
Abstract
At the histological level, tumor budding in colon cancer is the result of cells undergoing at least partial epithelial-to-mesenchymal transition. The microRNA 200 family is an important epigenetic driver of this process, mainly by downregulating zinc-finger E-box binding homeobox (ZEB) and transforming growth factor beta (TGF-β) expression. We retrospectively explored the expression of the miR200 family, and ZEB1 and ZEB2, and their relationship with immune resistance mediated through PD-L1 overexpression. For this purpose, we analyzed a series of 125 colon cancer cases and took samples from two different tumor sites: the area of tumor budding at the invasive front and from the tumor center. We found significant ZEB overexpression and a reduction in miR200 in budding areas, a profile compatible with epithelial-to-mesenchymal transition. In multivariate analysis of the cases with localized disease, low miR200c expression in budding areas, but not at the tumor center, was an adverse tumor-specific survival factor (hazard ratio: 0.12; 95% confidence interval: 0.03–0.81; p = 0.02) independent of the clinical stage of the disease. PD-L1 was significantly overexpressed in the budding areas and its levels correlated with a mesenchymal transition profile. These results highlight the importance of including budding areas among the samples used for biomarker evaluation and provides relevant data on the influence of mesenchymal transition in the immune resistance mediated by PD-L1 overexpression.
Similar content being viewed by others


Introduction
Tumor budding is defined as the presence of isolated cells or clusters of up to 5 cells at the invasive front of colon cancer tumors. It is a well-recognized independent predictor of lymph node metastasis in pT1 colorectal tumors and an independent predictor of survival in stage II patients [1,2,3,4]. Epithelial-to-mesenchymal transition has been postulated to be responsible for tumor bud formation [5,6,7,8]. In the consensus molecular classification of colon cancer, the mesenchymal subgroup (CMS4) is the most aggressive subtype, and displays upregulation of genes implicated in epithelial-to-mesenchymal transition and signatures associated with the activation of transforming growth factor receptor-β (TGF-β) [9].
MicroRNAs (miRNAs) are small, noncoding RNAs with important functions in development, and cell differentiation and proliferation [10]. Each miRNA targets multiple genes to negatively regulate their expression either through messenger RNA (mRNA) degradation or translation inhibition [10, 11]. The miR200 family comprises 5 members, located in two different clusters: cluster I in chromosome 1 containing miR200a, miR200b, and miR429, and cluster II in chromosome 12 containing miR200c and miR141 [12]. This family is known to be an epigenetic regulator of epithelial-to-mesenchymal transition, and is able to control the process through a feedback loop with the promoter regions of zinc-finger E-box binding homeobox (ZEB) and transforming growth factor beta (TGF-β) [13,14,15]. ZEB1 and ZEB2 are large transcription factors containing two zinc-finger domains and are important regulators of cadherin expression and epithelial-to-mesenchymal transition in colon cancer [16,17,18,19,20]. miR200 expression may be mediated by transcription factors such as ZEB or by epigenetic mechanisms. Moreover, methylation of the two miR200 promoter regions could be used to identify mesenchymal tumor subtypes, and has already been shown to be predictive of shorter disease-free survival rates in colon cancer patients [21].
In this work, we explored the expression of genes and miRNAs implicated in epithelial-to-mesenchymal transition: ZEB1, ZEB2, miR200a, miR200b, and miR200c, both in the center of tumors and at the invasive tumor front with the highest concentration of tumor buds. We related the expression of each molecule according to the sampled area, to patient survival rates, in a series of 125 colon cancer cases. Finally, we also evaluated the expression of PD-L1 in both areas to gain a better understanding of the mechanisms of immune resistance associated with tumor budding and epithelial-to-mesenchymal transition.
Materials and methods
We retrospectively analyzed 125 formalin-fixed paraffin-embedded (FFPE) colectomy specimens from colon cancer patients which had been received consecutively at the Department of Pathology in the Hospital Clínico Universitario of Valencia (Spain), over 15 months, from January 2012 to April 2013. Samples from tumors located below the peritoneal reflection were excluded.
Pathological evaluation
All the slides were reviewed by 2 pathologists (C.M. and S.N.F) and were classified according to the American Joint Committee on Cancer (AJCC) 8th edition guidelines [22]. The tumor differentiation (high or low), invasion pattern (infiltrative vs. expansive), and the presence of lymphovascular or perineural invasion were recorded in each case. Clinical and follow-up information was obtained from the medical history records of all the participants. The study protocol was approved by the Ethics Board at the INCLIVA Biomedical Research Institute. Tumor budding was assessed in a hotspot area of 0.785 mm2 on H&E sections, which was chosen after reviewing all the available slides containing invasive tumor sections and according to the recommendations of the international tumor budding consensus conference [7]. Each case was classified in one category: low, intermediate or high-grade tumor budding when having 0–4, 5–9, and 10 or more buds.
Immunohistochemical studies
Paraffin-embedded tumor sections were deparaffinized and the antigens were retrieved in a high pH citrate buffer in a pressure cooker (Dako). The immunohistochemical studies were done using an automated Autostainer Link 48 system (Dako, Glostrup, Denmark) with the following monoclonal antibodies: MLH1 (clone IR079, dilution 1:100. Dako), MSH2 (clone IR085, dilution 1:100. Dako), MSH6 (clone IR086, dilution 1:100. Dako), and PMS2 (clone IR087, dilution 1:100, Dako). PD-L1 immunostaining was carried out in a low pH citrate buffer (clone 28.8, prediluted, Dako). Only the complete loss of nuclear staining in combination with a positive internal control was accepted as the loss of mismatch repair (MMR) protein expression. Lymphocytes, stromal cells, and non-neoplastic epithelial cells were used as internal controls.
RNA isolation and molecular studies
RNA was purified from samples selected from two different tumor areas: from the center and at the invasion front, the latter including the tissue with the highest concentration of buds. Samples were deparaffinized using a deparaffinization solution (Qiagen, CA). Total RNA (including small RNAs) was purified using the AllPrep DNA/RNA FFPE kit (Qiagen, CA) following the manufacturer’s instructions. The RNA concentration was quantified using a NanoDrop spectrophotometer (NanoDrop). Reverse transcription (RT) was carried out using the TaqMan® MicroRNA Reverse Transcription Kit in presence of RNase inhibitor (Applied Biosystems, Foster City, CA). The RT reaction was multiplexed by creating a customized RT primer pool using the following primers: UAACACUGUCUGGUAACGAUGU for hsa-miR-200a-3p (MIMAT0000682), UAAUACUGCCUGGUAAUGAUGA for hsa-miR-200b-3p (MIMAT0000318), and UAAUACUGCCGGGUAAUGAUGGA for hsa-miR-200c (MIMAT0000617). A pre-amplification step for the RT-PCR studies was required using the following primers: UAACACUGUCUGGUAACGAUGU for ZEB1, UAAUACUGCCUGGUAAUGAUGA for ZEB2, and UAAUACUGCCGGGUAAUGAUGGA for PD-L1. The expression of genes and miRNAs were analyzed by quantitative real time-PCR (qRT-PCR) using TaqMan Gene Expression Assays for genes and TaqMan microRNA Assays for miRNAs (Applied Biosystems by Life Technologies, Carlsbad, California, USA). The samples were normalized to GAPDH and RNU6B for genes and miRNAs, respectively, and relative expression was calculated by using the comparative Ct method to obtain the fold-change value (ΔΔCt) [23].
Statistical analysis
The data were statistically analyzed using the software R, version 3.4.2 (Vienna, Austria); p-values of 0.05 or less were set as the threshold for statistical significance. The clinical and pathological characteristics of the patients were compared using the Chi-squared test or the Wilcoxon test for qualitative and quantitative variables, respectively; prior to this, the data were assessed for normality using the Shapiro–Wilk test. Tumor-specific survival was calculated from the time of colectomy to death from tumor-related causes or until the last known follow-up. Disease-free survival was defined as the period from the time of colectomy until metastasis or death. Survival was analyzed by comparing Kaplan–Meier curves using the log-rank test. Tumor-specific survival and disease-free survival were evaluated in non-metastatic (stages I to III) disease. Multivariate regression analysis was carried out using Cox proportional hazards models with stepwise selection, including any variables which significantly correlated with the survival probability in the univariate analysis.
Results
Clinicopathological features and tumor budding
The clinicopathological features of the data are summarized in Table 1; 30% of cases (n = 36) showed high-grade tumor budding, 32% (n = 39) intermediate-grade, and 38% (n = 47) low-grade. As shown in Table 2, we observed a significant association between high tumor budding grades and adverse prognostic factors in colon cancer: more advanced clinical stage (p = 0.03), higher tumor grade (p = 0.01), larger tumor size, or pT (p = 0.00), a higher number of positive lymph nodes, or pN (p = 0.00), and the presence of lymphovascular (p = 0.00) and perineural invasion (p = 0.00). Only 4 cases (3.2%) showed loss of MMR protein expression, and we found no relationship between MMR deficiency and tumor budding grade.
Expression analysis
The expression of miR200a, miR200b, and miR200c was significantly lower at the invasive front compared with the tumor center (p = 0.00 in all cases). Conversely, ZEB1 and ZEB2 were overexpressed at the invasive front (p = 0.00 and 0.02, respectively), especially in cases with an infiltrative growth pattern (p = 0.00). PD-L1 was also overexpressed at the invasive front with respect to the tumor center (p = 0.01), especially in cases with high-grade tumor budding compared to cases with low and intermediate-grade (p = 0.03). PD-L1 overexpression at the invasive front was correlated with lower levels of miR200a (p = 0.02) and miR200c (p = 0.03) expression, and higher levels of ZEB1 (p = 0.00) and ZEB2 (p = 0.00) expression (Fig. 1). However, we found no correlation between miR200a, miR200b, miR200c, ZEB1, or ZEB2 expression and tumor budding grade. Relative to the tumor centers, there was a significant decrease in miR200a, miR200b, and miR200c expression and increased ZEB1 and ZEB2 expression at the invasive fronts with the most tumor budding, but these differences were not significantly correlated to the tumor budding grade.

miR200, ZEB1, ZEB2, and PD-L1 expression in both the tumor center and at the invasive front. a–c miR200a, miR200b, and miR200c expression was lower in the invasive front compared with the tumor center (p = 0.00 in all cases, *Statistically significant). d–f ZEB1, ZEB,2 and PD-L1 were overexpressed at the invasive front with respect to the center (p = 0.00, p = 0.02, and p = 0.00, respectively, *Statistically significant), g Cases with high-grade tumor budding reached higher PD-L1 expression levels at the invasive front compared with cases with low and intermediate-grade tumor budding (p = 0.03). h–I miR200a and miR200c expression were inversely correlated with PD-L1 expression. j, k ZEB1 and ZEB2 expression directly correlated with PD-L1 expression
Immunohistochemical study of PD-L1
All the cases (5 of 5) with low-grade tumor budding showed positive PD-L1 immunoexpression restricted to peritumoral inflammatory cells in the invasive front, while the tumor cells were completely negative. However, 2 of 5 (40%) high-grade tumor budding cases presented positive immunostaining limited to the tumor buds at the invasive front, while the main tumor bulk was negative (Fig. 2).

Immunohistochemical study of PD-L1. a, b low-grade tumor budding case with positive immunoexpression restricted to peritumoral inflammatory cells at the invasive front; the tumor cells were completely negative (a: 10× and b: 20× magnification). c, d High-grade tumor budding case with positive immunostaining limited to the tumor buds at the invasive front; the tumor bulk was negative (c: 20× and b: 40× magnification)
Survival analysis
The median follow-up time was 43 months and 50% of all cases were followed for at least 50 months.
Tumor-specific survival in stages I, II, and III disease
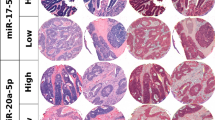
As shown in Fig. 3, the univariate analysis of cases in stages I to III showed that lower tumor-specific survival was significantly correlated with low (below the median) miR200c (p = 0.03) and miR200a (p = 0.04) expression at the invasive front. The clinical stage (p = 0.00) and the presence of lymphovascular invasion (p = 0.01) significantly correlated with tumor-specific survival. Multivariate analysis of stage I to III patients, showed that miR200c expression at the invasive front, followed only by the clinical stage, was an independent predictor of tumor-specific survival (Table 3).

Tumor-specific survival for patients in stage I to III, according to miR200c (a) and miR200a (b) expression
Disease-free survival in stage II and tumor-specific survival in stage III disease
We observed a significant association between the disease-free survival rate and miR200c expression at the invasive front in stage-II cases (p = 0.03). We did not perform multivariate analysis because the only variable significantly associated with disease-free survival found in the univariate analysis was miR200c expression. In stage-III cases, miR200c and miR200a expression significantly influenced tumor-specific survival (p = 0.01 and p = 0.05, respectively). In all cases, miR200 expression and survival only correlated when they were measured at the tumor front, and no other factors influenced the survival probability in stage-III disease. Multivariate analysis of cases in stage III revealed miR200c expression at the invasive front as the only independent predictor of tumor-specific survival (Table 3).
Discussion
According to our results, the colon cancer invasive front with the most tumor budding has an expression profile consistent with epithelial-to-mesenchymal transition: decreased miR200a, miR200b, and miR200c expression and increased ZEB1 and ZEB2 expression. Moreover, reduced miR200a and miR200c expression was associated with a significant decrease in the probability of tumor-specific survival, but only when these biomarkers were evaluated in the tumor fronts with the most budding activity and not in the tumor centers. This highlights the problem of intra-tumor heterogeneity and the importance of sample selection for molecular studies, especially in the identification of the CMS4 colon cancer molecular subtype, which has the worse prognosis [24]. These findings might be explained by the enrichment of cancer-associated fibroblasts at the invasive front because these act as TGF-β pathway activators, which influence neoplastic transformation, and probably induce tumor budding during epithelial-to-mesenchymal transition processes [8, 25, 26].
Immunohistochemical expression of PD-L1 has previously been used as a biomarker to identify subsets of patients with solid tumors who are more likely to respond to immune-checkpoint blockage inhibitors [27, 28]. In colon cancer, the only confirmed predictor of such a response is a MMR deficiency status which results in a log-fold increase in the tumor mutational burden [29,30,31,32,33]. PD-L1 expression in colon cancer is more frequent in cases with deficient MMR and has been associated with features of the serrated pathway such as BRAF mutations and a medullary morphology [34]. In microsatellite-stable colon cancer, the expression of PD-L1 is less frequent and varies between 3 and 40% according to the series [34, 35]. In our cases, PD-L1 was overexpressed at the tumor invasive front relative to the center, and this increase was significantly higher in tumors with high-grade tumor budding.
The immunohistochemical staining pattern of PDL1, and the absence of expression in the tumor bulk compared to the invasive edge, better illustrates these molecular findings. Interestingly, 40% of cases (2 of 5) with high-grade tumor budding displayed positive bud immunostaining while in low-grade tumor budding cases, tumor cells were completely negative and only peritumoral inflammatory cells were positive. Moreover, we observed a significant correlation between PD-L1 overexpression and an expression profile consistent with epithelial-to-mesenchymal transition: increased ZEB1 and ZEB2 and decreased miR200a and miR200c expression. These findings suggest that PD-L1 overexpression may be related to tumor cell evasion of the immune response during epithelial-to-mesenchymal transition. Expression of the immune checkpoint ligand PD-L1 has been demonstrated to activate the epithelial-to-mesenchymal transition program in model breast carcinoma tumor cells [36, 37]. In colon cancer, PD-L1 expression in tumor buds has been studied in a few cases, with similar results [38].
The main limitations of our study are its retrospective and exploratory design, which aimed to generate hypotheses, rather than confirm them. Based on these results, the invasive front of colon cancer seems to significantly contribute to the molecular tumor phenotype probably because of the presence of tumor buds in this area. Therefore, sample area selection is important to properly reflect intra-tumor heterogeneity, and the inclusion of the invasive edge in samples for molecular phenotyping and detection of biomarkers appears to be crucial. Both a decrease in miR200c expression and higher concentrations of tumor buds at the invasive front of colon cancer are negative survival factors in stage I to III patients and are predictors of relapse in stage II patients. Thus, inclusion of the invasive front and the tumor budding areas in the samples used for molecular studies (especially in the identification of the CMS4 molecular subtype) is imperative for two reasons: first, the expression profile of these areas, rather than in the tumor center, is consistent with epithelial-to-mesenchymal transition, and second, the expression of miR200 at the invasive front, but not in the tumor center, significantly influences the patient survival probability. Finally, PD-L1 is overexpressed in the tumor areas with an epithelial-to-mesenchymal transition expression profile: the invasive front and tumor budding cells.
References
Bosch S, Teerenstra S, De Wilt J, Cunningham C, Nagtegaal I. Predicting lymph node metastasis in pT1 colorectal cancer: a systematic review of risk factors providing rationale for therapy decisions. Endoscopy. 2013;45:827–34.
Ueno H, Mochizuki H, Hashiguchi Y, Shimazaki H, Aida S, Hase K et al. Risk factors for an adverse outcome in early invasive colorectal carcinoma. Gastroenterology. 2004;127:385–94.
Graham R, Vierkant R, Tillmans L, Wang A, Laird P, Weisenberger D et al. Tumor budding in colorectal carcinoma: confirmation of prognostic significance and histologic cutoff in a population-based cohort. Am J Surg Pathol. 2015;39:1340–6.
Koelzer V, Zlobec I, Lugli A. Tumor budding in colorectal cancer - Ready for diagnostic practice? Hum Pathol. 2016;47:4–19.
Zlobec I, Lugli A. Epithelial mesenchymal transition and tumor budding in aggressive colorectal cancer: tumor budding as oncotarget. Oncotarget. 2010;1:651–61.
Dan Grigore A, Kumar J, Jia D, Farach-Carson M, Levine H. Tumor Budding: the Name is EMT. Partial EMT. J Clin Med 2016; 29:51 https://doi.org/10.3390/jcm5050051
Lugli A, Kirsch R, Ajioka Y, Bosman F, Cathomas G, Dawson H et al. Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod Pathol. 2017;30:1–13. (2017)
De Smedt L, Palmans S, Andel D, Govaere O, Boeckx B, Smeets D et al. Expression profiling of budding cells in colorectal cancer reveals an EMT-like phenotype and molecular subtype switching. Br J Cancer. 2017;116:58–65.
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6.
Garzon R, Calin G, Croce C. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–79.
Macfarlane L, Murphy P. MicroRNA: biogenesis, function and role in cancer. Curr Genom. 2010;11:537–61.
Humphries B, Yang C. The microRNA-200 family: small molecules with novel roles in cancer development, progression and therapy. Oncotarget. 2015;6:6472–98.
Topalian S, Taube J, Anders R, Pardoll D. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87.
De Sousa E, Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij L et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–8.
Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. https://doi.org/10.1371/journal.pmed.1001453.
Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell- cell junctions. Nucleic Acids Res. 2005;24:6566–78.
Das S, Becker B, Hoffmann F, Mertz JE. Complete reversal of epithelial to mesenchymal transition requires inhibition of both ZEB expression and the Rho pathway. BMC Cell Biol. 2009; 10:94. https://doi.org/10.1186/1471-2121-10-94.
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28.
Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: At the crossroads of signal transduction in cancer. Int J Cancer. 2012;132:745–54.
Trinh A, Trumpi K, De Sousa E Melo F, Wang X, de Jong J, Fessler E et al. Practical and robust identification of molecular subtypes in colorectal cancer by immunohistochemistry. Clin Cancer Res. 2017;23:387–98.
Fessler E, Jansen M, De Sousa E Melo F, Zhao L, Prasetyanti P, Rodermond H et al. A multidimensional network approach reveals microRNAs as determinants of the mesenchymal colorectal cancer subtype. Oncogene. 2016;35:6026–37.
Amin M, Edge S, Greene F, Byrd D, Brookland R, Washington M et al. AJCC cancer staging manual. 8th ed. New York, NY: Springer; 2017.
Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2 delta delta C T method. Methods. 2001;25:402–8.
Ubink I, Elias S, Moelans C, Laclé M, Van Grevenstein W, Van Diest P et al. A novel diagnostic tool for selecting patients with mesenchymal-type colon cancer reveals intratumor subtype heterogeneity. J Natl Cancer Inst. 2017;109:djw303. https://doi.org/10.1093/jnci/djw303.
Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47:320–9.
Isella C, Terrasi A, Bellomo S, Petti C, Galatola G, Muratore A et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–9.
Chen J, Jiang C, Jin L, Zhang X. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016;27:409–16.
Valentini A, Di Pinto F, Cariola F, Guerra V, Giannelli G, Caruso M et al. PD-L1 expression in colorectal cancer defines three subsets of tumor immune microenvironments. Oncotarget. 2018;9:8584–96.
Amin M, Lockhart A. The potential role of immunotherapy to treat colorectal cancer. Expert Opin Investig Drugs. 2015;24:329–44.
Xiao Y, Freeman G. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015;5:16–8.
Sanchez-Castañón M, Er T, Bujanda L, Herreros-Villanueva M. Immunotherapy in colorectal cancer: what have we learned so far? Clin Chim Acta. 2016;460:78–87.
Diaz L, Le D. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Kim J, Park H, Cho N, Lee H, Kang G. Characterisation of PD-L1-positive subsets of microsatellite-unstable colorectal cancers. Br J Cancer. 2016;115:490–6.
Rosenbaum M, Bledsoe J, Morales-Oyarvide V, Huynh T, Mino-Kenudson M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod Pathol. 2016;29:1104–12.
Droeser R, Hirt C, Viehl C, Frey D, Nebiker C, Huber X et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur J Cancer. 2013;49:2233–42.
Alsuliman A, Colak D, Al-Harazi O, Fitwi H, Tulbah A, Al-Tweigeri T et al. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: significance in claudin-low breast cancer cells. Mol Cancer 2015;14:149. https://doi.org/10.1186/s12943-015-0421-2.
Noman M, Janji B, Abdou A, Hasmim M, Terry S, Tan T et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017;6:e1263412. https://doi.org/10.1080/2162402X.2016.1263412.
Prall F, Hühns M. PD-L1 expression in tumour buds of colorectal carcinoma. Histopathology. 2016;69:158–60.
Acknowledgements
Sara S. Oltra is funded by a FPU pre-doctoral fellowship (FPU13/04976) from MINECO (Spanish Government). Gloria Ribas is funded on a Miguel Servet II contract (CPII14-00013), Desamparados Roda is a recipient of the Joan Rodés Grant (Contract 16/00040), and Noelia Tarazona is a recipient of the Rio Hortega Grant (Contract 15/00246), all from the Instituto de Salud Carlos III. This work was supported by grant PI15/02180 from the Fondo de Investigaciones Sanitarias (Instituto de Salud Carlos III) from the Spanish Government and CIBERONC, awarded to Andrés Cervantes. Francisca Carrasco has a contract as a laboratory technician from CIBERONC. This study was partially supported by a grant from the Carlos III Health Institute (PI16/00395) awarded to Gloria Ribas, and FEDER funds.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Martinez-Ciarpaglini, C., Oltra, S., Roselló, S. et al. Low miR200c expression in tumor budding of invasive front predicts worse survival in patients with localized colon cancer and is related to PD-L1 overexpression. Mod Pathol 32, 306–313 (2019). https://doi.org/10.1038/s41379-018-0124-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0124-5
This article is cited by
-
Epithelial-to-mesenchymal transition in cancer progression: unraveling the immunosuppressive module driving therapy resistance
Cancer and Metastasis Reviews (2024)
-
Tumour budding in solid cancers
Nature Reviews Clinical Oncology (2021)
-
Elevated ZBTB7A expression in the tumor invasive front correlates with more tumor budding formation in gastric adenocarcinoma
Journal of Cancer Research and Clinical Oncology (2021)
-
Emerging role of tumor cell plasticity in modifying therapeutic response
Signal Transduction and Targeted Therapy (2020)
