-PCR), next-generation sequencing, and new multiplex gene fusion assays [9,10,11]. Of note, in contrast to the applications for carcinomas and melanomas, next-generation sequencing as of yet has limited value for soft tissue tumor diagnosis; FISH remains the most widely used molecular method for diagnosis in this class of tumors (Table 3) [11]. However, deciding which immunohistochemical markers to order requires an appropriate and limited differential diagnosis, which can be a challenge on a limited biopsy. Moreover, molecular genetic alterations are often shared by diverse tumor types; for this reason, most alterations have limited specificity (and must be integrated with clinical, histologic, and immunophenotypic findings) [12]. A common example is the EWSR1 gene, first described in Ewing sarcoma, but now known to be involved in gene rearrangements in a wide range of tumor types, not only sarcomas but also benign and intermediate soft tissue tumors [13]. Nonetheless, identifying an EWSR1 rearrangement by FISH can be sufficient to address a particular differential diagnosis and thereby render a specific diagnosis [14].
Approaching soft tissue tumors by first paying attention to the dominant cytology (spindle cell, epithelioid, pleomorphic, or round cell) is helpful to generate a differential diagnosis. Such a pattern-based diagnostic approach can facilitate ordering an appropriate panel of markers. In this review, selected novel diagnostic markers within each of these morphologic groups will be discussed.
Spindle cell tumors of soft tissue
In recent years, diagnosis of soft tissue tumors with spindle cell morphology (which show considerable morphologic overlap) has become much easier, as our understanding of the molecular pathogenetic basis for such tumors has increased. Recently developed diagnostic markers for spindle cell tumors are listed in Table 4. Two examples, solitary fibrous tumor and malignant peripheral nerve sheath tumor, are discussed in this section.
Solitary fibrous tumor
First recognized in the pleura, solitary fibrous tumor is now known to be an anatomically ubiquitous fibroblastic neoplasm, histologically characterized by “patternless” architecture, varying cellularity, prominent stromal collagen, and dilated, branching (“staghorn”) blood vessels. This constellation of features may not be recognized in a core needle biopsy. “Hemangiopericytoma” is an obsolete synonym for solitary fibrous tumor with uniform hypercellularity [15]. CD34 is positive in 95% of solitary fibrous tumors, but this marker has low specificity, as it is also expressed in other tumor types that may be confused with solitary fibrous tumor (e.g., spindle cell lipoma, soft tissue perineurioma, and dermatofibrosarcoma protuberans). Until recently, no specific diagnostic markers were available; distinguishing solitary fibrous tumor from other spindle cell neoplasms, especially spindle cell sarcomas such as synovial sarcoma and malignant peripheral nerve sheath tumor (in the case of hypercellular examples) was a particular diagnostic challenge in core needle and fine needle aspiration biopsies.
In 2013, several groups identified a recurrent, pathognomonic gene fusion in solitary fibrous tumor: NAB2-STAT6 [16,17,18,19]. This fusion gene is a consistent finding in solitary fibrous tumor, identified in nearly all cases. Since these genes reside in close proximity on chromosome 12q13, conventional FISH cannot be used as a diagnostic tool (i.e., the inversion resulting in this fusion cannot be visualized by such a technique). Fortunately, immunohistochemistry is an excellent surrogate for NAB2-STAT6 fusion: strong and diffuse nuclear staining for STAT6 is a highly sensitive and specific marker for solitary fibrous tumor [20, 21]. Monophasic synovial sarcoma, a close histologic mimic of cellular or malignant solitary fibrous tumors, is consistently negative for STAT6. Immunohistochemistry for STAT6 can be performed on core needle biopsies or cell blocks generated from fine needle aspiration biopsies (Fig. 1) [22].

Malignant solitary fibrous tumor. a Core needle biopsy of a large pelvic mass from a 73-year-old woman shows a highly cellular neoplasm with a focal area of pleomorphic cells and scattered thin-walled, dilated blood vessels. b The tumor is composed of sheets of ovoid cells with small amounts of cytoplasm. There was a high mitotic rate (not shown). c Immunohistochemistry for STAT6 shows strong and diffuse nuclear staining, confirming the diagnosis of solitary fibrous tumor. This finding reflects the presence of an underlying NAB2-STAT6 gene rearrangement
Malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumor may arise in patients with neurofibromatosis type 1, sporadically, or following therapeutic radiation therapy. When such tumors originate from a large nerve or a neurofibroma, or when a spindle cell sarcoma arises in a patient with neurofibromatosis type 1, the diagnosis is relatively straightforward. Without such associations, however, diagnosing malignant peripheral nerve sheath tumors is highly challenging, until recently relying in large part on recognition of distinctive histologic findings (and exclusion of histologic mimics such as monophasic synovial sarcoma) [23]. Characteristic histologic features of malignant peripheral nerve sheath tumor include a variegated appearance with alternating hypercellular and hypocellular areas (the former often with a tight, fascicular “fibrosarcoma”-like architecture), variable myxoid stroma, perivascular hypercellularity, and elongated, tapering nuclei; heterologous differentiation (most often rhabdomyoblastic) is observed in around 5% of cases (a helpful diagnostic clue). These features may not be appreciated in a limited biopsy specimen. Evidence of Schwann cell differentiation by immunohistochemistry for S100 protein or SOX10 is identified in less than 50% of cases (usually with limited staining, easily missed in a core needle biopsy) [24, 25].
In 2014, several groups identified SUZ12 and EED mutations in malignant peripheral nerve sheath tumors [26,27,28]. These genes encode proteins in the polycomb repressive complex 2 (PRC2), which plays a key role in epigenetic modification of chromatin through trimethylation of histone H3 at the lysine 27 residue. PRC2 alterations are found in up to 90% of malignant peripheral nerve sheath tumors; homozygous mutations (identified in around 65% of cases) result in loss of PRC2 function and loss of histone H3 lysine 27 trimethylation (H3K27me3). This can be assessed by immunohistochemistry, as specific monoclonal antibodies that only recognize H3K27me3 (but not histone H3 without lysine 27 trimethylation) are available [29,30,31]. The rate of H3K27me3 loss depends upon histologic grade: high-grade malignant peripheral nerve sheath tumors show loss of H3K27me3 in up to 85% of tumors, whereas low-grade malignant peripheral nerve sheath tumors show loss in only 35% of cases [29]. Nonetheless, immunohistochemistry for H3K27me3 has become the most sensitive and specific available marker for malignant peripheral nerve sheath tumor (since high-grade tumors are most often encountered in clinical practice). This marker can be helpful in biopsy specimens (Fig. 2); [32] however, it is important to recognize that loss of H3K27me3 is not entirely specific for malignant peripheral nerve sheath tumor, as a small subset of dedifferentiated liposarcomas and some spindle cell melanomas also show loss of H3K27me3 [33, 34].

Malignant peripheral nerve sheath tumor. a Core needle biopsy of a chest wall mass from a 47-year-old man shows a highly cellular, fascicular neoplasm with scattered thin-walled, dilated blood vessels. b The tumor is composed of tight fascicles of uniform spindle cells with even chromatin and scant cytoplasm. c Areas of the tumor contain collagenous stroma. Note the tapering nuclei. d Immunohistochemistry for H3K27me3 (histone H3 with lysine 27 trimethylation) shows loss of nuclear staining. Note the staining in the endothelial cell nuclei, which serve as a positive internal control. This tumor was negative for S100 protein and SOX10 (not shown); fluorescence in situ hybridization (FISH) was also negative for SS18 gene rearrangement (excluding monophasic synovial sarcoma)
Epithelioid tumors of soft tissue
A diverse array of rare soft tissue tumors shows epithelioid cytomorphology [35]. The possibility of metastatic carcinoma and metastatic melanoma must always be excluded before considering a soft tissue tumor in this category, since the former are so much more common. Paying attention to architecture, cytology, stroma, and anatomic location can be helpful to generate a differential diagnosis; however, the distinguishing histologic features may not be well represented in a limited biopsy specimen. In recent years, molecular genetic alterations for some sarcomas with epithelioid morphology have been identified; in many cases, immunohistochemistry may serve as a surrogate for such alterations (Table 5) [8]. One such example, epithelioid hemangioendothelioma, will be reviewed.
Epithelioid hemangioendothelioma
Originally believed to fall into the category of mesenchymal neoplasms of intermediate biologic potential, epithelioid hemangioendothelioma is now recognized to be a sarcoma, although this distinctive sarcoma is less aggressive than angiosarcomas [36]. Although epithelioid hemangioendothelioma shows endothelial differentiation, this tumor type is incapable of forming true vascular channels; its recognition is therefore challenging. Epithelioid hemangioendothelioma is composed of epithelioid cells, usually with bland nuclear morphology and eosinophilic, variably glassy cytoplasm with occasional intracytoplasmic vacuoles, arranged in cords and nests within a distinctive myxohyaline stroma. Epithelioid hemangioendothelioma may arise not only in soft tissue sites, but also in the liver, lungs, and bones; at these latter sites, multicentric presentation is common. Keratin expression in epithelioid hemangioendothelioma is a common finding; this feature, along with the histologic appearances and multifocality in anatomic locations where metastases are common, may easily lead to misdiagnosis as metastatic carcinoma, especially lobular breast carcinoma and poorly cohesive (including signet-ring-cell) gastric carcinomas.
Once the diagnosis is considered, demonstration of endothelial differentiation by immunohistochemistry with vascular markers such as CD31 and ERG is usually sufficient to render a specific diagnosis. However, in around 20% of cases, the degree of nuclear atypia is greater than conventional examples, often accompanied by increased mitotic activity and sometimes sheet-like areas; in such cases, the distinction from epithelioid angiosarcoma (which is more aggressive than epithelioid hemangioendothelioma and is more sensitive to chemotherapy) can be challenging, particularly in limited biopsies where areas of typical histology may not be evident [36].
In 2001, two cases of epithelioid hemangioendothelioma with a t(1;3)(p36.3;q25) translocation were published [37]. Ten years later in 2011, two independent groups identified the disease-defining gene rearrangement that results from this translocation: WWTR1-CAMTA1 [38, 39]. This gene fusion is a consistent finding in epithelioid hemangioendothelioma, found in 85–90% of cases. Immunohistochemistry for CAMTA1 serves as a useful surrogate for this gene rearrangement: nuclear staining for CAMTA1 is positive in epithelioid hemangioendotheliomas that harbor WWTR1-CAMTA1, whereas CAMTA1 is negative in epithelioid angiosarcomas, carcinomas, and other epithelioid mesenchymal neoplasms that may mimic epithelioid hemangioendothelioma (such as epithelioid sarcoma) [40, 41]. This marker is particularly helpful in liver core needle biopsies, where the subtle infiltration of epithelioid hemangioendothelioma cells among hepatocytes may be difficult to recognize (Fig. 3).

Epithelioid hemangioendothelioma of the liver. (a) Core needle biopsy of a liver mass from a 60-year-old woman who presented with multiple hepatic nodules. The liver parenchyma is infiltrated by a cellular neoplasm with hyalinized stroma. (b) Much of the biopsy consisted of nearly normal liver parenchyma with a subtle hypercellularity. (c) The hepatic sinusoids contain scattered hyperchromatic and multinucleated cells. (d) Immunohistochemistry for CAMTA1 shows nuclear staining in the atypical cells within the sinusoids. (e) Other areas of the biopsy show more typical cytoarchitectural features of epithelioid hemangioendothelioma, with cords of epithelioid cells containing occasional cytoplasmic vacuoles, embedded in a collagenous stroma. (f) CAMTA1 highlights the cord-like architecture. CAMTA1 expression correlates with the pathognomonic WWTR1-CAMTA1 gene fusion
Pleomorphic sarcomas of soft tissue
Despite significant histologic overlap, pleomorphic sarcomas are a diverse group of soft tissue sarcomas with marked differences in clinical behavior and metastatic potential [42]. As a whole, these aggressive sarcomas harbor complex cytogenetics, in most cases without specific, diagnostically useful molecular alterations. For this reason, careful attention to histology and application of immunohistochemistry to identify the line of differentiation remain the cornerstones of diagnosis for most tumor types in this category. For example, myxofibrosarcoma is recognized by areas of the tumor with myxoid stroma containing curvilinear blood vessels and scattered hyperchromatic, pleomorphic cells; in high-grade examples, however, the majority of the tumor may be devoid of myxoid stroma and indistinguishable from an undifferentiated pleomorphic sarcoma. In order to diagnose extraskeletal osteosarcoma, osteoid matrix being laid down by the malignant tumor cells must be identified; such a finding may be limited in extent, with the bulk of the tumor again indistinguishable from other pleomorphic sarcomas. Pleomorphic liposarcoma is defined by the presence of lipoblasts within an otherwise non-distinctive pleomorphic (or sometimes epithelioid) sarcoma. Each of these distinguishing histologic findings is rarely sampled in a core biopsy or fine needle aspiration biopsy. It is therefore often impossible to render a specific diagnosis in a limited biopsy sample. One notable exception is the relatively common sarcoma type dedifferentiated liposarcoma, for which useful diagnostic markers are available.
Dedifferentiated liposarcoma
Dedifferentiated liposarcoma most often arises at central body cavity sites, including the retroperitoneum, abdominal cavity, and pelvis. Despite the often highly pleomorphic morphology of this tumor type, dedifferentiated liposarcoma has much lower metastatic potential than other pleomorphic sarcomas (approximately 15%); nonetheless, most patients succumb to uncontrolled local recurrence, sometimes decades following first presentation. Dedifferentiated liposarcoma is defined by the usually abrupt transition from well-differentiated liposarcoma to a non-lipogenic sarcoma; the non-adipocytic component may show highly variable morphology (even within a single tumor), including pleomorphic, spindle cell, and epithelioid morphology; variable myxoid stroma; and heterologous elements in around 10% of cases (most often rhabdomyoblastic and chondro-osseous differentiation) [43]. Without sampling the well-differentiated liposarcoma component, dedifferentiated liposarcoma shows no distinctive histologic features. The non-lipogenic component shows variable expression of SMA and desmin and may therefore be confused with leiomyosarcoma (among other sarcoma types). In core needle biopsy specimens, recognition of dedifferentiated liposarcoma may be extremely challenging; the key point to remember is that a non-distinctive sarcoma of body cavity sites is most often dedifferentiated liposarcoma.
Fortunately, dedifferentiated (and well-differentiated) liposarcoma harbors distinctive molecular genetic alterations, in the form of high-level amplification of chromosome region 12q13~15, which includes the oncogenes MDM2 and CDK4 (among others) [44]. These amplification events are often contained within unusual forms of chromosomes: ring chromosomes and giant marker (rod) chromosomes, which can be identified by conventional karyotyping. MDM2 has become a defining marker for dedifferentiated liposarcoma (Fig. 4). Both FISH for MDM2 amplification and immunohistochemistry for MDM2 overexpression may be used in differential diagnosis; the former is more specific for dedifferentiated liposarcoma, since some other tumor types that might be considered in the differential diagnosis (e.g., malignant peripheral nerve sheath tumor and myxofibrosarcoma) also often express the MDM2 protein (but not secondary to amplification) [45, 46]. However, the combination of MDM2 and CDK4 expression is much more specific than MDM2 alone. Either type of testing may be extremely useful in the evaluation of a core needle biopsy or fine needle aspiration specimen; FISH for MDM2 can be performed on a cytologic smear and therefore requires limited cellular material (see Fig. 4) [47]. It has become relatively straightforward to diagnose dedifferentiated liposarcoma on limited biopsy material, so long as the possibility of this tumor type is considered.

Dedifferentiated liposarcoma. a This core needle biopsy of a large retroperitoneal mass from a 63-year-old man shows a variably cellular spindle cell neoplasm. b The tumor is composed of fascicles of mildly atypical, mitotically active spindle cells. The histologic appearances are not distinctive. c Immunohistochemistry for MDM2 shows strong nuclear staining. d Fluorescence in situ hybridization (FISH) performed on a cytologic smear from a corresponding fine needle aspiration shows multiple orange signals with a probe directed against chromosome 12q15 (MDM2) and two normal green signals with a probe directed against the chromosome 12 centromere, indicating high-level amplification of MDM2. This is a characteristic feature of dedifferentiated liposarcoma (and well-differentiated liposarcoma)
Round cell sarcomas of soft tissue
Round cell sarcomas are a diverse group of highly aggressive malignant neoplasms with a predilection for children and young adults. These sarcomas often require specific chemotherapeutic regimens; specific diagnosis is therefore critical. However, these sarcoma types share similar histologic features and can be extremely challenging to diagnose on a core needle biopsy or fine needle aspiration specimen. Paying attention to patient age and anatomic location can help narrow down the differential diagnosis, but diagnosis requires integration of these findings with histology, immunohistochemistry, and often molecular genetics [48]. Fortunately, many of these sarcomas harbor specific translocations, resulting in diagnostically useful gene rearrangements (Table 6) [48]. These gene fusions may be identified by FISH, RT-PCR, and other more recently developed techniques such as next-generation sequencing. Molecular genetic testing is routinely performed on biopsy specimens for the differential diagnosis of this class of sarcomas, more often than for other sarcoma groups discussed earlier in this review. One relatively recently identified sarcoma in this category, CIC-rearranged sarcoma, will be discussed.
CIC-rearranged sarcoma
CIC-rearranged sarcoma (also known as CIC-DUX4 sarcoma) is the most common “Ewing-like” (“undifferentiated”) round cell sarcoma that lacks EWSR1 gene fusions, representing at least 70% of sarcomas that were formerly relegated to this diagnostic wastebasket [49]. First identified in 2006 [50] and defined more completely in 2012 [49], CIC-rearranged sarcomas most often arise in the deep soft tissues of the extremities and trunk of young to middle-aged adults, although the age range and anatomic distribution is wide; unlike Ewing sarcoma, bone origin is rare [51]. CIC-rearranged sarcomas pursue a much more aggressive clinical course than Ewing sarcoma with worse outcomes, although these sarcoma types are currently treated in a similar fashion, since specific protocols for CIC-rearranged sarcomas have not yet been developed [51]. Although CIC-rearranged sarcomas are usually dominated by sheets of round cells and may therefore mimic Ewing sarcoma, this tumor type has distinguishing features, including mild nuclear variability, prominent nucleoli, and small amounts of pale or eosinophilic cytoplasm (Fig. 5); many tumors have focal areas of myxoid stroma and a minor spindle cell or epithelioid component, the latter sometimes with rhabdoid features. These findings contrast with Ewing sarcoma, which invariably shows remarkably uniform cytomorphology, with round nuclei, fine chromatin, indistinct nucleoli, and scant cytoplasm.

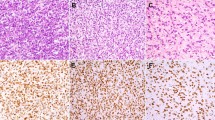
CIC-rearranged (CIC-DUX4) sarcoma. a A core needle biopsy from a thigh mass from a 37-year-old man shows a highly cellular neoplasm with large areas of necrosis. b The tumor is dominated by round cells with focal areas of scant myxoid stroma. c The tumor cells show nuclear variability with irregular nuclei and small amounts of eosinophilic cytoplasm. This degree of nuclear heterogeneity would not fit with Ewing sarcoma. By immunohistochemistry, the tumor cells show nuclear staining for both ETV4 (d) and WT1 (e), characteristic of CIC-rearranged sarcoma. Fluorescence in situ hybridization (FISH) shows separation (“break apart”) of the green and orange signals, corresponding to probes directed against the 5’ (centromeric) and 3’ (telomeric) sequences of the CIC gene, respectively. The yellow signals correspond to the normal copy of the CIC gene (with overlapping 5’ and 3’ probes)
In contrast to Ewing sarcoma, which shows strong and diffuse membranous staining for CD99 in essentially all cases, CIC-rearranged sarcomas often show patchy, heterogeneous staining for CD99, although diffuse staining may be observed in 20% of cases [51]. NKX2–2 expression also distinguishes Ewing sarcoma from CIC-rearranged sarcomas [52, 53]. Gene expression profiling has identified ETV4 and WT1 overexpression in CIC-rearranged sarcomas; [54] nuclear staining for these two markers by immunohistochemistry is helpful to support the diagnosis (see Fig. 5) [55, 56]. The most common gene fusion in CIC-rearranged sarcomas is CIC-DUX4; CIC-FOXO4 has been identified in rare cases [57, 58]. FISH to identify CIC rearrangement can be used to confirm the diagnosis (see Fig. 5). However, in around 15% of cases, FISH is negative, owing to cryptic rearrangements; upregulation of ETV4 is therefore more sensitive than FISH for CIC [59]. The optimal diagnostic approach for CIC-rearranged sarcomas is not yet entirely clear, although a combination of immunohistochemistry and FISH seems most promising.
Summary
Diagnosing soft tissue tumors in limited biopsy samples is a challenge for surgical pathologists. Fortunately, the molecular genetic alterations than underlie the pathogenesis of many of these tumor types continue to be identified, in some cases resulting in diagnostically useful FISH probes or immunohistochemical markers. Such markers have ushered in a new era in diagnostic immunohistochemistry; conventional lineage markers are now being supplanted in some cases with much more specific markers that recognize protein surrogates of molecular genetic alterations. It is therefore becoming easier to render a specific diagnosis in core needle biopsy or fine needle aspiration specimens. Unfortunately, this is not the case for all tumor types; in some cases, a specific diagnosis is not possible, and distinguishing benign soft tissue tumors from sarcomas remains our primary task.
References
Fletcher C, Bridge, JA, Hogendoorn, PCW, Mertens F. WHO Classification of tumours of soft tissue and bone. IARC Press, Lyon; 2013.
Singh HK, Kilpatrick SE, Silverman JF. Fine needle aspiration biopsy of soft tissue sarcomas: utility and diagnostic challenges. Adv Anat Pathol. 2004;11:24–37.
Strauss DC, Qureshi YA, Hayes AJ, et al. The role of core needle biopsy in the diagnosis of suspected soft tissue tumours. J Surg Oncol. 2010;102:523–9.
Layfield LJ, Schmidt RL, Sangle N, et al. Diagnostic accuracy and clinical utility of biopsy in musculoskeletal lesions: a comparison of fine-needle aspiration, core, and open biopsy techniques. Diagn Cytopathol. 2014;42:476–86.
Trieu J, Schlicht SM, Choong PF. Diagnosing musculoskeletal tumours: How accurate is CT-guided core needle biopsy? Eur J Surg Oncol. 2016;42:1049–56.
Coindre JM, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91:1914–26.
Deyrup AT, Weiss SW. Grading of soft tissue sarcomas: the challenge of providing precise information in an imprecise world. Histopathology. 2006;48:42–50.
Schaefer IM, Cote GM, Hornick JL. Contemporary sarcoma diagnosis, genetics, and genomics. J Clin Oncol. 2018;36:101–10.
Dal Cin P, Qian X, Cibas ES. The marriage of cytology and cytogenetics. Cancer Cytopathol. 2013;121:279–90.
Hwang DH, Garcia EP, Ducar MD, et al. Next-generation sequencing of cytologic preparations: An analysis of quality metrics. Cancer Cytopathol. 2017;125:786–94.
Tanas MR, Goldblum JR. Fluorescence in situ hybridization in the diagnosis of soft tissue neoplasms: a review. Adv Anat Pathol. 2009;16:383–91.
Antonescu CR, Dal Cin P. Promiscuous genes involved in recurrent chromosomal translocations in soft tissue tumours. Pathology. 2014;46:105–12.
Romeo S, Dei Tos AP. Soft tissue tumors associated with EWSR1 translocation. Virchows Arch. 2010;456:219–34.
Noujaim J, Jones RL, Swansbury J, et al. The spectrum of EWSR1-rearranged neoplasms at a tertiary sarcoma centre; assessing 772 tumour specimens and the value of current ancillary molecular diagnostic modalities. Br J Cancer. 2017;116:669–78.
Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281–92.
Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013;45:180–5.
Chmielecki J, Crago AM, Rosenberg M, et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet. 2013;45:131–2.
Mohajeri A, Tayebwa J, Collin A, et al. Comprehensive genetic analysis identifies a pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic imbalances, and a characteristic gene expression profile in solitary fibrous tumor. Genes Chromosomes Cancer. 2013;52:873–86.
Schweizer L, Koelsche C, Sahm F, et al. Meningeal hemangiopericytoma and solitary fibrous tumors carry the NAB2-STAT6 fusion and can be diagnosed by nuclear expression of STAT6 protein. Acta Neuropathol. 2013;125:651–8.
Doyle LA, Vivero M, Fletcher CD, et al. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol. 2014;27:390–5.
Cheah AL, Billings SD, Goldblum JR, et al. STAT6 rabbit monoclonal antibody is a robust diagnostic tool for the distinction of solitary fibrous tumour from its mimics. Pathology. 2014;46:389–95.
Tani E, Wejde J, Åström K, et al. FNA cytology of solitary fibrous tumors and the diagnostic value of STAT6 immunocytochemistry. Cancer Cytopathol. 2018;126:36–43.
Le Guellec S, Decouvelaere AV, Filleron T, et al. Malignant peripheral nerve sheath tumor is a challenging diagnosis: a systematic pathology review, immunohistochemistry, and molecular analysis in 160 patients from the French Sarcoma Group database. Am J Surg Pathol. 2016;40:896–908.
Nonaka D, Chiriboga L, Rubin BP. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol. 2008;32:1291–8.
Miettinen M, McCue PA, Sarlomo-Rikala M, et al. Sox10--a marker for not only schwannian and melanocytic neoplasms but also myoepithelial cell tumors of soft tissue: a systematic analysis of 5134 tumors. Am J Surg Pathol. 2015;39:826–35.
De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–51.
Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–32.
Zhang M, Wang Y, Jones S, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1170–2.
Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016;29:4–13.
Prieto-Granada CN, Wiesner T, Messina JL, et al. Loss of H3K27me3 expression is a highly sensitive marker for sporadic and radiation-induced MPNST. Am J Surg Pathol. 2016;40:479–89.
Cleven AH, Sannaa GA. Briaire-de Bruijn I, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol. 2016;29:582–90.
Mito JK, Qian X, Doyle LA, et al. Role of histone H3K27 trimethylation loss as a marker for malignant peripheral nerve sheath tumor in fine-needle aspiration and small biopsy specimens. Am J Clin Pathol. 2017;148:179–89.
Le Guellec S, Macagno N, Velasco V, et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: a study of 387 cases including mimicking lesions. Mod Pathol. 2017;30:1677–87.
Makise N, Sekimizu M, Kubo T, et al. Clarifying the distinction between malignant peripheral nerve sheath tumor and dedifferentiated liposarcoma: a critical reappraisal of the diagnostic utility of MDM2 and H3K27me3 status. Am J Surg Pathol. 2018;42:656–64.
James AW, Dry SM. Diagnostically challenging epithelioid soft tissue tumors. Surg Pathol Clin. 2015;8:309–29.
Antonescu C. Malignant vascular tumors--an update. Mod Pathol. 2014;27(Suppl 1):S30–38.
Mendlick MR, Nelson M, Pickering D, et al. Translocation t(1;3)(p36.3; q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am J Surg Pathol. 2001;25:684–7.
Tanas MR, Sboner A, Oliveira AM, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;3:98ra82.
Errani C, Zhang L, Sung YS, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50:644–53.
Shibuya R, Matsuyama A, Shiba E, et al. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology. 2015;67:827–35.
Doyle LA, Fletcher CD, Hornick JL. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol. 2016;40:94–102.
Goldblum JR. An approach to pleomorphic sarcomas: can we subclassify, and does it matter. Mod Pathol. 2014;27(Suppl 1):S39–46.
Dei Tos AP. Liposarcomas: diagnostic pitfalls and new insights. Histopathology. 2014;64:38–52.
Dei Tos AP, Doglioni C, Piccinin S, et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol. 2000;190:531–6.
Binh MB, Sastre-Garau X, Guillou L, et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol. 2005;29:1340–7.
Sirvent N, Coindre JM, Maire G, et al. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am J Surg Pathol. 2007;31:1476–89.
Mariño-Enríquez A, Hornick JL, Dal Cin P, et al. Dedifferentiated liposarcoma and pleomorphic liposarcoma: a comparative study of cytomorphology and MDM2/CDK4 expression on fine-needle aspiration. Cancer Cytopathol. 2014;122:128–37.
Antonescu C. Round cell sarcomas beyond Ewing: emerging entities. Histopathology. 2014;64:26–37.
Italiano A, Sung YS, Zhang L, et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer. 2012;51:207–18.
Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35; q13) translocation. Hum Mol Genet. 2006;15:2125–37.
Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41:941–9.
Yoshida A, Sekine S, Tsuta K, et al. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am J Surg Pathol. 2012;36:993–9.
Hung YP, Fletcher CD, Hornick JL. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: imperfect specificity for Ewing sarcoma. Mod Pathol. 2016;29:370–80.
Specht K, Sung YS, Zhang L, et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer. 2014;53:622–33.
Hung YP, Fletcher CD, Hornick JL. Evaluation of ETV4 and WT1 expression in CIC-rearranged sarcomas and histologic mimics. Mod Pathol. 2016;29:1324–34.
Le Guellec S, Velasco V, Pérot G, et al. ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol. 2016;29:1523–31.
Sugita S, Arai Y, Tonooka A, et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol. 2014;38:1571–6.
Solomon DA, Brohl AS, Khan J, et al. Clinicopathologic features of a second patient with Ewing-like sarcoma harboring CIC-FOXO4 gene fusion. Am J Surg Pathol. 2014;38:1724–5.
Kao YC, Sung YS, Chen CL, et al. ETV transcriptional upregulation is more reliable than RNA sequencing algorithms and FISH in diagnosing round cell sarcomas with CIC gene rearrangements. Genes Chromosomes Cancer. 2017;56:501–10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Rights and permissions
About this article

Cite this article
Hornick, J.L. Limited biopsies of soft tissue tumors: the contemporary role of immunohistochemistry and molecular diagnostics. Mod Pathol 32 (Suppl 1), 27–37 (2019). https://doi.org/10.1038/s41379-018-0139-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0139-y
This article is cited by
-
Soft Tissue Tumours—Study of a Short Panel of 8 Immunohistochemical Markers
Indian Journal of Surgery (2023)
-
Primary desmoplastic small round cell tumor of the submandibular gland: a case report and literature review
Diagnostic Pathology (2022)
-
Theranostic cells: emerging clinical applications of synthetic biology
Nature Reviews Genetics (2021)
-
Histone H3K27 dimethyl loss is highly specific for malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss
Modern Pathology (2019)
