
Abstract
Blockade of the interaction between PD-1 and its ligands PD-L1 has shown clinical efficacy across several tumor types, especially in mismatch-repair-deficient colorectal carcinoma. The aim of this study was to examine the pattern and cellular localization of PD-L1 expression in the different molecular subtypes of mismatch-repair-deficient colorectal cancers vs. their mismatch-repair-proficient counterparts. PD-L1/SATB2 double-antibody-immunohistochemistry was utilized to distinguish tumor cell from immune cell staining. We observed in our series of 129 colorectal adenocarcinomas that PD-L1 expression occurred primarily in tumor-associated-immune cells and most prominently at the tumor-stroma-interface of the invasive front. The level of invasive front immune cell staining was significantly higher in mismatch-repair-deficient tumors compared to mismatch-repair-proficient tumors (p < 0.001), but no difference was observed among the different subtypes of mismatch-repair-deficient tumors: Lynch syndrome-associated vs. MLH1-methylated vs. unexplained. While selected mismatch-repair-proficient tumors exhibited unusually high tumor-infiltrating-lymphocytes and had high level immune cell PD-L1 expression, a positive correlation between PD-L1 expression and high lymphocyte count was detected only in mismatch-repair-deficient tumors (r = 0.39, p < 0.001) and not in mismatch-repair-proficient tumors. Notably, true tumor cell PD-L1 expression in colorectal carcinoma was rare, present in only 3 of 129 tumors (2.3%): 2 MLH1-methylated and 1 mismatch-repair-proficient with high tumor-infiltrating-lymphocytes; and the staining in the tumor cells in all 3 was diffuse (>=50% of the tumor). These findings may serve to inform further efforts aiming to evaluate PD-L1 immunohistochemistry vis-à-vis molecular sub-classification as predictive biomarkers in the treatment of colorectal carcinoma.
Similar content being viewed by others


Introduction
Recently, several clinical trials have demonstrated utility of mismatch repair deficiency as a predictive marker for response to anti-PD-1 therapy in solid tumors, particularly in colorectal carcinoma, signifying a major advancement in cancer immunotherapy [1,2,3]. However, not all mismatch-repair-deficient cancers respond to anti-PD-1 immunotherapy, the reported overall objective response rate being around 36% [2, 4]. Thus, there exists a need for even better predictive biomarkers within (and beyond) mismatch-repair-deficient colorectal cancers [4, 5].
PD-L1 immunohistochemistry has shown predictive utility in anti-PD-1 therapy in some tumor types, such as non-small cell lung carcinoma [6]. In the case of colorectal cancer, analysis of two recent clinical trials [2, 3] failed to demonstrate a positive correlation between PD-L1 expression and response, and suggested that PD-L1 immunohistochemistry was not a predictive biomarker [3]. However, this conclusion was based on a very low cutoff value (5% and 1% tumor cell PD-L1 expression in the two studies, respectively), which may be suboptimal.
In this study, we evaluated a series of molecularly defined mismatch-repair-deficient- and proficient-colorectal carcinomas, including a selected group of mismatch-repair-proficient tumors that had unusually high tumor-infiltrating lymphocytes. We also utilized double-antibody immunohistochemistry to assess tumor cell vs. immune cell staining. Our goal was to characterize the extent and cellular composition of PD-L1 staining in the various molecular subtypes of colorectal carcinoma. We hypothesized that the results from such an analysis would serve to guide further efforts that aim to evaluate PD-L1 immunohistochemistry in parallel with molecular sub-classification as predictive biomarkers in this tumor type both within and outside the context of clinical trials.
Materials and methods
Case selection and molecular classification
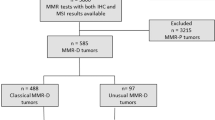
The study was approved by the institutional board review. Cases were retrieved from the databases of the pathology department and the clinical genetics service of a single tertiary care cancer center between July 2006 and July 2015. The cases were accrued as part of the institutional protocol to perform mismatch repair protein immunohistochemistry on all colorectal cancers that fulfilled the Bethesda guidelines (2006–2013) or a more relaxed set of criteria (aged 70 or younger or tumor with suggestive morphology irrespective of age, 2014 onwards). This protocol was set up with the goal of detecting Lynch syndrome.
The mismatch repair protein immunohistochemistry testing was performed on whole sections. Antibodies included MLH1 (clone G168–728, diluted 1:250; BD PharMingen or clone M1, ready-to-use, Ventana), MSH2 (clone G219.1129, ready-to-use, Cell Marque), MSH6 (clone 44, ready-to-use, Ventana), and PMS2 (clone A16.4, diluted 1:100, BD Biosciences). As guided by the individual patient’s clinical scenario, selected tumors then underwent further testing as necessary: (1) PCR microsatellite instability testing using the Promega Analysis System; (2) MLH1 promoter methylation via testing of five CpG sites within the MLH1 promoter (in region −209 to −181 from the transcription start site) and calculating the degree of methylation for each site as follows: methylation % = peak height of methylated (C)/(peak height of methylated [C] + peak height nonmethylated [T]) × 100. Methylation status was graded as “present,” when all five CpG sites were methylated above 10% [1]; and (3) Germline mutation testing, performed as part of the clinical diagnostic work-up and according to standard methodologies.
Colorectal carcinoma cases that have undergone mismatch repair protein immunohistochemistry and appropriate molecular work-up and have tissue blocks available for further testing were included in our study. The patients’ clinical information was retrieved from the hospital information system.
Tumor pathology
Only adenocarcinomas primary to colon or rectum were included. H&E sections were reviewed for the following pathological variables: (1) tumor differentiation: well/moderately differentiated vs. poorly differentiated (defined as less or more than 50% of the tumor showing non-glandular/solid growth, medullary or medullary like patterns were counted as solid growth); (2) presence of non-conventional histological patterns: mucinous (in ≥ 50% of the tumor), medullary or with medullary features [7] (in ≥ 20% of the tumor); (3) presence or absence of lymphovascular invasion; (4) presence or absence of perineural invasion; (5) tumor-infiltrating lymphocytes: recorded as count per HPF (HPF refers to 40x objective lens and 10x ocular lens) averaged from counts of five consecutive HPFs in areas with highest tumor-infiltrating lymphocyte density; (6) peritumoral lymphocyte aggregates: recorded as the total number of aggregates observed under 4x objective lens and 10x ocular lens; and (7) tumor pT and pN status. The final TNM status was determined based on combined pathological and clinical findings.
Single-antibody and double-antibody immunohistochemistry for PD-L1 expression
PD-L1 immunohistochemistry was performed on whole sections. We used a monoclonal antibody against PD-L1 (clone E1L3N, dilution 1:250; Cell Signaling Technology, Danvers, MA, USA) on the Leica Biosystems’ Bond III platform (Leica Biosystems, Buffalo Grove, IL, USA). The dilution factor of this antibody was optimized by using liver parenchyma as negative control and tonsil tissue as positive control as recommended. In addition to antibody concentration, we also optimized target retrieval buffer. After optimizing the immunohistochemical conditions, 4 micron thick sections on super-frost plus slides were baked in an oven for 20 min at 70 °C; and deparaffinized. Target retrieval was performed in standard Epitope Retrieval Solution 2 (Catalog # AR 9640, pH 9.0) slightly basic buffer, for 30 min. The primary antibody was diluted by 1:100 with a final concentration of 0.2 μg/ml. The incubation time for the primary antibody was 30 min. The slides were rinsed with buffer, followed by incubation with the secondary antibody and final development with the 3,3′-Diaminobenzidine Map Kit (Leica Biosystems, Inc., Catalog # DS9800) for 7 min. The Leica’s Bond Polymer Refine detection kit (Leica Biosystems, Inc., Catalog # DS9800) was used for detection. Appropriate positive and negative controls were employed.
Double-antibody immunohistochemistry was performed on all tumors that were regarded as potentially showing tumor cell PD-L1 staining on single antibody testing. To select a nuclear marker to label the carcinoma cells, we first tested all cases with 2 candidate markers: CDX2 (clone CDX2.88, dilution 1:100; BioGenex) and SATB2 (clone EP281, dilution 1:400; Cell Marque). We found a 100% sensitivity of SATB2, but only 82% of CDX2 in staining the cancer cells in these cases (in keeping with previous reports [8, 9]). Thus, SATB2 was chosen for double staining with PD-L1. Cells that exhibited nuclear staining for SATB2 with concurrent membranous staining for PD-L1 were regarded as PD-L1 positive tumor cells.
Statistical analysis
Descriptive and comparative statistics were performed using JMP®, statistical software, Version < 10 > (SAS Institute, Cary, NC). Continuous variables were compared using the Student t-test. Categorical variables were compared using χ2 or the Fisher exact test depending on the number of observations. Correlation between PD-L1 and tumor-infiltrating lymphocytes/HPF was estimated using Spearman’s rank method and p values were generated using a reference distribution approximated by an Edgeworth series approximation [10]. Nonparametric regression was performed using a LOWESS smoother [11].
Results
Tumor characteristics
The study cohort included 129 formalin-fixed paraffin-embedded primary colorectal carcinoma specimens from 129 patients. These cases were classified by mismatch repair protein immunohistochemistry and subsequent molecular studies as described in Materials and Methods. They were molecularly classified into five groups: mismatch-repair-deficient—Lynch syndrome (n = 23), mismatch repair-deficient—unexplained (n = 13), mismatch-repair-deficient—sporadic (n = 37), mismatch-repair-proficient—tumor-infiltrating lymphocytes high (n = 17), and mismatch-repair-proficient—consecutive (n = 39). These groups were designated as A–E. Specific definitions for each group are outlined in Table 1.
The 17 “mismatch-repair-proficient—tumor-infiltrating lymphocytes high” tumors, group D, were mismatch-repair-proficient tumors selected based on the presence of unusually high tumor-infiltrating lymphocyte count > 10/HPF. All 17 tumors were tested by both mismatch-repair protein immunohistochemistry and microsatellite instability testing, and both tests showed them to be mismatch-repair-proficient (protein expression normal and microsatellite stable). The 39 “mismatch-repair-proficient—consecutive” cases, group E, represented a consecutive series of mismatch-repair-proficient cancers with no pre-selection of tumor-infiltrating lymphocyte content.
The clinical and pathological characteristics of all cases are summarized in Table 2. As expected, various pathological features including tumor location, tumor differentiation, histological patterns, and lymphocytic infiltrates, differed significantly between mismatch-repair-deficient- and proficient-tumors. The tumor-infiltrating lymphocyte count was high across all three groups of mismatch-repair-deficient tumors; by design, it was also high in the group of “mismatch-repair-proficient—tumor-infiltrating lymphocytes high”. The highest tumor-infiltrating lymphocyte count was observed in the mismatch-repair-deficient—sporadic group. In contrast, the consecutive series of mismatch-repair-proficient tumors (group E) had very low tumor-infiltrating lymphocyte counts, significantly lower than that of all other groups (p < 0.0001).
PD-L1 expression was primarily localized to tumor-associated immune cells at the tumor-stroma interface along the invasive front
PD-L1 expression was assessed on invasive carcinoma in whole tissue sections. Some PD-L1 expression (any amount) was present in 70% of the tumors: up to 89% (65/73) in mismatch-repair-deficient tumors, 76% (13/17) in mismatch-repair-proficient—tumor-infiltrating lymphocytes high tumors, and 31% (12/39) in the mismatch-repair-proficient—consecutive tumors (Fig. 1). The staining intensity varied but all positive cases had a degree of intensity easily recognizable as being positive. Therefore, while the intensity was graded as 0, 1+, 2+ and 3+, all cases that had at least 1+ staining were considered to be positive.

Histogram illustrating the distribution of PD-L1 positivity across the entire cohort of cases. Positivity was measured as staining in tumor-associated immune cells at tumor-stroma-interface of the invasive front
Across all tumor groups, the PD-L1 staining appeared most prominent in the tumor-stroma interface along the invasive front (Fig. 2a, b). Positivity within tumor nests away from the tumor-stroma interface was observed, but it invariably co-existed with positivity at the tumor-stroma interface at the invasive front.

PD-L1 expression at the tumor-stroma interface along the invasive front in a colonic adenocarcinoma. Scanning view of PD-L1 immunohistochemical stain shows a band of positively stained cells at tumor-stroma interface along the deepest invasive front (A, outlined by arrows). The boxed area in (A) is magnified in (B), revealing conspicuous brown labeling of cells along the tumor border and also extending upward into the nests of tumor (B)
As summarized in Table 3, the types of cells that were positive on single antibody (PD-L1) immunohistochemistry were easily recognizable as either tumor cell or immune cell in 73% (37/51) mismatch-repair-deficient cancers and 100% (12/12) mismatch-repair-proficient ones. On the other hand, 27% (14/51) of the positively stained mismatch-repair-deficient cancers exhibited varied degrees of ambiguity in the types of positively stained cells (Fig. 3a, b). The ambiguity was most apparent in the mismatch-repair-deficient–sporadic cancers.

Immunohistochemistry highlights the lack of tumor cell PD-L1 staining and the presence of immune cell PD-L1 staining in a colonic adenocarcinoma. H&E shows that the tumor has a solid growth pattern (A). Single antibody PD-L1 stain shows positively stained cells both at the tumor-stroma interface and within the tumor cell nest; some positively stained cells, especially those within the tumor cell nest (such as the arrow indicated ones), show nuclear features difficult to distinguish from that of the tumor cells (B). By double-antibody immunohistochemistry (PD-L1 + SATB2), it becomes clear that the PD-L1 positive cells (red chromogen, red-arrow indicating two example cells) are all negative for SATB2; in contrast, all cells that are positive for SATB2 (i.e., tumor cells) are negative for PD-L1 (C)
Double-antibody immunohistochemistry (PD-L1 + SATB2), performed on all ambiguous cases (n = 14) as well as 15 control cases (eight mismatch-repair-deficient and seven mismatch-repair-proficient), revealed that all control cases that were deemed to have either unequivocal immune cell or tumor cell staining on single antibody stain were all correctly classified, whereas in all 14 ambiguous cases, the positivity appeared entirely attributed to immune cell staining (with no convincing PD-L1 staining in SATB2 positive tumor cells). It was observed that some positive immune cells were deep seated within the tumor nests and were difficult to distinguish from the tumor cells (Fig. 3c).
By a “single-antibody followed by double-antibody” approach, true tumor cell staining was observed in three tumors only (3/129, 2%), two mismatch-repair-deficient—sporadic, and one mismatch-repair-proficient—tumor-infiltrating lymphocytes high. The clinicopathological features of these three tumors are summarized in Table 4. All three tumors had diffuse tumor cell staining beyond the tumor-stroma interface in ≥ 50% of the tumor (Fig. 4a-c).

A colonic adenocarcinoma showing diffuse tumor cell positivity for PD-L1. H&E section shows that the tumor grows in irregular sheets and nests (A). Single antibody PD-L1 stain shows diffuse positivity in the tumor cell nests (B). Double-antibody immunohistochemistry (PD-L1 + SATB2) further confirms that the SATB-2 positive cells, i.e., tumor cells, have clearly demonstrable PD-L1 labeling (red chromogen) (C)
The level of immune cell PD-L1 expression at invasive front was significantly higher in mismatch-repair-deficient tumors, but with no difference across their different molecular subtypes
As illustrated in Fig. 5, the expression of PD-L1 in immune cells at tumor invasive front was significantly higher in mismatch-repair-deficient tumors when compared to mismatch-repair-proficient—consecutive tumors (p < 0.001), but no difference was detected across the three types of mismatch-repair-deficient tumors (groups A–C, p = 0.32). The mismatch-repair-proficient—tumor-infiltrating lymphocytes high tumors showed high level PD-L1 expression comparable to mismatch-repair-deficient tumors (with no statistical difference), but significantly higher than mismatch-repair-proficient-consecutive tumors (p < 0.001).

Distribution of PD-L1 positivity across the groups of tumors: A, mismatch repair-deficient—Lynch syndrome associated; B, mismatch-repair-deficient—unexplained; C, mismatch-repair-deficient—MLH1-methylated; D, mismatch-repair-proficient—tumor-infiltrating lymphocyte-high; E, mismatch-repair-proficient—consecutive. There is high expression in all mismatch-repair-deficient tumors and in mismatch-repair-proficient—tumor-infiltrating-lymphocyte-high tumors (p not significant), but distinctly low expression in mismatch-repair-proficient—consecutive tumors (p < 0.001)
Immune cell PD-L1 expression at invasive front correlated with tumor-infiltrating lymphocytes in mismatch-repair-deficient tumors but not in mismatch-repair-proficient tumors
As a group, mismatch-repair-deficient tumors demonstrated a significant correlation between the level of immune cell PD-L1 expression and the level of tumor-infiltrating lymphocytes (Fig. 6). When breaking down to mismatch-repair-deficient subtypes, the correlation remained significant in the sporadic group (group 3), which had the highest tumor-infiltrating lymphocyte count (Table 2).

Correlation between immune cell PD-L1 expression at invasive front and tumor-infiltrating lymphocyte count (A and B). A positive correlation is demonstrated in mismatch-repair-deficient tumors (groups A–C, p < 0.001), but not in mismatch-repair-proficient tumors (groups D and E, respectively). Designations for the groups: A, mismatch-repair-deficient—Lynch syndrome associated; B, mismatch-repair-deficient—unexplained C, mismatch-repair-deficient—MLH1-methylated; D, mismatch-repair-proficient—tumor-infiltrating lymphocyte-high; E, mismatch-repair-proficient—consecutive
On the other hand, in the selected group of mismatch-repair-proficient—tumor-infiltrating lymphocytes high tumors, while the levels of PD-L1 expression (Fig. 5) and tumor-infiltrating lymphocyte (Table 2) were similar to that of mismatch-repair-deficient tumors, no correlation between immune cell PD-L1 expression and tumor-infiltrating lymphocyte was detected.
Discussion
In this study, we demonstrated that in colorectal carcinoma, the expression of PD-L1 was primarily localized to tumor-associated immune cells, most prominent at the tumor-stroma interface along the invasive front. True tumor cell expression was a rare event. Additionally, we observed that immune cell PD-L1 expression was significantly higher in mismatch-repair-deficient colorectal carcinomas when compared to mismatch-repair-proficient tumors, but there was no difference among the different mismatch repair deficiency molecular subtypes. Intriguingly, some mismatch-repair-proficient tumors harbored unusually high tumor-infiltrating lymphocytes and these tumors tended to have high immune cell PD-L1 expression similar to mismatch-repair-deficient tumors. However, while there existed a direct correlation between PD-L1 expression and tumor-infiltrating lymphocyte level in mismatch-repair-deficient tumors, this correlation was not found in mismatch-repair-proficient tumors.
The lack of real tumor cell immunohistochemical expression of PD-L1 in colorectal carcinoma contrasts sharply with what occurs in non-small cell lung carcinoma, where tumor cell staining contributes rather significantly to the PD-L1 positivity. In fact, the majority of published studies on lung carcinomas consider tumor cell staining exclusively in the definition of PD-L1 expression [6]; and two of the four immunohistochemistry assays currently approved by FDA as diagnostic tests in advanced non-small cell lung carcinoma—Dako PD-L1 22C3 pharmDx (pembrolizumab), Dako PD-L1 28–8 pharmDx (nivolumab)—defined PD-L1 positivity solely on tumor cell positivity.
This difference may at least in part be explained by the different presumptive mechanisms underlying the upregulation of PD-L1 in different tumors. In non-small cell lung carcinoma, an “intrinsic” oncogene-driven mechanism is believed to be at play [12]. The epidermal growth factor receptor (EGFR) gene has particularly been implicated. Activating EGFR mutations were associated with increased PD-L1 expression in surgically resected non-small cell lung carcinomas and ectopic expression of mutant EGFR in bronchial epithelial cells induced PD-L1 expression [13, 14]. Inhibition of EGFR signaling by the EGFR tyrosine kinase inhibitor erlotinib downregulated surface expression of PD-L1 in EGFR mutation-positive non-small cell lung carcinoma cells, but not in the EGFR wild-type cells [14]. In contrast, in colorectal carcinoma, although some studies suggest the presence of PD-L1 expression in tumor cells [15, 16], evidence supporting an “intrinsic” mechanism as seen in the lung is lacking.
Interestingly, in colorectal carcinoma, when tumor cell staining for PD-L1 does occur, it is conspicuous. In the three tumors (3/129, 2%) we observed, the positive staining in the tumor cells was uniformly diffuse and easily recognizable on single antibody immunohistochemistry. While all three occurred in elderly patients and were right-sided tumors recapitulating a clinical profile of sporadic MLH1-methylated microsatellite unstable cases, one of the three was mismatch repair-proficient. Notably, this mismatch repair proficient case belonged to the “mismatch repair-proficient—tumor-infiltrating lymphocytes high” group. Furthermore, it is also notable that two were indeed sporadic MLH1-methylated cancers and that represented a 5% frequency of this phenomenon in this group (2/37). All three were treated with surgical resection alone. One patient with positive lymph node fared worse than the other two who had stage 2 disease with no nodal metastasis.
In contrast to the low frequency of tumor cell staining, the presence of at least focal immune cell staining is rather common in colorectal carcinoma. Some positivity at the tumor-stroma interface was seen in 70% of our cases, including up to 89% of mismatch-repair-deficient tumors and 31% of consecutive mismatch-repair proficient tumors. This is indeed concordant with the study by Overman et al. (CheckMate 142) [3], who also observed that all of their 68 colorectal cancers tested had at least “rare” immune cell PD-L1 staining (35% with “rare” positive immune cells, and 65% had “intermediate” or “numerous” positive immune cells). In their study, the cases enrolled [3] were all determined to have mismatch repair deficiency in the tumor either by immunohistochemistry or by PCR microsatellite testing, although a subset showed discordant mismatch repair status by subsequent centralized testing.
An important observation that emerged from our double-antibody immunohistochemistry is that the PD-L1 positive immune cells may be intimately associated with tumor cells either at the periphery of tumor cell nests (i.e., tumor-stroma interface) or in some cases deep within the nests. These immune cells (mostly macrophages) can be easily mis-interpreted as tumor cell staining. Indeed, 19% of our mismatch-repair-deficient tumors (including 27% of sporadic tumors) showed ambiguous labeling. Only when double-antibody immunohistochemistry was employed that highlighted the SATB2 positive tumor cell nuclei and not immune cell nuclei did the true nature of the PD-L1 positive cells become clear.
Also of importance are our observations that (1) immune cell PD-L1 expression was high across different subtypes of mismatch-repair-deficient tumors (Lynch syndrome associated or sporadic), and (2) some mismatch-repair-proficient tumors may have high tumor-infiltrating lymphocytes and express high level PD-L1 as well, but the correlation between the two was not as direct and tight as it was in mismatch-repair-deficient tumors.
These observations seem in line with an “extrinsic”, immune cell mediated, PD-L1 upregulation mechanism in colorectal carcinoma. It has been suggested [17] and widely supported [4] that the hypermutator phenotype endowed by microsatellite instability results in neo-antigens that induce an active immune microenvironment featuring two opposing forces: an immune-stimulatory force represented by increased cytotoxic effector T lymphocytes and an immune inhibitory force including upregulated PD-1/PD-L1 checkpoint. In our study, we not only demonstrated the parallel existence of high tumor-infiltrating lymphocytes and high PD-L1 expression in immune cells in mismatch-repair-deficient cancers, but also observed that this intensified immune cell interaction may exist in some mismatch-repair-proficient tumors as well (as demonstrated by the “mismatch-repair-proficient—tumor-infiltrating lymphocytes high” group). As these tumors do not show mismatch repair deficiency either by immunohistochemistry or PCR microsatellite testing, it is likely that some other mechanism is driving the vigorous immune response in them. To this effort, the lack of a direct correlation between tumor-infiltrating lymphocytes and PD-L1 expression in these tumors further suggests that the mechanism may be complex and not always reflected in the amount of tumor-infiltrating lymphocytes alone. Mutations in the exonuclease domain of polymerase epsilon catalytic subunit (POLE) gene could potentially be at play here, as POLE mutations have been shown to occur in mismatch-repair-proficient colorectal carcinomas, and these tumors are ultramutated and may have variably increased tumor-infiltrating lymphocytes and upregulated PD-L1 [18, 19]. Efforts are ongoing to explore these mechanisms.
Specific mechanisms notwithstanding, our observations carry practical implications. The most optimal criteria for evaluating PD-L1 immunohistochemistry as a predicative marker, for example, need further consideration. Qualitatively, caution is needed in scoring PD-L1 staining in colorectal cancer to not mis-interpret immune cell staining as tumor cell staining. Lack of awareness of this interpretation pitfall may have affected the accuracy of previous data. It is possible that studies that reported very high frequencies of tumor cell staining [15, 20] might have regarded immune cell staining as tumor cell staining. Similarly, when employing a “1% (or 5%) tumor cell staining” as the cutoff, those “ambiguous” cases as highlighted in our study may have been inconsistently scored as either tumor cell or non-tumor cell staining, resulting in accruement of cases with different PD-L1 expression status. Thus, future efforts on assessing the predictive utility of PD-L1 immunohistochemistry in colorectal carcinoma should take such caveats into consideration. The fact that some albeit rare mismatch-repair-proficient tumors may also show increased tumor-infiltrating lymphocytes and have high PD-L1 expression also deserves attention, as they may also potentially benefit from immunotherapy.
This work has limitations. First, the study samples included only colorectal primary carcinomas. As the immune system is dynamic and inducible, temporal and spatial differences may exist. As such, the ideal sample to be evaluated should be the tumor that is being treated, which in most situations is the metastasis. Additionally, we used a PD-L1 antibody (clone E1L3N, Cell Signaling Technology) that is different from the current FDA approved antibodies. However, this antibody (clone E1L3N) has been validated against clone 22C3 (pharmDx) and was found to be comparable [21, 22].
As we move forward with immunotherapy in colorectal carcinoma, it is imperative that heightened attention be given to the identification of the most reliable predictive biomarkers [1, 2, 23]. In further determining the specific utility, or lack thereof, of PD-L1 immunohistochemistry as a predictive tool or a component of a predictive “tool box”, data from this study are likely informative. Most notably, as we design further studies or clinical trials, it is worth taking into consideration that, in colorectal carcinoma, high PD1/PD-L1 expression is neither a phenomenon limited to mismatch-repair-deficient tumors nor a finding consistently present in all tumors within the mismatch-repair-deficient group.
References
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509–20.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–91.
Basile D, Garattini SK, Bonotto M, Ongaro E, Casagrande M, Cattaneo M, et al. Immunotherapy for colorectal cancer: where are we heading? Expert Opin Biol Ther 2017;17:709–21.
Le DT, Hubbard-Lucey VM, Morse MA, Heery CR, Dwyer A, Marsilje TH, et al. A Blueprint to Advance Colorectal Cancer Immunotherapies. Cancer Immunol Res 2017;5:942–9.
Brody R, Zhang Y, Ballas M, Siddiqui MK, Gupta P, Barker C, et al. PD-L1 expression in advanced NSCLC: Insights into risk stratification and treatment selection from a systematic literature review. Lung Cancer 2017;112:200–15.
Lee LH, Yantiss RK, Sadot E, Ren B, Calvacanti MS, Hechtman JF, et al. Diagnosing colorectal medullary carcinoma: interobserver variability and clinicopathological implications. Hum Pathol 2017;62:74–82.
Lin F, Shi J, Zhu S, Chen Z, Li A, Chen T, et al. Cadherin-17 and SATB2 are sensitive and specific immunomarkers for medullary carcinoma of the large intestine. Arch Pathol Lab Med 2014;138:1015–26.
Inaguma S, Lasota J, Wang Z, Felisiak-Golabek A, Ikeda H, Miettinen M. Clinicopathologic profile, immunophenotype, and genotype of CD274 (PD-L1)-positive colorectal carcinomas. Mod Pathol 2017;30:278–85.
Best D.J. RD. Algorithm AS 89: The Upper Tail Probabilities of Spearman's Rho. Applied Statistics 1975;24:377–9.
Cleveland WS. LOWESS: A Program for Smoothing Scatterplots by Robust Locally Weighted Regression. The American Statistician 1981;35:54.
Ritprajak P, Azuma M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol 2015;51:221-8.
Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013;3:1355–63.
Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol 2014;25:1935–40.
Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol 2014;75:348–53.
Lee LH, Cavalcanti MS, Segal NH, Hechtman JF, Weiser MR, Smith JJ, et al. Patterns and prognostic relevance of PD-1 and PD-L1 expression in colorectal carcinoma. Mod Pathol 2016;29:1433–42.
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015;5:43–51.
Shia J, Schultz N, Kuk D, Vakiani E, Middha S, Segal NH, et al. Morphological characterization of colorectal cancers in The Cancer Genome Atlas reveals distinct morphology-molecular associations: clinical and biological implications. Mod Pathol 2017;30:599–609.
Gong J, Wang C, Lee PP, Chu P, Fakih M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J Natl Compr Canc Netw 2017;15:142–7.
Wu Y, Chen M, Wu P, Chen C, Xu ZP, Gu W. Increased PD-L1 expression in breast and colon cancer stem cells. Clin Exp Pharmacol Physiol 2017;44:602–4.
Gaule P, Smithy JW, Toki M, Rehman J, Patell-Socha F, Cougot D, et al. A Quantitative Comparison of Antibodies to Programmed Cell Death 1 Ligand 1. JAMA Oncol 2016. https://doi.org/10.1001/jamaoncol.2016.3015
Rimm DL, Han G, Taube JM, Yi ES, Bridge JA, Flieder DB, et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non-Small Cell Lung Cancer. JAMA Oncol 2017;3:1051–8.
Maleki Vareki S, Garrigos C, Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev Oncol Hematol 2017;116:116–24.
Acknowledgements
This study was supported in part by National Cancer Institute grant P30 C008748 and by the Rome Milio Lynch Syndrome Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
ZKS reports spouse (Ophthalmology) consulting for Genentech/Roche, Novartis, Regeneron, Biomarin, Fortress Bio. JFH received honorarium for a lecture from Bayer/ Loxo. NHS has received research funding from Roche/Genentech, Pfizer, Merck, BMS, MedImmune/AstraZeneca, and Incyte. He is on the advisory board/Consulting for Roche/Genentech, Merck, BMS, MedImmune/AstraZeneca, Boehringer Ingelheim, Pfizer, Pieris, PsiOxus, Synlogic, Aduro, Kyn Therapautics, PureTech Ventures, Horizon Pharma, EMD Serono, Gritstone Oncology, Chugai, IFM therapeutics, and Imugene. JJS has received travel support for fellow education from Intuitive Surgical Inc. and is an advisor for Endogenesis, Inc. AC has received research funding from Amgen, Abbivie, and Seattle Genetics, and done consult for Bayer, Purdue Pharma. JGA has received honorarium from Intuitive Surgical and Johnson and Johnson, and consulting fee (or payment) from Medtronics. LBS received a commercial research support from Taiho Pharmaceuticals. LAD is a founder and shareholder of PapGene and Personal Genome Diagnostics (PGDx) and a consultant for Merck, PGDx and Phoremost. The first two of these companies, as well as other companies, have licensed technologies from Johns Hopkins University, on which LAD is an inventor. These licenses and relationships are associated with equity or royalty payments to LAD. He is also a member of the board of directors of PGDx and Jounce Therapeutics. The terms of these arrangements are being managed by Johns Hopkins and Memorial Sloan Kettering in accordance with their conflict of interest policies. In the past 3 years, LAD did consult for one-time engagements for Amgen, Caris, Lyndra, Genocea Biosciences, Genentech, Illumina and Cell Design Labs. DSK is a consultant and equity holder of Paige.AI, and a consultant of Merck. He receives royalties from American Registry of Pathology and UpToDate. JS received travel support for attending an advisory board meeting for BMS, on a topic different from the subject matter of this study. All other authors declare no commercial or financial relationships that could be construed as a potential conflict of interest.
Rights and permissions
About this article

Cite this article
Liu, S., Gӧnen, M., Stadler, Z.K. et al. Cellular localization of PD-L1 expression in mismatch-repair-deficient and proficient colorectal carcinomas. Mod Pathol 32, 110–121 (2019). https://doi.org/10.1038/s41379-018-0114-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0114-7
This article is cited by
-
DNA damage repair and cancer immunotherapy
Genome Instability & Disease (2023)
-
Programmed death-ligand 1 expression in the immune compartment of colon carcinoma
Modern Pathology (2022)
-
Hypermutated phenotype in gliosarcoma of the spinal cord
npj Precision Oncology (2021)
-
Exhaustion in tumor-infiltrating Mucosal-Associated Invariant T (MAIT) cells from colon cancer patients
Cancer Immunology, Immunotherapy (2021)
